
Abstract
Background
Sphaerothecum destruens is an obligate intracellular fish parasite which has been identified as a serious threat to freshwater fishes. Taxonomically, S. destruens belongs to the order Dermocystida within the class Ichthyosporea (formerly referred to as Mesomycetozoea), which sits at the animal-fungal boundary. Mitochondrial DNA (mtDNA) sequences can be valuable genetic markers for species detection and are increasingly used in environmental DNA (eDNA) based species detection. Furthermore, mtDNA sequences can be used in epidemiological studies by informing detection, strain identification and geographical spread.
Methods
We amplified the entire mitochondrial (mt) genome of S. destruens in two overlapping long fragments using primers designed based on the cox1, cob and nad5 partial sequences. The mt-genome architecture of S. destruens was then compared to close relatives to gain insights into its evolution.
Results
The complete mt-genome of Sphaerothecum destruens is 23,939 bp in length and consists of 47 genes including 21 protein-coding genes, 2 rRNA, 22 tRNA and two unidentified open reading frames. The mitochondrial genome of S. destruens is intronless and compact with a few intergenic regions and includes genes that are often missing from animal and fungal mt-genomes, such as, the four ribosomal proteins (small subunit rps13 and 14; large subunit rpl2 and 16), tatC (twin-arginine translocase component C), and ccmC and ccmF (cytochrome c maturation protein ccmC and heme lyase).
Conclusions
We present the first mt-genome of S. destruens which also represents the first mt-genome for the order Dermocystida. The availability of the mt-genome can assist the detection of S. destruens and closely related parasites in eukaryotic diversity surveys using eDNA and assist epidemiological studies by improving molecular detection and tracking the parasite’s spread. Furthermore, as the only representative of the order Dermocystida, its mt-genome can be used in the study of mitochondrial evolution of the unicellular relatives of animals.

Similar content being viewed by others


Background
Introduced parasites can cause significant population declines in susceptible species and generalist parasites in particular, are more likely to be introduced, established and expand their host range [1, 2]. The eukaryotic parasite Sphaerothecum destruens is considered a true generalist [1] that can infect and cause high mortalities in freshwater fish species; including commercially important species such as carp and Atlantic salmon [3, 4]. Sphaerothecum destruens has been recorded in North America [5,6,7], Europe [8,9,10,11,12] and China [10]. Sana et al. [10] provided data to support that S. destruens was introduced to Europe from China along with the accidental introduction of the invasive fish, topmouth gudgeon Pseudorasbora parva. Gozlan et al. [9] has identified P. parva as a reservoir host for S. destruens, i.e. the parasite can be maintained in P. parva and can be transmitted to other fish species whilst not causing disease and mortality in P. parva. Since its introduction to Europe, P. parva has spread to at least 32 countries from its native range in China [13] and S. destruens has been detected in at least 5 introduced P. parva populations [8, 10, 12, 14].
Sphaerothecum destruens is an asexually reproducing intracellular parasite with a direct life-cycle which involves the release of infective spores to the environment through urine and seminal fluids [15]. The spores can survive and release free-living zoospores in the environment at temperatures ranging from 4 °C to 30 °C [16]. The ability for environmental persistence and its generalist nature, places this parasite as a potential risk to fish biodiversity [17]. Thus, efficient detection of this parasite is essential. Molecular detection using the 18S rRNA gene is currently the most efficient detection method compared to traditional histology [18]. However, due to the thickened cell wall of S. destruens, molecular detection in hosts with low parasite numbers can be difficult [15]. Developing more molecular markers such as mitochondrial DNA markers could improve detection, as there are multiple copies of mitochondria per cell (but note that there are also multiple copies of 18S rRNA genes per cell as well). Furthermore, mitochondrial genes are increasingly used for environmental DNA (eDNA)-based metabarcoding detection and so sequencing the mt-genome of this fish parasite could increase its detection in eDNA-based metabarcoding studies.
In addition to the importance of S. destruens as a potential disease risk for freshwater fishes, its taxonomic position is also evolutionarily important, as it belongs to the class Ichthyosporea (formerly referred to as Mesomycetozoea) which sits at the animal-fungal boundary (Fig. 1) [19]. The class Ichthyosporea consists of two orders, Dermocystida and Ichthyophonida with S. destruens grouping within the former [15, 19]. Phylogenomic studies placed S. destruens in a new clade termed as “Teretosporea” comprised of Ichthyosporea and Corallochytrium limacisporum [20]. Teretosporea was found to be the earliest-branching lineage in the Holozoa [20] and so can be used to provide clues into the origins of higher organisms and mtDNA evolution. Ichthyosporea are difficult to culture, therefore genetic information is often scarce. For example, mitochondrial DNA sequences are lacking for all members of the order Dermocystida.

A schematic representation of the phylogenetic position of Sphaerothecum destruens (reconstructed from [19, 20]). Sphaerothecum destruens belongs to the order Dermocystida which belongs to the class Ichthyosporea. Its taxonomic position is between fungi and animals (Metazoa). Due to the lack of mitochondrial genomes in close relatives, the mitochondrial genome of S. destruens was compared to Amoebidium parasiticum (Ichthyophonida), Ministeria vibrans (Filasterea), Capsaspora owczarzaki (Filasterea), Monosiga brevicollis (Choanoflagellatea) and Oscarella carmela (Demospongiae, Metazoa)
Here, we have sequenced and present the first complete mt-genome of a species of the Dermocystida, S. destruens, in order to develop new tools for the parasite’s detection and provide insights into the parasite’s genome architecture evolution.
Methods
DNA extraction and sequencing of Sphaerothecum destruens mitochondrial DNA
The S. destruens spores used were obtained from S. destruens culture in EPC cells [4]. Sphaerothecum destruens reproduces asexually so the cultured spores represent clones of a single organism. The partial 18S rRNA gene from this culture has also been sequenced confirming that this is a culture of S. destruens ([4]; GenBank: MN726743). Total genomic DNA was isolated from S. destruens spores using the DNeasy Blood and tissue kit (Qiagen, Hilden, Germany). All the steps were performed per manufacturer’s guidelines and DNA was eluted in 100 µl elution buffer and quantified using the Nanodrop (Thermo Fisher Scientific, Waltham, USA). A number of universal mtDNA primers for Metazoa and degenerate primers specific for cnidarians were used to amplify short gene fragments of S. destruens mtDNA. The primer pairs were successful in amplifying the short gene fragments of cox1 [21], cob [22] and nad5 [23] of S. destruens mtDNA. The mitochondrial fragments spanning the cob-cox1 and cox1-nad5 were amplified using the primer pairs LR-COB-F (5′-ATG AGG AGG GTT TAG TGT GGA TAA TGC-3′) and LR-COX1-R (5′-GCT CCA GCC AAC AGG TAA GGA TAA TAA C-3′); LR-COX1-R3 (5′-GTT ATT ATC CTT ACC TGT GTT GGC TGG AGC-3′) and LR-NAD5-R1 (5′-CCA TTG CAT CTG GCA ATC AGG TAT GC-3′), respectively, with two long PCR kits; Long range PCR kit (Thermo Fisher Scientific) and LA PCR kit (Takara, Clontech, Kasatsu, Japan). The PCR cycling conditions for the mitochondrial fragments were: cob-cox1: 94 °C for 2 min, 10× (94 °C for 20 s, 58 °C for 30 s, 68 °C for 7 min), 25× (94 °C for 20 s, 58 °C for 30 s, 68 °C for 7 min (increment of 5 s/cycle) 68 °C for 10 min; and cox1–nad5 94 °C for 1 min, 16× (94 °C for 20 s, 60 °C for 20 s, 68 °C for 8 min) 19× (94 °C for 20 s, 60 °C, for 20 s, 68 °C for 8 min) 68 °C for 12 min.
The remaining regions of the mitochondrial genome were amplified with the modified step-out approach [24]. The step-out primer used the primers Step-out3 (5′-AAC AAG CCC ACC AAA ATT TNN NAT A-3′) coupled with the species-specific primers LR-cob-R2 (5′-TCA ACA TGC CCT AAC ATA TTC GGA AC-3′) and LR-nad5-R4 (5′-TGG GGC AAG ATC CTC ATT TGT-3′). The PCR cycling conditions were as follows: 94 °C for 1 min, 1× (94 °C for 20 s, 30 °C for 2 min, 68 °C for 8 min), pause to add species-specific primers, 16× (94 °C for 20 s, 65 °C (decrement of 0.3 °C per cycle) for 20 s, 68 °C for 8 min), 19× (94 °C for 20 s, 60 °C for 20 s, 68 °C for 8 min (increment of 15 s per cycle), 68 °C 12 min. Small DNA fragments of up to 1500 bp were directly sequenced. The long fragments which were 12,986 bp and 7048 bp in length were sequenced by primer walk (Beckman Coulter Genomics, Fullerton, USA).
Gene annotation
Gene annotation of the mitochondrial genome of S. destruens was performed using the automated annotation tool MFannot (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl), followed by visual inspection. Gene annotation was further checked by examining the amino acid sequences of the genes. Genes were translated using the mold, protozoan, and coelenterate mitochondrial code and the mycoplasma/spiroplasma code and aligned with homologous proteins using Clustal W with default options (Gap open cost: 15 and Gap extend cost: 6.66). The 22 tRNA genes were further scanned and secondary structures were generated with MITOS [25]. The annotation for the tatC gene was further checked by predicting its secondary structure and comparing it to the secondary structure of two homologous proteins from Monosiga brevicollis and Oscarella carmela.
tRNA phylogenetic analysis
tRNA replication was further investigated through phylogenetic analysis using the identified tRNAs from S. destruens and the reported tRNAs from its closest relative A. parasiticum (GenBank: AF538045 and AF538046; but note that the two species belong to two different orders). Prior to phylogenetic analysis, all tRNA sequences were modified [24]. Specifically, all tRNA sequences had their anticodon sequence and variable loops deleted and CCA was added to all tRNA sequences in which it was missing. The sequences were then aligned using Muscle in Seaview [25, 26] followed by visual inspection. A neighbour-joining tree was constructed in MegaX [27], using 1000 bootstraps and p-distance to calculate evolutionary distance with pairwise deletion option for a total of 56 sequences (22 from S. destruens and 24 from A. parasiticum (GenBank: AF538045 and AF538046).
Results
Gene content and organization
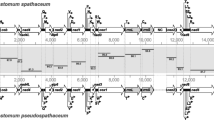
The mitochondrial genome of S. destruens was 23,939 bp long with an overall A+T content of 71.2% (Fig. 1). A list of gene order, gene length, and intergenic spacer regions of S. destruens mtDNA is given in Table 1. The nucleotide composition of the entire S. destruens mtDNA sequences is 40.8% thymine, 31% adenine, 19.7%, guanine and 8.5% cytosine (detailed nucleotide composition is listed in Table 2). It consisted of a total of 47 genes including protein-coding genes (21), rRNA (2) and tRNA (22) and two unidentified open reading frames (ORFs), with all genes encoded by the same strand in the same transcriptional orientation (Fig. 2).

The complete mitochondrial genome for Sphaerothecum destruens. All genes are encoded in the same transcriptional orientation. 22 tRNA genes (pink), 2 rRNA genes (red), 19 protein coding genes (yellow), 2 open reading frames (ORFs) (orange)) and 2 non-coding regions (NCR) (blue) are labelled. Twenty-two transfer RNA genes are designated with single-letter amino acid code: A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; G, glycine; H, histidine; I, isoleucine, K, lysine; L, leucine; M, methionine; N, asparagine; P, proline; R, arginine; S, serine; T, threonine; V, valine; W, tryptophan; Y, tyrosine. Three methionine (M) and two serine (S) and arginine (R) tRNA genes are labelled along with their anticodon sequence
The standard proteins encoded by mitochondria include 13 energy pathway proteins, including subunits 6, 8 and 9 of ATP synthase (atp6, atp8 and atp9), three subunits of cytochrome c oxidase (cox1, cox2 and cox3), apocytochrome b (cob) and NADH dehydrogenase subunits 1–6 and 4L (nad1, nad2, nad3, nad4, nad5, nad-6 and nad4L). Genes involved in mRNA translation were the small and large subunit rRNAs (rrns and rrnl). The S. destruens mtDNA included genes that are usually absent from standard animal and fungal mtDNAs such as four ribosomal proteins (small subunit rps13 and 14; large subunit rpl2 and 16), tatC (twin-arginine translocase component C), ccmC and ccmF (cytochrome c maturation protein ccmC and heme lyase). The mitochondrial genome of S. destruens was intronless and compact with a few intergenic regions. The longest intergenic region was 357 bp and occurred between tatC and nad2. Several neighbouring genes overlapped by 1–46 nucleotides (Table 1, Fig. 2).
The tatC gene (also known as mttB and ymf16) is present in M. brevicollis (Choanoflagellatea) and also reported in only one other animal mt-genome that of O. carmela (sponge) (Table 3; [28, 29]). This protein, a component of twin-arginine translocase (tat) pathway, is involved in the transport of fully folded proteins and enzyme complexes across lipid membrane bilayers and is usually present in prokaryotes, chloroplasts and some mitochondria [30]. The tatC gene in S. destruens is 660 bp long and utilizes GTG as its initiation codon. The derived amino acid sequence of S. destruens tatC is most similar to M. brevicollis tatC (21%) (Choanoflagellatea) followed by Reclinomonas americana (19%) (Jakobid) and O. carmela (16%) (Porifera, Metazoa) (Table 4). Secondary structure analysis using TNHMM [31] indicated that the tatC gene of S. destruens has 6 predicted transmembrane helices at similar locations with the predicted six transmembrane helices for M. brevicollis and O. carmela (Additional file 1: Figure S1). The ccmF protein also known as yejR is involved in Heme c maturation (protein maturation) and ccmC (also known as yejU) plays role in heme delivery (protein import).
Codon usage
Among 21 protein coding genes, 14 genes (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad2, nad3 nad4, nad4l, rps14, rpl16 and ccmC) were inferred to use ATG as initiation codon, 5 genes (nad5, nad6, ccmF, tatC and rps13) used GTG as a start codon and the remaining rpl2 was initiated with TTG. Ten proteins were terminated with the stop codon TAA (atp6, atp8, atp9, cox1, cox2, cox3, nad6, ccmC, rps13, rps14), and nine genes used the stop codon TAG (nad1, nad2, nad3, nad4, nad5, cob, tatC, ccmF and rpl16).
Ribosomal RNA and transfer RNA genes
Genes for the small and large subunits for mitochondrial rRNAs (rrnS and rrnL, respectively) were present. They were separated by four tRNA genes (trnA, trnI, trnM and trnR2). The rrns and rrnl (1369 and 2449 bp) had sizes approximately similar to those in M. brevicollis (1596 and 2878 bp) and A. parasiticum (1385 and 3053 bp). These sizes were comparable to their eubacterial homologs (1542 and 2904 bp in Escherichia coli).
Twenty-two tRNA genes, including three copies of trnM, were identified in S. destruens mtDNA. The tRNA genes had a length range of 71–80 bp and their predicted secondary structures had a clover leaf shape (Fig. 3). Three copies of trnM (methionine, CAT) had the same length (71 bp) and had the same anticodon - CAT. trnM1 was at 1713 bp from trnM2, whereas trnM2 and trnM3 were adjacent (Fig. 2). Two serine and two arginine tRNA genes were differentiated by their anticodon sequence trnS1 (GCT) and trnS2 (TGA), which were 70% similar, and trnR1 (ACG) and trnR2 (TCT) which were 63% similar. All the tRNA secondary structures had a dihydrouridine (DHU) arm, a pseudouridine (TΨC) arm and an anticodon stem, except for trnS1 (GCT) that had an additional short variable loop. The TΨC and D-loop was comprised of 7 and 7–10 nucleotides, respectively (Fig 3).

The predicted secondary structures of 22 tRNAs of Sphaerothecum destruens mitochondrial DNA generated in MITOS [25] The tRNA stands for trnA (transfer RNA alanine), trnL (transfer RNA leucine), trnM1-3 (transfer RNA methionine), trnC (transfer RNA cysteine), trnD (transfer RNA aspartic acid), trnE (transfer RNA glutamic acid), trnG (transfer RNA glycine), trnH (transfer RNA histidine), trnI (transfer RNA isoleucine), trnK (transfer RNA lysine), trnP (transfer RNA proline), trnR1-2 (transfer RNA arginine), trnS1-2 (transfer RNA serine), trnV (transfer RNA valine), trnW (transfer RNA tryptophan), trnY (transfer RNA tyrosine), trnN (transfer RNA asparagine) and trnT (transfer RNA threonine)
Non-coding regions
The total length of the non-coding regions was 842 bp and was comprised of 32 intergenic sequences ranging in size from 1 to 357 bp. Only two intergenic regions had lengths greater than 100 bp: (i) the non-coding region 1 (NCR 1) was 357 bp long and was located between the tatC and nad2 genes; and (ii) the non-coding region 2 (NCR 2) was 117 bp and was located between the trnL and ccmF genes (Fig. 2).
tRNA phylogenetic analysis
The phylogenetic analysis of the tRNAs of S. destruens and A. parasiticum showed that the majority of tRNAs grouped by species with few interspecies grouping (Fig. 4). The phylogenetic results suggest that some of the tRNA genes of S. destruens could have evolved by gene recruitment; these genes were trnV (TAC) and trnL (TAG); indicated by the black arrow in Fig. 4. For A. parasiticum gene recruitment is suggested for trnM, trnI, trnV, trnT and trnA, white arrow in Fig. 4, as already suggested by Lavrov & Lang [32].

Neighbour-joining treed based on pairwise distances among tRNA genes from S. phaerothecum destruens (SD) and Amoebidium parasiticum (AP, AF538045; AF*, AF538046) Nucleotides for anticodons and the variable loops were excluded from the analysis. Portions of the tree discussed in the text are indicated by the black and white arrows. Only bootstrap values above 50 are shown
Discussion
The mt-genome of Sphaerothecum destruens is remarkably compact when compared to other unicellular organisms in similar taxonomic positions and shows the presence of gene overlaps and an absence of both long intergenic regions and repeat sequences. The mt-genome of S. destruens has the highest coding portion, 96.4%, among the unicellular relatives of animals, with other members showing much smaller coding regions, e.g. M. brevicollis (47%) and A. parasiticum (20%). In addition, S. destruens had extensive gene loss especially for ribosomal proteins compared to species within the Filasterea and Choanoflagellatea with only four ribosomal genes in its mitochondrial genome and only 22 tRNAs.
The presence of the tatC in S. destruens represents the first record of this gene within the class Ichthyosporea. TatC has also been reported in M. brevicollis, a choanoflagellate representing the closest unicellular relatives to multicellular animals, and in multicellular animals such as the sponge O. carmella [29]. The tatC gene (also known as ymf16 and mttB) codes for the largest subunit of the twin-arginine transport system pathway and functions in the transport of fully folded proteins and enzyme complexes across membranes [33]. Support for its presence within the S. destruens mt-genome was based on sequence similarity and secondary structure comparisons to homologous proteins in M. brevicollis and O. carmela (Additional file 1: Figure S1). All three homologous tatC proteins have a Met initiation codon; with the tatC from S. destruens and M. brevicolis also having the same amino acids following the initiation codon (Ser and Lys). The overall amino sequence similarity between the tatC in S. destruens and its homologues in M. brevicollis and O. carmella was 21% and 16%, respectively, and all homologous genes had predicted secondary structures encompassing 6 transmembrane domains consistent with their transmembrane localisation.
Ten genes displayed overlapping regions, with these regions ranging from 1 to 46 nucleotides. Similar levels of gene overlaps have been described in other species [34, 35]. The tRNA trnN and rnl genes overlap by 46 nucleotides. The overlap is supported by the percentage similarity between the rnl sequences of S. destruens and M. brevicollis, which is 54% (Table 4). The genes nad3 and tatC overlap by 31 nucleotides and are 44% similar (Table 4). As transcription of the S. destruens mitochondrial genome has not been examined, the transcription mechanisms for these proteins can only be hypothesised. A potential mechanism could be the transcription mechanism described for ATPase subunits in mammalian mitochondrial genomes [36].
The closest relative to S. destruens which has its mt-genome partially sequenced is A. parasiticum which is a member of the order Icthyophonida within the class Ichthyosporea [19]. In contrast to the mt-genome of S. destruens, the mt-genome of A. parasiticum is large (> 200 kbp) and consists of several hundred linear chromosomes [37]. To date, only 65% of the mt-genome of A. parasiticum has been sequenced [37]. In comparison to A. parasiticum, the mt-genome of S. destruens is at least eight times smaller with all genes encoded by a single circular strand in the same transcriptional orientation. There is a remarkable difference in the coding portion of the genomes between both species with only 20% of the mt-genome of A. parasiticum coding for proteins compared to 93% in S. destruens. The mt-genome of S. destruens contains 47 intron-less genes (including two ORFs) while the mt-genome of A. parasiticum intron and gene rich with 44 identified genes and 24 ORFs [37].
Both S. destruens and A. parasiticum use the mitochondrial UGA (stop) codons to specify tryptophan and have multiple copies of the trnM gene. These observed tRNA gene replications are also reported in M. brevicollis, C. owczarzaki and M. vibrans [29, 32, 37]. Similar to M. brevicollis, the mitochondrial tRNAs in S. destruens did not have a truncated D or T loop structure. The trnS of A. parasiticum [28], M. brevicollis [28] and S. destruens does not have a nucleotide at position 8, which connects the aminoacyl and D stems of trnS, and in position 26 there is a pyrimidine (uracil) instead of a purine. The trnS gene in S. destruens also has an adenine instead of uracil in the second nucleotide of its D-loop.
Phylogenetic analysis of the available tRNA sequences of S. destruens and A. parasiticum suggests that some tRNAs of both species could have evolved by gene recruitment. For S. destruens these are trnV and trnL. Gene recruitment is a process by which a gene is recruited from one isoaccepting group to another changing the tRNA identity [32]. Gene recruitment has been previously reported in A. parasiticum for trnM, trnI, and trnV [32]. It is important to note that due to the lack of mitochondrial genomes from close phylogenetic relatives of S. destruens, the results of this phylogenetic analysis are limited and must be interpreted with caution. In S. destruens, trnM1 and trnM3 share a higher nucleotide similarity, 70%, in comparison to trnM2 which is 54% and 63%, respectively. The trnM replication in S. destruens could represent different functions of the methionine tRNAs in protein synthesis and initiation of translation [38]; however, the functional significance remains unknown.
Conclusions
Mitochondrial DNA sequences can be valuable genetic markers for species detection and are increasingly used in eDNA-based species detection. This is the first record of the mt-genome of S. destruens, an important pathogen to freshwater fishes, and the first mt-genome for the order Dermocystida. The availability of this mt-genome should help in the detection of S. destruens and closely related parasites in eukaryotic diversity surveys using eDNA. Due to the abundance of mitochondria within cells, mitochondrial DNA could also be used in epidemiological studies by improving molecular detection and tracking the spread of this parasite across the globe [11]. Furthermore, as the only sequenced representative of the order Dermocystida, its mt-genome can be used in the study of the mitochondrial evolution of the unicellular relatives of animals.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional file. The generated mitochondrial DNA has been submitted to the GenBank database under the accession number MG832660.
Abbreviations
- ccmC:
-
cytochrome c maturation protein
- ccmF:
-
cytochrome c heme lyase subunit
- cob:
-
cytochrome b
- cox1:
-
cytochrome c oxidase subunit 1
- eDNA:
-
environmental deoxy ribonucleic acid
- mtDNA:
-
mitochondrial DNA
- mt-genome:
-
mitochondrial genome
- nad5:
-
NADH dehydrogenase subunit 5
- NCR:
-
non-coding region
- nt:
-
nucleotide
- ORF:
-
open reading frame
- rRNA:
-
ribosomal ribonucleic acid
- rrnl:
-
large subunit ribosomal RNA
- rrns:
-
small subunit ribosomal RNA
- tatC:
-
twin-arginine translocase component C
- trnM:
-
transfer RNA methionine
- trnR:
-
transfer RNA arginine
- trnS:
-
transfer RNA serine
- trnI:
-
transfer RNA isoleucine
- trnV:
-
transfer RNA valine
- trnT:
-
transfer RNA threonine
- trnA:
-
transfer RNA valine
References
Andreou D, Gozlan RE. Associated disease risk from the introduced generalist pathogen Sphaerothecum destruens: management and policy implications. Parasitology. 2016;143:1204–10.
Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–94.
Andreou D, Arkush KD, Guégan JF, Gozlan RE. Introduced pathogens and native freshwater biodiversity: a case study of Sphaerothecum destruens. PLoS ONE. 2012;7:e36998.
Paley R, Andreou D, Bateman K, Feist S. Isolation and culture of Sphaerothecum destruens from Sunbleak (Leucaspius delineatus) in the UK and pathogenicity experiments in Atlantic salmon (Salmo salar). Parasitology. 2012;139:904–14.
Arkush KD, Frasca S, Hedrick RP. Pathology associated with the rosette agent, a systemic protist infecting salmonid fishes. J Aquat Anim Health. 1998;10:1–11.
Harrell LW, Elston RA, Scott TM, Wilkinson MT. A significant new systemic disease of net-pen reared chinook salmon (Oncorhynchus tshawytscha) brood stock. Aquaculture. 1986;55:249–62.
Hedrick R, Friedman C, Modin J. Systemic infection in Atlantic salmon Salmo salar with a Dermocystidium-like species. Dis Aquat Organ. 1989;7:171–7.
Ercan D, Andreou D, Sana S, Öntaş C, Baba E, Top N, et al. Evidence of threat to European economy and biodiversity following the introduction of an alien pathogen on the fungal-animal boundary. Emerg Microbes Infec. 2015;4:e52.
Gozlan RE, St-Hilaire S, Feist SW, Martin P, Kent ML. Biodiversity: disease threat to European fish. Nature. 2005;435:1046.
Sana S, Williams C, Hardouin EA, Blake A, Davison P, Pegg J, et al. Phylogenetic and environmental DNA insights into emerging aquatic parasites: implications for risk management. Int J Parasitol. 2018;48:473–81.
Spikmans F, van Tongeren T, van Alen TA, van der Velde G, den Camp H. High prevalence of the parasite Sphaerothecum destruens in the invasive topmouth gudgeon Pseudorasbora parva in the Netherlands, a potential threat to native freshwater fish. Aquat Invasions. 2013;8:355–60.
Sana S, Hardouin EA, Gozlan RE, Ercan D, Tarkan AS, Zhang T, et al. Origin and invasion of the emerging infectious pathogen Sphaerothecum destruens. Emerg Microbes Infec. 2017;6:e76.
Gozlan RE, Andreou D, Asaeda T, Beyer K, Bouhadad R, Burnard D, et al. Pan-continental invasion of Pseudorasbora parva: towards a better understanding of freshwater fish invasions. Fish Fish. 2010;11:315–40.
Boitard PM, Charrier A, Labrut S, Jamin M. First detection of Sphaerothecum destruens in salmonids in France. Bull Eur Assoc Fish Pat. 2017;37:198–204.
Arkush KD, Mendoza L, Adkison M, Hedrick RP. Observations on the life stages of Sphaerothecum destruens ng, n. sp., a mesomycetozoean fish pathogen formally referred to as the rosette agent. J Eukaryot Microbiol. 2003;50:430–8.
Andreou D, Gozlan R, Paley R. Temperature influence on production and longevity of Sphaerothecum destruens zoospores. J Parasitol. 2009;95:1539–41.
Al-Shorbaji FN, Gozlan RE, Roche B, Robert Britton J, Andreou D. The alternate role of direct and environmental transmission in fungal infectious disease in wildlife: threats for biodiversity conservation. Sci Rep. 2015;5:10368.
Mendonca HL, Arkush KD. Development of PCR-based methods for detection of Sphaerothecum destruens in fish tissues. Dis Aquat Organ. 2004;61:187–97.
Adl SM, Bass D, Lane CE, Lukes J, Schoch CL, Smirnov A, et al. Revisions to the classification, nomenclature, and diversity of eukaryotes. J Eukaryot Microbiol. 2019;66:4–119.
Torruella G, de Mendoza A, Grau-Bové X, Antó M, Chaplin MA, del Campo J, et al. Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr Biol. 2015;25:2404–10.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit 1 from diverse metazoan invertebrates. Mol Mar Biol Biotech. 1994;5:294–9.
Boore JL, Brown WM. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol. 2000;17:87–106.
Lavrov DV, Wang X, Kelly M. Reconstructing ordinal relationships in the Demospongiae using mitochondrial genomic data. Mol Phylogenet Evol. 2008;49:111–24.
Widmann J, Harris JK, Lozupone C, Wolfson A, Knight R. Stable tRNA-based phylogenies using only 76 nucleotides. RNA. 2010;16:1469–77.
Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004;5:113.
Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Burger G, Lavrov DV, Forget L, Lang BF. Sequencing complete mitochondrial and plastid genomes. Nat Protoc. 2007;2:603–14.
Wang X, Lavrov DV. Mitochondrial genome of the homoscleromorph Oscarella carmela (Porifera, Demospongiae) reveals unexpected complexity in the common ancestor of sponges and other animals. Mol Biol Evol. 2007;24:363–73.
Lee PA, Tullman-Ercek D, Georgiou G. The bacterial twin-arginine translocation pathway. Annu Rev Microbiol. 2006;60:373–95.
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Lavrov DV, Lang BF. Mitochondrial genomes in unicellular relatives of animals. In: Wells RD, Bond JS, Klinman J, Masters BSS, Bell E, editors. Molecular life sciences. New York: Springer; 2016. p. 1–4.
Berks BC, Palmer T, Sargent F. The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol. 2003;47:187–254.
Anderson S, Bankier AT, Barrell BG, Debruijn MHL, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65.
Burger G, Plante I, Lonergan KM, Gray MW. The mitochondrial DNA of the amoeboid protozoan Acanthamoeba castellani complete sequence, gene content and genome organization. J Mol Biol. 1995;245:522–37.
Feagin JE. Mitochondrial genome diversity in parasites. Int J Parasitol. 2000;30:371–90.
Burger G, Forget L, Zhu Y, Gray MW, Lang BF. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci USA. 2003;100:892–7.
Popova OV, Mikhailov KV, Nikitin MA, Logacheva MD, Penin AA, Muntyan MS, et al. Mitochondrial genomes of Kinorhyncha: trnM duplication and new gene orders within animals. PLoS One. 2016;11:e0165072.
Lavrov DV, Forget L, Kelly M, Lang BF. Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol Biol Evol. 2005;22:1231–9.
Burger G, Gray MW, Forget L, Lang BF. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol Evol. 2013;5:418–38.
Acknowledgements
We would like to thank two anonymous reviewers for their help in improving this manuscript. We especially thank Dr Dong Zhang for his help with gene annotation.
Funding
This study received financial support from the Fusion Funds of Bournemouth University, Poole, UK.
Author information
Authors and Affiliations
Contributions
SS, DA, EAH designed the study. SS performed all work. SS, DA and EAH performed analyses. SS, DA, EAH, RP and TZ wrote the manuscript. RP provided cultures of Sphaerothecum destruens. All authors commented on the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The project received overall ethical approval by Bournemouth University, Poole, UK (2016, ID: 8905).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Secondary structure analysis and comparison of tatC gene of Sphaerothecum destruens with Monosiga brevicollis and Oscarella carmela usingTNHMM [31].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sana, S., Hardouin, E.A., Paley, R. et al. The complete mitochondrial genome of a parasite at the animal-fungal boundary. Parasites Vectors 13, 81 (2020). https://doi.org/10.1186/s13071-020-3926-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-3926-5