
Abstract
Background
Wolbachia are intracellular bacterial endosymbionts found in most insect lineages. In mosquitoes, the influence of these endosymbionts on host reproduction and arboviral transmission has spurred numerous studies aimed at using Wolbachia infection as a vector control technique. However, there are several knowledge gaps in the literature and little is known about natural Wolbachia infection across species, their transmission modes, or associations between various Wolbachia lineages and their hosts. This study aims to address these gaps by exploring mosquito-Wolbachia associations and their evolutionary implications.
Methods
We conducted tissue-specific polymerase chain reaction screening for Wolbachia infection in the leg, gut and reproductive tissues of wild mosquitoes from Singapore using the Wolbachia surface protein gene (wsp) molecular marker. Mosquito-Wolbachia associations were explored using three methods—tanglegram, distance-based, and event-based methods—and by inferred instances of vertical transmission and host shifts.
Results
Adult mosquitoes (271 specimens) representing 14 genera and 40 species were screened for Wolbachia. Overall, 21 species (51.2%) were found positive for Wolbachia, including five in the genus Aedes and five in the genus Culex. To our knowledge, Wolbachia infections have not been previously reported in seven of these 21 species: Aedes nr. fumidus, Aedes annandalei, Uranotaenia obscura, Uranotaenia trilineata, Verrallina butleri, Verrallina sp. and Zeugnomyia gracilis. Wolbachia were predominantly detected in the reproductive tissues, which is an indication of vertical transmission. However, Wolbachia infection rates varied widely within a mosquito host species. There was no clear signal of cophylogeny between the mosquito hosts and the 12 putative Wolbachia strains observed in this study. Host shift events were also observed.
Conclusions
Our results suggest that the mosquito-Wolbachia relationship is complex and that combinations of transmission modes and multiple evolutionary events likely explain the observed distribution of Wolbachia diversity across mosquito hosts. These findings have implications for a better understanding of the diversity and ecology of Wolbachia and for their utility as biocontrol agents.
Graphical Abstract

Similar content being viewed by others


Background
Wolbachia are intracellular endosymbiotic bacteria that alter host reproduction [1]. They are widespread in arthropods, infecting a wide range of insect, crustacean, and nematode species [2, 3]. In some cases, Wolbachia exist in a mutualistic relationship with their hosts [4,5,6]. However, Wolbachia are most often recognised as reproductive manipulators that bias the sex ratio of the host offspring towards the production of more infected females [7, 8]. This reproductive manipulation is commonly achieved through four phenotypes—male killing [9], feminisation [10, 11], parthenogenesis [12, 13], and cytoplasmic incompatibility [14, 15]—which increase the endosymbiont’s reproductive success [16]. Owing to their strong influence on host reproduction, an increasing amount of research is being dedicated to exploring the impacts of reproductive endosymbionts on host population dynamics and evolution [17, 18], especially in medically important insects such as mosquitoes. The promising use of Wolbachia to alter both mosquito reproduction [19] and arboviral transmission [20] has prompted the deployment of novel Wolbachia-infected mosquitoes for population replacement and suppression [21].
Several countries, including Singapore, have started to employ Wolbachia as biocontrol agents of mosquitoes by releasing infected mosquitoes [22,23,24]. However, the presence of naturally occurring endosymbionts in wild mosquito populations has not been adequately assessed. The release of mosquitoes artificially infected with Wolbachia might have a profound impact on closely interacting wild mosquito populations through various transmission modes. For instance, horizontal transmission of an introduced Wolbachia strain may result in manipulation of the reproductive biology of non-target species, which could potentially result in an unintentional population crash, opening up niches for other vector species [25]. Another possible effect of this type of biocontrol method is the increased likelihood of co-infections with other naturally occurring Wolbachia strains or other endosymbionts, such as Cardinium, Rickettsia, and Spiroplasma. These co-infections may result in a synergistic effect on mosquito host fitness and future transmission of endosymbionts [26,27,28,29]. Without a detailed characterisation of Wolbachia prevalence and diversity among wild mosquitoes, the ecological risk of releasing artificially infected mosquitoes might be overlooked. Therefore, bearing the precautionary principle in mind, it is important to investigate the natural occurrences of Wolbachia.
There is also a need to discern the main mode of infection transmission among mosquitoes. Although Wolbachia are mainly thought to be vertically transmitted [15, 30], there have been accounts of horizontal transmissions into wild populations through parasitism [31, 32], or through proximity to infected individuals [33]. Wolbachia may not be strictly localised in germline tissues, as they have also been detected in somatic tissues such as the gastrointestinal tract and haemolymph [34,35,36]. The detection of Wolbachia in the gastrointestinal tract suggests that they could be horizontally transmitted through uptake from the environment or host sharing [34, 37, 38], whereas their detection in non-gastrointestinal somatic tissues, such as those of jointed appendages, could indicate horizontal bacterial genome integration into the host genome [36]. Currently, detection of Wolbachia in mosquitoes is mostly achieved through conventional polymerase chain reaction (PCR) methods using DNA extracted from an entire individual or its abdomen [39,40,41,42,43,44,45,46,47]. This limits our ability to identify the site of endosymbiont infection within an individual (tissue tropism). Tissue-specific screening of Wolbachia is necessary to provide insights and infer the extent of vertical and horizontal transmission.
It has been proposed that host mitochondrial DNA (mtDNA) and Wolbachia are maternally co-transmitted within the cytoplasm [17, 48], which suggests a congruency between host mtDNA and Wolbachia phylogenies—a consequence of cytoplasmic hitchhiking driven by endosymbiont transmission [17]. In insect systems such as bedbugs where vertical transmission has been established to be the main mode of transmission, Wolbachia exhibit clear patterns of cophylogeny with their hosts, with few instances of host shifting or multiple infections within a single host species [49, 50]. In contrast, cophylogeny is not apparent among nematodes and bees, and numerous acquisitions of Wolbachia infections through horizontal transmission as well as losses have been shown in these diversified host lineages [51, 52]. The modes of Wolbachia transmission among mosquitoes have not been well established, nor has the extent of multiple infections within mosquito hosts or host shifting of these bacteria.
There is presently no comprehensive analysis of the evolutionary associations between Wolbachia and their mosquito host species. An understanding of host-endosymbiont associations will not only further our ability to discern the mode of transmission which influences Wolbachia diversity, but will also allow for an evaluation of Wolbachia host specificity, speciation, and their ability to establish in new hosts. All of this is key to understanding the diversity and ecology of Wolbachia, and their utility in biocontrol methods.
This study has three major research objectives. First, to examine the prevalence and diversity of Wolbachia among wild mosquitoes from Singapore. Second, to determine the tissue tropism of Wolbachia infection in mosquitoes using a tissue-specific PCR screening method. Finally, to reconstruct the evolutionary associations between Wolbachia and their mosquito hosts to provide a basis for an understanding of host-endosymbiont evolution.
Methods
Adult mosquito collection and identification
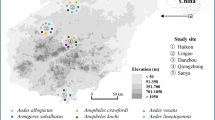
Mosquito samples were collected from 12 localities across Singapore between March 2018 and November 2019 (Fig. 1a). Three methods were employed to collect the samples: CO2-baited Centers for Disease Control and Prevention traps, sweep-netting using hand-held fan traps, and larval sampling [53]. For the latter, dipping was carried out at streams and ponds and pipettes were used to collect larvae from various microhabitats, including tree holes, plant axils, and artificial containers. Thereafter, the field-collected larvae were reared to adults in an incubator maintained at 26 °C and 70% relative humidity, under a 12:12-h (day:night) photoperiod. Larvae were fed with pulverised fish food (TetraMin Granules) daily. Mosquitoes were identified using relevant taxonomic keys and descriptions [54,55,56,57,58,59]. A subset of individuals from commonly sampled species was selected and preserved in phosphate-buffered saline solution at – 80 °C for subsequent dissection step.

Map of sampling sites and diagrammatic images of Aedes aegypti and its dissected tissues. a Various mosquito collection localities across Singapore and their respective coordinates, b mosquito leg, c gut, d female reproductive tissue (left) and male reproductive tissue (right)
Tissue-specific dissection
Tissue-specific dissection was carried out on each adult mosquito sample to isolate the leg, gut, and reproductive tissues (Fig. 1b–d). To prevent the contamination of tissues with bacteria on the external surface of the mosquito, the leg was removed first before isolating the gut and reproductive tissues. All dissection equipment and microscope slides were thoroughly wiped with 70% ethanol before commencing dissection of the next sample. Dissected tissues were individually placed into a 96-well plate on ice to prevent DNA degradation.
DNA extraction, PCR amplification, and sequencing
DNA extraction of each dissected tissue was performed using 7 μl of QuickExtract DNA Extraction Solution (Lucigen, Madison, USA) in a thermocycler (Eppendorf, Hamburg, Germany) with the following protocol: 65 °C for 18 min, followed by 98 °C for 2 min, ending with cooling on ice for at least 10 min. All dissected tissues were screened for Wolbachia infections following single-primer PCR protocols described by Martin et al. [26] with slight modifications to the cycle conditions. The Wolbachia surface protein gene (wsp) general primers, wsp81F (5′-TGGTCCAATAAGTGATGAAGAAACTAGCT-3′) and wsp691R (5′-AAAAATTAAACGCTACTCCAGCTTCTGCAC-3′), were used in this study [60]. In addition, a fragment of the cytochrome c oxidase subunit I (cox1) gene of the mosquito hosts was also amplified using primers LCO1498 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) [61]. This served to confirm host identity and acted as an internal control. We used DNA from known Wolbachia-infected Nasonia specimens as positive controls for this study.
All PCR procedures were performed in reaction mixtures consisting of 12.5 μl of GoTaq G2 Green Mastermix (Promega, Madison, USA), 1 μl of 1 mg ml−1 bovine serum albumin, 0.184 μl of 25 mM magnesium chloride, 1.5 μl of extracted DNA, and 1.5 μl each of 5 μM wsp forward and reverse primers for Wolbachia PCR screens or 1.0 μl each of 5 μM LCO1498 and HCO2198 primers for cox1 PCRs. Double-distilled water was used to top up the reaction mixture to a final volume of 25 μl. PCR amplification of positive and negative controls was also conducted simultaneously.
PCR conditions were as follow: 94 °C for 5 min, followed by 35 cycles of 95 °C for 30s, 55 °C for 45s, and 72 °C for 1 min, with a final elongation step of 72 °C for 10 min. Amplicons were separated by gel electrophoresis on 2% agarose gel stained with GelRed (Biotium, Fremont, USA) and visualised under a ultraviolet transilluminator (Syngene, Cambridge, UK). PCR products were purified using SureClean Plus (Bioline, London, UK) following the manufacturer's protocol. Samples were sequenced by First Base Laboratories (Axil Scientific, Singapore), using a 3730XL DNA Analyzer (Applied Biosystems, Waltham, USA). Obtained sequences were edited and aligned using Geneious Prime (version 2019.2.3) (https://geneious.com). Similarities with publicly available sequences were assessed using the Basic Local Alignment Search Tool (BLAST) [62].
Statistical analyses
To test if there were significant differences in Wolbachia infection across the different mosquito tissues, Cochran’s Q-test was carried out. As a follow-up, McNemar’s post hoc test was employed to identify the tissue pairs that differed significantly in infection. Individuals for which the internal control (cox1 gene) was not amplified successfully for any of the three dissected tissues were excluded from this statistical analysis. The effect of sex on host infection was also tested using binary logistics regression with sex as a categorical dependent variable and infection outcome as a binary independent variable. Logistic regression was conducted on a subset that only included species that had a roughly similar representation of both sexes, i.e. for every species included, the number of individuals of the less common sex was proportionally at least 60% of the number of individuals of the more common sex. This was to prevent a biased analysis due to a dataset with unequal representation of the sexes. Statistical significance was determined as P < 0.05. All statistical analyses were performed in R version 3.6.2 [63] with packages nonpar [64], rcompanion [65], and ISLR [66].
Sequence analyses
Multiple alignment of consensus sequences was carried out using the ClustalW algorithm with default settings (gap penalty = 15, gap extension penalty = 6.66) [67], in software MEGA X [68]. Mosquito cox1 sequences generated in this study were aligned with 61 reference cox1 barcodes of identified local mosquitoes from Chan et al. [53]. For wsp sequences, the generated sequences were aligned with 54 available wsp sequences of known Wolbachia strains obtained from GenBank [69]. Short sequence reads (< 500 base pairs) were excluded.
Neighbour-joining (NJ) phylogenetic trees for mosquito hosts and Wolbachia were reconstructed using the sequenced cox1 gene fragment and the wsp gene, respectively. cox1 sequences from previous publications were not included because a comparison of the genetic relationships between the hosts was not the aim of this research. Instead, 54 wsp sequences from GenBank were included in the construction of the Wolbachia NJ tree. The NJ tree reconstruction was performed with the Kimura two-parameter model as the nucleotide substitution model in MEGA X [68]. Internal gaps were treated as indels and terminal gaps as missing for wsp sequences. Bootstrap probabilities were estimated by generating 1000 bootstrap replicates. We designated two biting midge species, Culicoides asiana (KJ162955.1) and Culicoides wadai (KT352425.1), as outgroups for the host NJ tree construction. Due to the lack of an appropriate endosymbiont outgroup [51], the Wolbachia NJ tree was midpoint rooted.
When possible, Wolbachia strains were classified into supergroups and putative strains using 97% bootstrap probability as a threshold [60]. Wolbachia surface protein sequences that did not have 97% bootstrap support were evaluated on a case-by-case basis. For example, sequences which clustered closely together and had a relatively high support value (> 90%) were deemed as originating from the same putative strain.
Putative strains which were infectious to only one host species were categorized as ‘specialists’ and those which infected two or more hosts as ‘generalists’. Then, the standardised phylogenetic host specificity (SPS) score of each generalist strain was calculated by adapting the method outlined by Poulin et al. [70] and Kembel et al. [71]. SPS measures the degree of phylogenetic relatedness among host species infected by the same endosymbiont strain. It also tests for significance by comparing it with null models generated with 999 replicates of random host-endosymbiont associations. A positive SPS value with a high P-value (P > 0.95) indicates a high degree of host flexibility where Wolbachia infect hosts which are phylogenetically even. A negative SPS value with low P-value (P < 0.05) suggests a low degree of host flexibility where the infected hosts are phylogenetically clustered together. SPS scores were calculated using R package picante [71].
Evolutionary analyses of the mosquito-Wolbachia relationship
Three distinct methods were used to explore the evolutionary associations between mosquito hosts and their Wolbachia endosymbionts. The analyses were carried out using pruned phylogenies where each species is represented by a single individual.
First, using the software TreeMap 3.0 [72], a tanglegram was created between host and endosymbiont NJ trees to visualise mosquito-Wolbachia associations. A tanglegram is useful as a pictorial representation of the interactions between two phylogenies [73]. TreeMap also seeks to minimise the entanglement between the two trees to provide a clearer visualisation of the phylogenetic relationship between host and endosymbiont [72].
Second, ParaFit Global test, a distance-based method, was employed to quantitatively estimate congruence between the host and endosymbiont phylogenetic trees by comparing genetic distances among infected host species and the Wolbachia strains [74]. The null hypothesis for this test states that the associations between host and endosymbiont trees are random, whereas the alternative hypothesis suggests that there are strong associations between hosts and parasites, which are indicated by phylogenetic distances. Significance was tested by comparing the observed associations between host and endosymbiont with randomised associations generated with 5000 permutations. The respective host-endosymbiont associations which contributed significantly to the ParaFit Global statistics were also identified by performing a Parafit Link test. ParaFit tests were performed with the Cailliez correction to correct for negative eigenvalues generated [75] using R package ape [76].
Third, an event-based analysis was performed in Jane 4.0 [77] to map out potential evolutionary events of the endosymbiont in relation to the host phylogeny [78]. Five evolutionary events were considered: co-speciation (host and endosymbiont speciate simultaneously), duplication (intra-host speciation), duplication with host shift (endosymbiont host shifts), loss (host speciates but endosymbiont fails to establish in one of the new lineages), failure to diverge (host speciates and endosymbiont remains in both lineages). As each event is expected to have differing likelihoods, default cost values were attached to each of the events. Jane 4.0 determined the best reconstruction of evolutionary events by minimising the overall cost. The following cost-scheme regime was used with 100 generations and a population size of 300: co-speciation = 0, duplication = 1, duplication with host shift = 2, loss = 1, and failure to diverge = 1 [79]. As a follow-up, random tip mapping (randomisation of host-endosymbiont associations) was carried out for 50 iterations, to determine if the overall cost of reconstruction was significantly lower than expected by chance. If 5% or fewer of the random solutions have costs lower than the reconstructed coevolution phylogeny, there is support for the coevolution of the hosts and endosymbionts through co-speciation.
Results
Prevalence of Wolbachia in wild-caught mosquitoes
A total of 271 adult mosquitoes, representing 40 species and 14 genera, were collected from 12 localities in Singapore (Fig. 1a). Overall, infection prevalence was moderate with 119 out of 271 (43.9%) individuals screening positive for Wolbachia (Table 1). In total, 21 (51.2%) species were positive for Wolbachia. According to our knowledge, Wolbachia infection in seven of these species is reported here for the first time (Table 1). Wolbachia were detected in all genera except for Aedeomyia, Anopheles and Mimomyia (i.e. 11 out of 14 genera; 78.6%). Five out of the seven Aedes species collected (71.4%) were positive for Wolbachia, while in the genus Culex, five out of 16 species (31.3%) were positive. Some of the screened species in the genera Aedes and Culex that were positive for Wolbachia, such as Aedes albopictus and Culex quinquefasciatus, are medically important vector species.
The infection rates varied across the mosquito species. Notably, there was variation in the percentage of infection between species that are epidemiologically related. For instance, Wolbachia infection was not detected in Aedes aegypti. However, infection was moderately high (56.8%) for Aedes albopictus. There was also a difference in the infection rate of two closely related species, Culex pseudovishnui (86.4%) and Culex vishnui (0%) [53].
Locality did not seem to play a role in the Wolbachia infection of mosquito hosts. Among species that have a wide range across Singapore, the percentage of infection was consistent in populations across different habitats. For example, the infection percentage was consistently high for Cx. pseudovishnui, while consistently low for Malaya genurostris. Based on our results, species identity was a better predictor of infection status than locality.
Based on a data subset containing 153 individuals (45.8% males) representing 12 mosquito species, sex was a significant explanatory variable, and there was a significantly lower infection prevalence in males than females (odds ratio 0.434; binary logistics regression: Z = – 2.48, df = 151, P = 0.013).
Tissue tropism of Wolbachia infection in mosquitoes
Among the 159 successfully amplified cox1 sequences, Wolbachia infection was mainly observed in the reproductive tissues. Among the reproductive tissues of 159 dissected individuals, 42.1% (n = 67) were infected. Percentage infection was lower in the gut (5.7%, n = 9) and leg (3.1%, n = 5) tissues. The difference in percentage infection across the three dissected tissues was statistically significant (Cochran’s Q-test: Q = 109.5, df = 2, P < 0.0001). The percentage of infection in the reproductive tissues was significantly higher than in the gut (McNemar’s post hoc test: P < 0.0001) and leg tissues (McNemar’s post hoc test: P < 0.0001), but the difference in percentage of infection between the gut and leg tissues was not significant (McNemar’s post hoc test: P = 1.0). Notably, the amplicon size of wsp in the gut and leg tissues tended to be shorter than 400 base pairs.
Wolbachia diversity among mosquito fauna from Singapore
Following Zhou et al. [60], all wsp sequences obtained in this study can be broadly classified into A and B Wolbachia supergroups. Out of 21 infected species, six were infected with supergroup A, ten with supergroup B, and one species, Ae. albopictus, was infected with both supergroups (Fig. 2). Infection of the remaining four species (Culex tritaeniorhynchus, Tripteroides sp., Verrallina butleri, and Verrallina sp.) was unclassified due to short sequences (< 400 base pairs) or sequence alignment issues during sequences analyses. The analysed wsp sequences were also clustered into 12 putative strains: ‘Wol 1’ to ‘Wol 12’. Four (Wol 1, Wol 2, Wol 3, and Wol 8) out of the 12 putative strains could be matched to previously typed strains [60, 80]. Wolbachia strains from this study are also closely related to those isolated from other insect groups (Fig. 2). For instance, Wol 9 and Wol 10 are closely related to the Wolbachia strains harboured by Drosophila spp. (bootstrap value > 99%).

Wolbachia neighbour-joining (NJ) tree constructed with the Wolbachia surface protein gene (wsp). All analysed sequences generated from this study (bold) were broadly classified into Wolbachia supergroups A or B and clustered into 12 putative strains (‘Wol 1’–‘Wol 12’). The number of sequences of each putative strain is indicated within parentheses. Also included are 54 sequences obtained from GenBank. Taxa are labelled as the host from which the Wolbachia strain was isolated, followed by the strain name. The NJ tree was mid rooted due to a lack of appropriate outgroups [45]. Bootstrap probability (generated with 1000 replicates) higher than 50% is indicated on the tree. Genbank accession number of each sequence is indicated within brackets
Host specificity of Wolbachia strains
The degree of host specificity varied across the 12 putative strains. Seven out of the 12 strains (Wol 2, Wol 4, Wol 5, Wol 6, Wol 8, Wol 10, and Wol 12) were considered as specialists. These strains were host specific and were only detected in one host species each (Fig. 3). The remaining five strains were considered as generalists as they were found in more than one host. Amongst the generalists, Wol 3 was found in the highest number of host species, i.e. three, Coquillettidia crassipes, Mansonia indiana, and Culex sitiens. The SPS scores revealed that Wol 1 had the lowest degree of host flexibility (SPS test: Z = – 1.41, P = 0.049). Wol 7 had the highest degree of host flexibility, but this was not statistically significant (SPS test: Z = 0.07, P = 0.779) (Table 2).

Tanglegram of mosquito cox1 NJ tree compared to the Wolbachia endosymbiont NJ tree. Mosquito host species that harboured Wolbachia infection are indicated in red. Specialist Wolbachia strains are in bold. Grey lines represent the associations between hosts and endosymbionts. A red line indicates the host-endosymbiont association that was significant in the Global ParaFit test of congruence between host and endosymbiont phylogenies (Parafit Link test: ParaFit Link = 0.045, P = 0.029)
Evolutionary relationship between mosquitoes and Wolbachia
We recorded 18 counts of mosquito-Wolbachia associations in wild-caught mosquitoes from Singapore. A visualisation of these associations using a tanglegram showed patterns of broad associations (Fig. 3). For instance, the clade which consists of Aedes species was observed to be mostly associated with Wolbachia supergroup A. In contrast, other species, especially the clade representing various Culex species, had numerous associations with Wolbachia supergroup B.
The distance-based quantitative test showed that mosquito and Wolbachia phylogenies were weakly congruent at the global level (ParaFit Global test: ParaFit Global = 0.006, P = 0.048). Among the numerous host-endosymbiont links, only the association between Mansonia indiana and Wol 3 was statistically significant (ParaFit Link test: ParaFit Link = 0.045, P = 0.029) (Fig. 3).
The event-based analysis between mosquito and Wolbachia phylogenies resulted in a reconstructed output of one co-speciation event, three counts of duplication, seven counts of duplication with host shift, 29 losses, and six counts of failure to diverge, amounting to a total cost of 52 (Fig. 4). Interestingly, the number of duplications with a host shift and losses was much greater than co-speciation events. Notably, multiple host shift events tend to follow after loss events occurring earlier in the evolutionary history of the endosymbiont. For example, we see instances of consecutive host shifts to new hosts that were not previously infected (Fig. 4, red arrows). Additionally, based on random tip mapping, 14% of the random solutions had lower costs than the reconstructed output. Overall, there was support for multiple host shift events and losses of Wolbachia among the mosquitoes, and no clear signal for mosquito-Wolbachia cophylogeny.

Least-cost evolutionary reconstruction between mosquito (black) and Wolbachia (blue) phylogenies achieved using Jane 4.0. In total, one co-speciation event (open circle), three counts of duplication (grey dot), seven counts of duplication with host shift (black dot with an arrow pointing outwards), 29 losses (dotted line), and six counts of failure to diverge (squiggly line) were mapped out. Red arrows indicate periods where multiple host shifts occurred in succession
Discussion
Detection of Wolbachia infection and distribution in wild mosquitoes
In this study, the PCR-based Wolbachia screening method had a high positive detection rate with 86.3% of all sequenced amplicons having successful BLAST matches to Wolbachia. This suggests that the conventional PCR method used is adequate for Wolbachia detection. Even if the study had been carried out without the additional DNA sequencing step, observed amplicon bands would likely have indicated true positives.
Our results indicate that Wolbachia are widespread across members of the family Culicidae. To our knowledge, Wolbachia infections have not been previously reported in seven of the mosquito species collected in this study. Overall, the percentage infection of screened individuals was 43.9%, which was largely congruent with percentages reported in past studies from the Oriental region, i.e. 31% infection in Malaysia [81], 26.4% in Sri Lanka [39], and 61.6% in Thailand [82]. At the species level, previous studies reported Wolbachia infection in 40% of all tested mosquito species in India [83], 18.2% in Sri Lanka [39], 51.7% in Taiwan [84], and between 28.1% and 37.8% in Thailand [82, 85]. Our study showed that 51.2% of all tested species were infected with Wolbachia, which is generally higher than the percentage reported in most studies. This was likely due to the broad range of species tested, including those from the genera Malaya, Verrallina, and Zeugnomyia [85]. It is also possible that infection prevalence may vary across geographical regions.
Wolbachia detection in three medically important mosquito genera, Culex, Anopheles, and Aedes, was highly consistent with that of past studies. These genera are responsible for the transmission of vector-borne diseases such as filariasis, malaria and arboviral diseases [86]. Within the genus Culex, Wolbachia infection has been reported to be variable across its member species [39, 46, 82, 84]. In this study, infections were observed only in five out of 16 Culex species. We noticed moderately high Wolbachia infection in Cx. quinquefasciatus (62.5%), which is a member of the Culex pipiens complex responsible for the transmission of filariasis in Singapore [86, 87]. Surprisingly, no Wolbachia infection was observed in Cx. vishnui–which has been found to harbour Japanese encephalitis virus in Southeast Asia [89]–although it is closely related to Cx. pseudovishnui [88] in which the rate of Wolbachia infection was high. However, studies in India and Thailand showed a reverse pattern, with Wolbachia infection present in Cx. vishnui but not in Cx. pseudovishnui [39, 85]. As the two species are morphologically similar [53], DNA barcoding was conducted to aid morphological identification, and thus avoid any misidentification. The results lend further support to possible variation in infection prevalence between geographically distant populations.
We did not detect Wolbachia in any of the wild-caught Anopheles species (18 individuals representing three species), many of which are potential malaria vectors [86]. This is largely consistent with previous reports from different countries [39, 90, 91]. The absence of Wolbachia in Anopheles mosquitoes is thought to be due to the unsuitability of Anopheles reproductive tissues for Wolbachia establishment [84, 85]. However, there have been recent reports of Wolbachia detected in wild Anopheles mosquitoes from West Africa [42, 92, 93] and Malaysia [94]. Knowledge of natural Wolbachia infections in Anopheles mosquitoes is important for malaria control strategies [93], hence more wild-caught Anopheles samples should be screened in Singapore to determine more accurately their infection status.
Wolbachia were not detected in Ae. aegypti, the primary vector of dengue in Southeast Asia [87]. Conversely, Wolbachia infection was moderately high in the secondary vector Ae. albopictus. These results are highly consistent with those of past studies, which reported an absence of infection in wild Ae. aegypti [21, 95], but found stable infection in wild Ae. albopictus [96]. Although Ae. aegypti and Ae. albopictus belong to the same subgenus, Stegomyia, and occupy similar ecological niches [97], they are rarely found in the same locality, [43, 98, 99], as also observed in this study. This could imply a certain degree of competitive exclusion between the two species, preventing them from occupying the same space. There is evidence that symbionts may influence a host’s resource acquisition and specificity, which may ultimately lead to competitive exclusion between closely related host species with differing symbiont infections [100, 101]. However, research on Wolbachia-induced competitive exclusion is scarce except for a few studies on heterogonic gall wasps [102], grasshoppers [103], and gall-inducing aphids [104]. Given the widespread influence of Wolbachia, future research should explore potential cases of Wolbachia-induced competitive exclusion between closely related species of mosquitoes as this may have major implications for an understanding of their symbioses and speciation.
Additionally, although Ae. aegypti is frequently artificially infected with Wolbachia for biocontrol purposes [105,106,107,108,109], our findings suggest that infected Ae. aegypti might not be stably maintained in the wild. This may be advantageous for vector population suppression as the cytoplasmic-incompatibility effect of any artificially introduced Wolbachia strain will likely be fully manifested in the uninfected native population [21]. However, this also implies that this type of biocontrol method may have low long-term effectiveness if the infection cannot be naturally sustained in the wild population. The detection of natural Wolbachia infection in wild Ae. aegypti, therefore, has huge implications for vector control programmes [21]. Not only does it inform the selection of a suitable Wolbachia strain prior to its field release, but it can also be used to gauge the long-term effectiveness of a specific vector control programme.
Interestingly, the sex of the mosquitoes had an effect on their Wolbachia infection status. This could be an artefact of various Wolbachia-induced reproductive phenotypes, such as parthenogenetic and male-killing ones, resulting in offspring that are largely female [15]. If this were true, over multiple generations with vertical Wolbachia transmission, one should observe an increasing proportion of females that are infected. Hence, the phenomenon observed here could be a consequence of reproductive manipulation by Wolbachia and vertical transmission.
While we were unable to statistically test for the effects of locality on infection status due to uneven and small sample sizes of the respective species across different localities, our results suggest that mosquitoes found in localities across Singapore have roughly equal chances of harbouring Wolbachia. This also suggests that underlying physiological factors and phylogenetic relatedness in mosquitoes contribute more to their infection by Wolbachia than the habitat in which they are found.
The reproductive effect of Wolbachia can be masked or enhanced by other reproductive endosymbionts such as Cardinium, Rickettsia, and Spiroplasma [7, 26,27,28,29]. Unfortunately, we were unable to detect these endosymbionts due to a high degree of false positives with the PCR-based screening methods used here (Additional file 1). This was likely due to using primers that are not optimised for screening mosquito-specific endosymbionts [110,111,112]. As a result, co-infections with various reproductive endosymbionts, which would have provided greater insights into the synergistic effects of co-infections on mosquito evolution, could not be identified among the wild mosquitoes examined here. There is, hence, a need to develop and optimise alternative screening methods, such as multilocus sequence typing (MLST) techniques, especially for the detection of Cardinium, Rickettsia, and Spiroplasma in mosquitoes.
Tissue tropism of Wolbachia infection in mosquitoes
Wolbachia were detected mainly in the reproductive tissues, which agrees with results from studies across multiple insect groups [15, 84, 113], and suggests that Wolbachia are mainly vertically transmitted. Interestingly, through the course of this study, there was significant variation in reproductive traits (testis and ovary length) across and within species. These reproductive traits did not vary significantly with Wolbachia infection status, even after accounting for phylogenetic relatedness (see Additional file 2).
Infection in the gut and leg tissues was detected, albeit infrequently. This is not surprising, as previous studies have also detected Wolbachia in those tissues [34,35,36, 114]. Interestingly, the nucleotide sequences from gut and leg infections tend to be shorter in length. Considering that Wolbachia are unlikely to survive extracellularly for a long period of time [35], the small amplicon size suggests potential horizontal integration of the Wolbachia genome into the host genome for a few species. This phenomenon has been observed in several Wolbachia hosts [115, 116], and mosquito species such as Ae. aegypti and Cx. quinquefasciatus [117, 118]. A recent study showed that horizontal integration of the Wolbachia genome into the host genome can have implications for sex determination and evolution. This is evident in the common pillbug Armadillidium vulgare, and results in the formation of a new sex chromosome [119]. Researchers have also proposed that horizontal gene transfer between an endosymbiont and host can result in evolutionary innovation where new functional genes arise in both host and bacteria [117, 118].
Future research should explore the relative importance of each transmission method with relation to host-endosymbiont ecology and evolution. Tissue-specific screening methods such as those used here can be used in other arthropods, especially when the mode of transmission is not clear. Currently, most Wolbachia screening is conducted on ground specimens or specimens in their entirety [39,40,41]. In these cases, researchers are unable to determine tissue tropism of Wolbachia infection, which could provide clues to its mode of transmission. Thus, adopting tissue-specific screening methods would enable researchers to verify or refute the commonly reported assumption that Wolbachia is transmitted vertically [15, 30].
Diversity and host-specificity of Wolbachia strains
Not only does the wsp molecular marker allow successful detection of Wolbachia infection across numerous taxa, it also enables strain genotyping and evolutionary comparison between detected Wolbachia strains [60]. In this study, Wolbachia wsp sequences were clustered into 12 putative Wolbachia strains falling within supergroup A or B. This is consistent with the results of previous studies that looked at Wolbachia infections in mosquitoes [39, 80, 85]. Each mosquito host species was only infected by strains belonging to supergroups A or B, with the exception of Ae. albopictus, which harboured both. Infection with more than one strain (superinfection of wild Ae. albopictus with Wolbachia supergroups A and B) has been previously reported, and this phenomenon was commonly observed to be fixed in the examined populations due to strong cytoplasmic incompatibility effects [120, 121]. This suggests stable vertical transmission of both strains in Ae. albopictus. Additionally, only four out of 12 putative strains were identified as previously typed Wolbachia strains reported by Zhou et al. [60] and Ruang-Areerate et al. [80]—Wol 1, Wol 2, Wol 3, and Wol 8 were identified as wPip, wAlbB, wCra, and wRi strain, respectively.
Host specificity is thought to be a characteristic of the ancestral Wolbachia strain, with host flexibility reported mainly in Wolbachia supergroups A and B [122]. In our study, we found a combination of specialists and generalists, with more of the former. A study of mosquitoes from Taiwan showed a similar pattern [84]. In beetles, a mixture of Wolbachia supergroup A host-specific and host-flexible strains within a population has also been reported [49]. While our estimates of specialists and generalists might vary with greater sampling effort, the higher numbers of specialists observed can be explained by the process of reciprocal selection between host and endosymbiont over evolutionary time [123]. This is also known as Red Queen dynamics, where the endosymbiont constantly adapts to its host to ensure continued establishment in the same host [124]. An alternative, generalist strategy can also be maintained in a population. It ensures survival in an environment where resources (i.e. hosts) are rarely found [123]. However, there are generally more instances of host specialists than generalists across numerous parasitic and endosymbiotic taxa [125,126,127].
The SPS scores revealed that host flexibility among the generalists varied greatly. Understanding Wolbachia host specificity has huge implications, especially for the optimisation of Wolbachia biocontrol strategies. Not only should researchers select strains that can effectively limit pathogen replication [128], they should also select strains for their host specificity. This is not possible without the screening of a wide variety of species or closely related species, which was achieved in this study. Using a host-specific strain will decrease the likelihood of host shift to non-target species, and thereby minimise the overall ecological risk of a strategy.
Evolutionary relationships between mosquitoes and Wolbachia
Host-Wolbachia relationships are often understudied and limited to a few taxa [52]. Studies have shown that the evolutionary associations between Wolbachia and their insect hosts do vary across taxa [49,50,51,52, 129]. Likewise, our exploratory analyses of mosquito hosts and their Wolbachia infection support such a complex relationship, with neither co-speciation nor host shifting fully accounting for evolutionary association in these lineages.
Based on the tanglegram, a broad association pattern between mosquitoes and Wolbachia strains was observed (Fig. 3). Aedes mosquitoes tended to be associated with Wolbachia supergroup A, while other mosquito species, particularly of the genus Culex, were largely associated with Wolbachia supergroup B. This showed that closely related Wolbachia strains are likely to establish themselves in related hosts. There might have been radiation of Wolbachia in these clades after their respective initial establishment. Nevertheless, the observed variations in host-endosymbiont associations make us question the mosquito-Wolbachia association pattern.
The ParaFit analysis showed weak support for congruency between host and endosymbiont phylogenies. Among the 18 host-Wolbachia associations, only the link between Mansonia indiana and Wol 3 showed a significant association (Fig. 3). This was interesting considering that Wol 3 was largely host flexible. Given that this was the only significant association, it is worth carrying out further genus-specific study on Mansonia spp. to elucidate coevolutionary patterns within a group of closely related mosquito species. It is possible that the degree to which Wolbachia co-evolve with their mosquito hosts varies across different taxonomic levels [74]. The analyses carried out thus far suggest that mosquito-Wolbachia associations are likely random at higher taxonomic levels, and that mosquito-Wolbachia co-speciation occurs at finer phylogenetic resolution (i.e. similar to patterns seen in diffuse coevolution).
The event-based analysis performed in Jane 4.0 (Fig. 4) indicated that co-speciation events were infrequent as compared to other evolutionary events. We noticed a greater proportion of host shifts and numerous losses. Interestingly, the least cost coevolutionary reconstruction indicated multiple consecutive host shifts occurring near the tips of the cladogram. This suggests that co-speciation does not fully explain the evolutionary associations between mosquito hosts and Wolbachia. Instead, recent host shifting through horizontal transmission seems to promote Wolbachia diversification. This lends greater support to the idea that horizontal transmission between distantly related species is possible [32, 33, 130].
Furthermore, losses, which represent endosymbiont extinction events that occurred upon host speciation, seem to dominate the evolutionary history of Wolbachia. Extinction events are believed to be frequent in host-endosymbiont systems [123], due to either evolution of resistance in the host or declining host population size, which result in the inability of highly specialised endosymbionts to establish themselves [131, 132]. Additionally, losses could potentially influence endosymbiont evolution through the creation of vacant niches [131]. The observed losses followed by host shifts in the mosquito-Wolbachia relationship are possible consequences of vacant niche exploitation by generalists. Perhaps this enabled successful endosymbiont invasion due to minimal intra-strain competition. If this were true, horizontal Wolbachia transmission and losses may play a bigger role in accounting for Wolbachia diversity than previously thought.
As this was an exploratory study, we were unable to determine the exact mechanism behind the diversity and evolutionary associations of Wolbachia. The presence of numerous specialists could be a sign of mosquito-Wolbachia coevolution since coevolution is fundamentally reciprocal selection between host and endosymbiont which gives rise to micro-evolutionary changes [133]. The numerous host shifts and losses might have, however, blurred the effects of vertical transmission over a long evolutionary period [52]. Thus, co-speciation might have occurred within smaller clades of Wolbachia and mosquitoes, but at higher taxa levels, horizontal transmission and loss events are more likely the prominent force driving Wolbachia evolution.
Strengths, limitations, and future directions
The three distinct methods employed here to explore evolutionary associations have both strengths and limitations. The tanglegram allows for clear visualisation of host-endosymbiont association without taking into account any evolutionary relationships, but there have been calls for careful interpretation of the results generated using this method as the degree of entanglement may not necessarily represent phylogenetic congruence [134]. The Global ParaFit test seeks to address this limitation by testing for global congruency with an unbiased, statistical approach [74]. The event-based method enables the evaluation of potential evolutionary events that might have occurred throughout an endosymbiont’s evolutionary history such as co-speciation, duplication, and host shifting. This last method, however, cannot fully differentiate a topological congruence from an evolutionary event [135]. Without knowledge of the time of divergence for both symbiont and host, a co-phylogenetic pattern may be better explained by ecological factors (as compared to co-speciation) given that bacterial lineages often evolve faster than their hosts [136, 137], and the high likelihood of host shifts among closely related species [133].
The Wolbachia wsp gene has been shown to provide well-resolved phylogenies [60], and this study provides an exploratory snapshot of the evolutionary associations between mosquito hosts and their Wolbachia endosymbionts. There is, of course, a potential caveat, since only a single gene was used to construct the respective phylogenetic trees. To obtain a more accurate phylogeny, future studies could adopt MLST [17, 51], or whole-genome shotgun sequencing [52]. The former could potentially characterise putative Wolbachia strains that cannot be distinguished with wsp gene primers.
Notwithstanding their limitations, the employment of various analytical methods allows for a comprehensive examination of the evolutionary associations between Wolbachia and mosquito hosts which are presently lacking in the literature. The scope of future studies that examine the evolution of medically important vector species could be narrowed to the Aedini tribe, as this would provide greater statistical power for the examination of mosquito-endosymbiont associations.
Conclusion
To our knowledge, this is the first study to examine Wolbachia infections in wild mosquitoes in Singapore. We detected 12 putative strains of Wolbachia among 40 mosquito species, and recorded infections in seven species for which, to our knowledge, Wolbachia infections have not been previously reported. By employing a tissue-specific PCR screening method, we were able to observe that the Wolbachia infections were preferentially located in the reproductive tissues, which provides support for vertical transmission as the main mode of infection transmission. However, even if Wolbachia infection is mainly transmitted vertically, this is unlikely to fully explain the observed diversity of Wolbachia and why closely related Wolbachia lineages were found in distantly related mosquito species. Hence, this study also served as an exploratory study which examined mosquito-Wolbachia evolutionary associations across a wide range of host mosquito species through three evolutionary analyses. Overall, we propose that the evolutionary associations between mosquito hosts and Wolbachia are consequences of both vertical and horizontal transmission and various evolutionary events.
Availability of data and materials
The datasets generated and/or analysed during this study are available in the Dryad repository, https://doi.org/10.5061/dryad.zs7h44j63. Sequence data that support the findings of this study have been deposited in Genbank with the accession codes MT645167–MT645184.
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- cox1:
-
Cytochrome c oxidase subunit I gene
- MLST:
-
Multilocus sequence typing
- mtDNA:
-
Mitochondrial DNA
- NJ:
-
Neighbour joining
- PCR:
-
Polymerase chain reaction
- SPS:
-
Standardised phylogenetic host specificity
- wsp :
-
Wolbachia surface protein gene
References
Weeks AR, Reynolds KT, Hoffmann AA, Tracy K, Ary R, Hoffmann A. Wolbachia dynamics and host effects: what has (and has not) been demonstrated? Trends Ecol Evol. 2002;17:257–62.
Duron O, Bouchon D, Boutin S, Bellamy L, Zhou L, Engelstädter J, et al. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 2008;6:1–12.
Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS ONE. 2012;7:7–9.
Zchori-Fein E, Borad C, Harari AR. Oogenesis in the date stone beetle, Coccotrypes dactyliperda, depends on symbiotic bacteria. Physiol Entomol. 2006;31:164–9.
Moran NA, Mccutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–90.
Fenn K, Blaxter M. Are filarial nematode Wolbachia obligate mutualist symbionts? Trends Ecol Evol. 2004;19:163–6.
Zchori-Fein E, Perlman SJ. Distribution of the bacterial symbiont Cardinium in arthropods. Mol Ecol. 2004;13:2009–16.
Weeks AR, Turelli M, Harcombe WR, Reynolds KT, Hoffmann AA. From parasite to mutualist: rapid evolution of Wolbachia in natural populations of Drosophila. PLoS Biol. 2007;5:e114.
Jiggins FM, Hurst GDD, Majerus MEN. Sex ratio distortion in Acraea encedon (Lepidoptera: Nymphalidae) is caused by a male-killing bacterium. Heredity (Edinb). 1998;81:87–91.
Rousset F, Bouchon D, Pintureau B, Juchault P, Solignac M. Wolbachia endosymbionts responsible for various alterations of sexuality in arthropods. Proc R Soc B Biol Sci. 1992;250:91–8.
Richard FJ. Symbiotic bacteria influence the odor and mating preference of their hosts. Front Ecol Evol. 2017;5:143.
Weeks AR, Breeuwer JAJ. Wolbachia-induced parthenogenesis in a genus of phytophagous mites. Proc R Soc B Biol Sci. 2001;268:2245–51.
Ma WJ, Schwander T. Patterns and mechanisms in instances of endosymbiont-induced parthenogenesis. J Evol Biol. 2017;30:868–88.
Moretti R, Calvitti M. Male mating performance and cytoplasmic incompatibility in a wPip Wolbachia trans-infected line of Aedes albopictus (Stegomyia albopicta). Med Vet Entomol. 2013;27:377–86.
Werren J, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–51.
Tseng SP, Wetterer JK, Suarez AV, Lee CY, Yoshimura T, Shoemaker DW, et al. Genetic diversity and Wolbachia infection patterns in a globally distributed invasive ant. Front Genet. 2019;10:1–15.
Atyame CM, Delsuc F, Pasteur N, Weill M, Duron O. Diversification of Wolbachia endosymbiont in the Culex pipiens mosquito. Mol Biol Evol. 2011;28:2761–72.
Kajtoch Ł, Kotásková N. Current state of knowledge on Wolbachia infection among Coleoptera: a systematic review. PeerJ. 2018;6:e4471.
Bourtzis K, Dobson SL, Xi Z, Rasgon JL, Calvitti M, Moreira LA, et al. Harnessing mosquito–Wolbachia symbiosis for vector and disease control. Acta Trop. 2014;132:150–63.
Blagrove MSC, Arias-goeta C, Di GC, Failloux A, Sinkins SP. A Wolbachia wMel transinfection in Aedes albopictus is not detrimental to host fitness and inhibits chikungunya virus. PLoS Negl Trop Dis. 2013;7:e2152.
Ross P. An elusive endosymbiont: does Wolbachia occur naturally in Aedes aegypti? Ecol Evol. 2020;10:1581–91.
Nguyen TH, Le NH, Nguyen TY, Vu SN, Tran ND, Le TN, et al. Field evaluation of the establishment potential of wmelpop Wolbachia in Australia and Vietnam for dengue control. Parasites Vectors. 2015;8:1–14.
Nazni WA, Hoffmann AA, NoorAfizah A, Cheong YL, Mancini MV, Golding N, et al. Establishment of Wolbachia strain wAlbB in Malaysian populations of Aedes aegypti for dengue control. Curr Biol. 2019;29:4241–8.
National Environment Agency. Wolbachia-Aedes mosquito suppression strategy. 2018. https://www.nea.gov.sg/corporate-functions/resources/research/wolbachia-aedes-mosquito-suppression-strategy/frequently-asked-questions. Accessed 13 Sept 2020.
Iturbe-Ormaetxe I, Walker T, O’Neill SL. Wolbachia and the biological control of mosquito-borne disease. EMBO Rep. 2011;12:508–18.
Martin OY, Puniamoorthy N, Gubler A, Wimmer C, Bernasconi MV. Infections with Wolbachia, Spiroplasma, and Rickettsia in the Dolichopodidae and other Empidoidea. Infect Genet Evol. 2013;13:317–30.
White JA, Kelly SE, Cockburn SN, Perlman SJ, Hunter MS. Endosymbiont costs and benefits in a parasitoid infected with both Wolbachia and Cardinium. Heredity. 2011;106:585–91.
Zhang YK, Chen YT, Yang K, Qiao GX, Hong XY. Screening of spider mites (Acari: Tetranychidae) for reproductive endosymbionts reveals links between co-infection and evolutionary history. Sci Rep. 2016;6:1–9.
Engelstädter J, Telschow A, Yamamura N. Coexistence of cytoplasmic incompatibility and male-killing-inducing endosymbionts, and their impact on host gene flow. Theor Popul Biol. 2008;73:125–33.
Engelstädter J, Hurst GDD. The ecology and evolution of microbes that manipulate host reproduction. Annu Rev Ecol Evol Syst. 2009;40:127–49.
Heath BD, Butcher RDJ, Whitfield WGF, Hubbard SF. Horizontal transfer of Wolbachia between phylogenetically distant insect species by a naturally occurring mechanism. Curr Biol. 1999;9:313–6.
Ahmed MZ, Li S, Xue X, Yin X, Ren S. The intracellular bacterium Wolbachia uses parasitoid wasps as phoretic vectors for efficient horizontal transmission. PLoS Pathog. 2015;11:e1004672.
Li S, Ahmed MZ, Lv N, Shi P, Wang X, Huang J-L, et al. Plant-mediated horizontal transmission of Wolbachia between whiteflies. ISME J. 2017;11:1019–28.
Frost CL, Pollock SW, Smith JE, Hughes WOH. Wolbachia in the flesh: symbiont intensities in germ-line and somatic tissues challenge the conventional view of Wolbachia transmission routes. PLoS ONE. 2014;9:e95122.
Pietri JE, DeBruhl H, Sullivan W. The rich somatic life of Wolbachia. Microbiol Open. 2016;5:923–36.
Dobson SL, Bourtzis K, Braig HR, Jones BF, Zhou W, Rousset F, et al. Wolbachia infections are distributed throughout insect somatic and germ line tissues. Insect Biochem Mol Biol. 1999;29:153–60.
Espino CI, Gómez T, González G, Brazil Do Santos MF, Solano J, Sousa O, et al. Detection of Wolbachia bacteria in multiple organs and feces of the triatomine insect Rhodnius pallescens (Hemiptera, Reduviidae). Appl Environ Microbiol. 2009;75:547–50.
Andersen SB, Boye M, Nash DR, Boomsma JJ. Dynamic Wolbachia prevalence in Acromyrmex leaf-cutting ants: potential for a nutritional symbiosis. J Evol Biol. 2012;25:1340–50.
Nugapola NWNP, De Silva WAPP, Karunaratne SHPP. Distribution and phylogeny of Wolbachia strains in wild mosquito populations in Sri Lanka. Parasites Vectors. 2017;10:1–8.
Sunish IP, Rajendran R, Paramasivan R, Dhananjeyan KJ, Tyagi BK. Wolbachia endobacteria in a natural population of Culex quinquefasciatus from filariasis endemic villages of south India and its phylogenetic implication. Trop Biomed. 2011;28:569–76.
Thongsripong P, Chandler JA, Green AB, Kittayapong P, Wilcox BA, Kapan DD, et al. Mosquito vector-associated microbiota: metabarcoding bacteria and eukaryotic symbionts across habitat types in Thailand endemic for dengue and other arthropod-borne diseases. Ecol Evol. 2018;8:1352–68.
Niang EHA, Bassene H, Makoundou P, Fenollar F, Weill M, Mediannikov O. First report of natural Wolbachia infection in wild Anopheles funestus population in Senegal. Malar J. 2018;17:1–6.
Kulkarni A, Yu W, Jiang J, Sanchez C, Karna AK, Martinez KJL, et al. Wolbachia pipientis occurs in Aedes aegypti populations in New Mexico and Florida, USA. Ecol Evol. 2019;9:6148–56.
Leggewie M, Krumkamp R, Badusche M, Heitmann A, Jansen S, Schmidt-Chanasit J, et al. Culex torrentium mosquitoes from Germany are negative for Wolbachia. Med Vet Entomol. 2018;32:115–20.
Bozorg-Omid F, Oshaghi MA, Vahedi M, Karimian F, Seyyed-Zadeh SJ, Chavshin AR. Wolbachia infection in West Nile Virus vectors of northwest Iran. Appl Entomol Zool. 2020;55:105–13.
Jeffries CL, Tantely LM, Raharimalala FN, Hurn E, Boyer S, Walker T. Diverse novel resident Wolbachia strains in culicine mosquitoes from Madagascar. Sci Rep. 2018;8:1–15.
Shaikevich E, Bogacheva A, Rakova V, Ganushkina L, Ilinsky Y. Wolbachia symbionts in mosquitoes: intra- and intersupergroup recombinations, horizontal transmission and evolution. Mol Phylogenet Evol. 2019;134:24–34.
Hurst GDD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc R Soc B Biol Sci. 2005;272:1525–34.
Sontowski R, Bernhard D, Bleidorn C, Schlegel M, Gerth M. Wolbachia distribution in selected beetle taxa characterized by PCR screens and MLST data. Ecol Evol. 2015;5:4345–53.
Balvín O, Roth S, Talbot B, Reinhardt K. Co-speciation in bedbug Wolbachia parallel the pattern in nematode hosts. Sci Rep. 2018;8:1–9.
Lefoulon E, Bain O, Makepeace BL, D’Haese C, Uni S, Martin C, et al. Breakdown of coevolution between symbiotic bacteria Wolbachia and their filarial hosts. PeerJ. 2016;4:e1840.
Gerth M, Röthe J, Bleidorn C. Tracing horizontal Wolbachia movements among bees (Anthophila): a combined approach using multilocus sequence typing data and host phylogeny. Mol Ecol. 2013;22:6149–62.
Chan A, Chiang L-P, Hapuarachchi HC, Tan C-H, Pang S-C, Lee R, et al. DNA barcoding: complementing morphological identification of mosquito species in Singapore. Parasites Vectors. 2014;7:1–12.
Rattanarithikul R, Harbach RE, Harrison BA, Panthusiri P, Coleman RE, Richardson JH. Illustrated keys to the mosquitoes of Thailand. VI. Tribe Aedini. Southeast Asian J Trop Med Public Health. 2010;41:1–225.
Rattanarithikul R, Harbach RE, Harrison BA, Panthusiri P, Coleman RE. Illustrated keys to the mosquitoes of Thailand. V. Genera Orthopodomyia, Kimia, Malaya, Topomyia, Tripteroides, and Toxorhynchites. Southeast Asian J Trop Med Public Health. 2007;38:1.
Rattanarithikul R, Harrison BA, Panthusiri P, Peyton EL, Coleman RE. Illustrated keys to the mosquitoes of Thailand. III. Genera Aedeomyia, Ficalbia, Mimomyia, Hodgesia, Coquillettidia, Mansonia, and Uranotaenia. Southeast Asian J Trop Med Public Health. 2006;37:1–85.
Rattanarithikul R, Harrison BA, Panthusiri P, Coleman RE. Illustrated keys to the mosquitoes of Thailand. I. Background; geographic distribution; lists of genera, subgenera, and species; and a key to the genera. Southeast Asian J Trop Med Public Health. 2005;36:1–80.
Rattanarithikul R, Harbach RE, Harrison BA, Panthusiri P, Jones JW. Illustrated keys to the mosquitoes of Thailand. II. Genera Culex and Lutzia. Southeast Asian J Trop Med Public Health. 2005;36:1–97.
Rattanarithikul R, Harrison BA, Harbach RE, Panthusiri P, Coleman RE. Illustrated keys to the mosquitoes of Thailand. IV. Anopheles. Southeast Asian J Trop Med Public Health. 2006;37:1–128.
Zhou W, Rousset F, O’Neill S. Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc R Soc Lond B. 1998;265:509–15.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Madden T. The BLAST sequence analysis tool. NCBI Handbook. 2nd Ed. Bethesda: National Center for Biotechnology Information (US); 2013.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. 2019.
Sweet L. Nonpar: a collection of nonparametric hypothesis tests. R package version 1.0.1. 2017. https://CRAN.R-project.org/package=nonpar. Accessed 23 Jun 2020.
Mangiafico S. Rcompanion: functions to support extension education program evaluation. R package version 2.3.7. 2019. https://CRAN.R-project.org/package=rcompanion. Accessed 23 Jun 2020.
James G, Witten D, Hastie T, Tibshirani R. ISLR: data for an introduction to statistical learning with applications in R. R package version 1.2. 2017. https://CRAN.R-project.org/package=ISLR. Accessed 23 Jun 2020.
Thompson JD, Higgins DG, Gibson TJ. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44:D7-19.
Poulin R, Krasnov BR, Mouillot D. Host specificity in phylogenetic and geographic space. Trends Parasitol. 2011;27:355–61.
Kembel SW, Cowan PD, Helmus M, Cornwell W, Morlon H, Ackerly D, et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics. 2010;26:1463–4.
Charleston M. TreeMap 3b. 2011. https://sites.google.com/site/cophylogeny. Accessed 23 Jun 2020.
Matsen FA, Billey SC, Kas A, Konvalinka M. Tanglegrams: a reduction tool for mathematical phylogenetics. IEEE/ACM Trans Comput Biol Bioinforma. 2018;15:343–9.
Legendre P, Desdevises Y, Bazin E. A statistical test for host-parasite coevolution. Syst Biol. 2002;51:217–34.
Balbuena JA, Míguez-Lozano R, Blasco-Costa I. PACo: a novel Procrustes application to cophylogenetic analysis. PLoS ONE. 2013;8:e61048.
Paradis E, Schliep K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–8.
Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol. 2010;5:1–10.
Charleston M. Jungles: a new solution to the host/parasite phylogeny reconciliation problem. Math Biosci. 1998;149:191–223.
Li YM, Shivas RG, Cai L. Cryptic diversity in Tranzscheliella spp. (Ustilaginales) is driven by host switches. Sci Rep. 2017;7:43549.
Ruang-Areerate T, Kittayapong P, Baimai V, O’Neill SL. Molecular phylogeny of Wolbachia endosymbionts in Southeast Asian mosquitoes (Diptera: Culicidae) based on wsp gene sequences. J Med Entomol. 2003;40:1–5.
Noor-Shazleen-Husnie MM, Emelia O, Ahmad-Firdaus MS, Zainol-Ariffin P, Aishah-Hani A. Detection of Wolbachia in wild mosquito populations from selected areas in Peninsular Malaysia by loop-mediated isothermal amplification (LAMP) technique. Trop Biomed. 2018;35:330–46.
Wiwatanaratanabutr I. Geographic distribution of wolbachial infections in mosquitoes from Thailand. J Invertebr Pathol. 2013;114:337–40.
Ravikumar H, Ramachandraswamy N, Sampathkumar S, Prakash BM, Huchesh HC, Uday J, et al. A preliminary survey for Wolbachia and bacteriophage WO infections in Indian mosquitoes (Diptera: Culicidae). Trop Biomed. 2010;27:384–93.
Tsai K-H, Lien J-C, Huang C-G, Wu W-J, Chen W-J. Molecular (sub)grouping of endosymbiont Wolbachia infection among mosquitoes of Taiwan. J Med Entomol. 2004;41:677–83.
Kittayapong P, Baisley KJ, Baimai V, O’Neill SL. Distribution and diversity of Wolbachia infections in Southeast Asian mosquitoes (Diptera: Culicidae). J Med Entomol. 2000;37:340–5.
Lam-Phua SG, Yeo H, Lee RML, Chong CS, Png AB, Foo SY, et al. Mosquitoes (Diptera: Culicidae) of Singapore: updated checklist and new records. J Med Entomol. 2019;56:103–19.
Foster WA, Walker ED. Mosquitoes (Culicidae). In: Mullen G, Durden L, editors. Medical and Veterinary Entomology. New York: Academic Press; 2018. p. 261–325.
Yeo G, Wang Y, Chong SM, Humaidi M, Lim XF, Mailepessov D, et al. Characterization of fowlpox virus in chickens and bird-biting mosquitoes: a molecular approach to investigating avipoxvirus transmission. J Gen Virol. 2019;100:838–50.
Vythilingam I, Oda K, Chew TK, Mahadevan S, Vijayamalar B, Morita K, et al. Isolation of Japanese encephalitis virus from mosquitoes collected in Sabak Bernam, Selangor, Malaysia in 1992. J Am Mosq Control Assoc. 1995;11:94–8.
de Oliveira CD, Gonçalves DS, Baton LA, Shimabukuro PHF, Carvalho FD, Moreira LA. Broader prevalence of Wolbachia in insects including potential human disease vectors. Bull Entomol Res. 2015;105:305–15.
Shaikevich E, Bogacheva A, Ganushkina L. Dirofilaria and Wolbachia in mosquitoes (Diptera: Culicidae) in central European Russia and on the Black Sea coast. Parasite. 2019;26:1–12.
Baldini F, Segata N, Pompon J, Marcenac P, Robert Shaw W, Dabiré RK, et al. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nat Commun. 2014;5:1–7.
Gomes FM, Hixson BL, Tyner MDW, Ramirez JL, Canepa GE, Alves e Silva TL, et al. Effect of naturally occurring Wolbachia in Anopheles gambiae s.l. mosquitoes from Mali on Plasmodium falciparum malaria transmission. Proc Natl Acad Sci. 2017;114:12566–71.
Wong ML, Liew JWK, Wong WK, Pramasivan S, Mohamed Hassan N, Wan Sulaiman WY, et al. Natural Wolbachia infection in field-collected Anopheles and other mosquito species from Malaysia. Parasites Vectors. 2020;13:1–15.
Gloria-Soria A, Chiodo TG, Powell JR. Lack of evidence for natural Wolbachia infections in Aedes aegypti (Diptera : Culicidae). J Med Entomol. 2018;55:1354–6.
Kittayapong P, Baimai V, O’Neill SL. Field prevalence of Wolbachia in the mosquito vector Aedes albopictus. Am J Trop Med Hyg. 2002;66:108–11.
Lounibos LP, Juliano SA. Where vectors collide: the importance of mechanisms shaping the realized niche for modeling ranges of invasive Aedes mosquitoes. Biol Invasions. 2018;20:1913–29.
Chan KL, Chan YC, Ho BC. Aedes aegypti (L.) and Aedes albopictus (Skuse) in Singapore city. 4. Competition between species. Bull World Health Organ. 1971;44:643–9.
Coon KL, Brown MR, Strand MR. Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Mol Ecol. 2016;25:5806–26.
Brucker RM, Bordenstein SR. Speciation by symbiosis. Trends Ecol Evol. 2012;27:443–51.
Janson EM, Stireman JO, Singer MS, Abbot P. Phytophagous insect-microbe mutualisms and adaptive evolutionary diversification. Evol Int J Org Evol. 2008;62:997–1012.
Schuler H, Egan SP, Hood GR, Busbee RW, Driscoe AL, Ott JR. Diversity and distribution of Wolbachia in relation to geography, host plant affiliation and life cycle of a heterogonic gall wasp. BMC Evol Biol. 2018;18:1–15.
Martínez-Rodríguez P, Bella JL. Chorthippus parallelus and Wolbachia: overlapping orthopteroid and bacterial hybrid zones. Front Genet. 2018;9:604.
Amit L, Ben-Shlomo R, Chiel E. Are microbial symbionts involved in the speciation of the gall-inducing aphid, Slavum wertheimae? Arthropod Plant Interact. 2017;11:475–84.
Hancock PA, White VL, Callahan AG, Godfray CHJ, Hoffmann AA, Ritchie SA. Density-dependent population dynamics in Aedes aegypti slow the spread of wMel Wolbachia. J Appl Ecol. 2016;53:785–93.
Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ, et al. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature. 2011;476:450–5.
McMeniman CJ, Lane RV, Cass BN, Fong AWC, Sidhu M, Wang YF, et al. Stable introduction of a life-shortening Wolbachia infection into the mosquito Aedes aegypti. Science. 2009;323:141–4.
Ant TH, Herd CS, Geoghegan V, Hoffmann AA, Sinkins SP. The Wolbachia strain wAu provides highly efficient virus transmission blocking in Aedes aegypti. PLoS Pathog. 2018;14:1–19.
Fraser JE, De Bruyne JT, Iturbe-Ormaetxe I, Stepnell J, Burns RL, Flores HA, et al. Novel Wolbachia-transinfected Aedes aegypti mosquitoes possess diverse fitness and vector competence phenotypes. PLoS Pathog. 2017;13:1–19.
Davis MJ, Ying Z, Brunner BR, Pantoja A, Ferwerda FH. Rickettsial relative associated with papaya bunchy top disease. Curr Microbiol. 1998;36:80–4.
Majerus TMO, Schulenburg JH, Majerus MEN, Hurst GDD. Molecular identification of a male-killing agent in the ladybird Harmonia axyridis (Pallas) (Coleoptera: Coccinellidae). Insect Mol Biol. 1999;8:551–5.
Weeks AR, Velten R, Stouthamer R. Incidence of a new sex-ratio-distorting endosymbiotic bacterium among arthropods. Proc R Soc Lond B. 2003;270:1857–65.
Goodacre SL, Martin OY. Modification of insect and arachnid behaviours by vertically transmitted endosymbionts: infections as drivers of behavioural change and evolutionary novelty. Insects. 2012;3:246–61.
Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasites Vectors. 2013;6:1–12.
McNulty SN, Abubucker S, Simon GM, Mitreva M, McNulty NP, Fischer K, et al. Transcriptomic and proteomic analyses of a Wolbachia-free filarial parasite provide evidence of trans-kingdom horizontal gene transfer. PLoS ONE. 2012;7:1–12.
Kondo N, Nikoh N, Ijichi N, Shimada M, Fukatsu T. Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc Natl Acad Sci. 2002;99:14280–5.
Klasson L, Kambris Z, Cook PE, Walker T, Sinkins SP. Horizontal gene transfer between Wolbachia and the mosquito Aedes aegypti. BMC Genomics. 2009;10:1–9.
Woolfit M, Iturbe-Ormaetxe I, McGraw EA, O’Neill SL. An ancient horizontal gene transfer between mosquito and the endosymbiotic bacterium Wolbachia pipientis. Mol Biol Evol. 2009;26:367–74.
Leclercq S, Thézé J, Chebbi MA, Giraud I, Moumen B, Ernenwein L, et al. Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genome. Proc Natl Acad Sci. 2016;113:15036–41.
Afizah AN, Roziah A, Nazni WA, Lee HL. Detection of Wolbachia from field collected Aedes albopictus Skuse in Malaysia. Indian J Med Res. 2015;142:205–10.
Kittayapong P, Baisley KJ, Sharpe RG, Baimai V, O’Neill SL. Maternal transmission efficiency of Wolbachia superinfections in Aedes albopictus populations in Thailand. Am J Trop Med Hyg. 2002;66:103–7.
Gerth M, Gansauge MT, Weigert A, Bleidorn C. Phylogenomic analyses uncover origin and spread of the Wolbachia pandemic. Nat Commun. 2014;5:1–7.
de Vienne DM, Refrégier G, López-Villavicencio M, Tellier A, Hood ME, Giraud T. Cospeciation vs. host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol. 2013;198:347–85.
Price PW, Westoby M, Rice B, Atsatt PR, Fritz RS, Thompson JN, et al. Parasite mediation in ecological interactions. Annu Rev Ecol Syst. 1986;17:487–505.
Ikeda-Ohtsubo W, Brune A. Cospeciation of termite gut flagellates and their bacterial endosymbionts: Trichonympha species and “Candidatus Endomicrobium trichonymphae”. Mol Ecol. 2009;18:332–42.
Drès M, Mallet J. Host races in plant-feeding insects and their importance in sympatric speciation. Philos Trans R Soc B Biol Sci. 2002;357:471–92.
Giraud T, Refrégier G, Le Gac M, de Vienne DM, Hood ME. Speciation in fungi. Fungal Genet Biol. 2008;45:791–802.
Bian G, Xu Y, Lu P, Xie Y, Xi Z. The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog. 2010;6:e1000833.
Ahmed MZ, Breinholt JW, Kawahara AY. Evidence for common horizontal transmission of Wolbachia among butterflies and moths. BMC Evol Biol. 2016;16:1–16.
Le Clec’h W, Chevalier FD, Genty L, Bertaux J, Bouchon D, Sicard M. Cannibalism and predation as paths for horizontal passage of Wolbachia between terrestrial isopods. PLoS ONE. 2013;8:e60232.
Ricklefs RE. Evolutionary diversification, coevolution between populations and their antagonists, and the filling of niche space. Proc Natl Acad Sci. 2010;107:1265–72.
de Castro F, Bolker BM. Parasite establishment and host extinction in model communities. Oikos. 2005;111:501–13.
de Vienne DM, Giraud T, Shykoff JA. When can host shifts produce congruent host and parasite phylogenies? A simulation approach. J Evol Biol. 2007;20:1428–38.
de Vienne DM. Tanglegrams are misleading for visual evaluation of tree congruence. Mol Biol Evol. 2019;36:174–6.
Chen R, Wang Z, Chen J, Jiang LY, Qiao GX. Insect-bacteria parallel evolution in multiple-co-obligate-aphid association: a case in Lachninae (Hemiptera: Aphididae). Sci Rep. 2017;7:1–9.
Degnan PH, Lazarus AB, Brock CD, Wernegreen JJ. Host-symbiont stability and fast evolutionary rates in an ant-bacterium association: cospeciation of Camponotus species and their endosymbionts Candidatus blochmannia. Syst Biol. 2004;53:95–110.
Moran NA. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc Natl Acad Sci. 1996;93:2873–8.
Acknowledgements
We would like to thank the following individuals for their assistance in the field: Ita Liana Abdul Rahman, Javier Jun Heng Tham, Ming Kai Tan, Muhammad Zulhilmi bin Zainal, Nicole Li Ying Lee and Persis Chan. We are also grateful to John Werren and Philip Bellomio from the Werren Lab at the University of Rochester for the Wolbachia positive controls. We thank the National Parks Board for the permit (NP/RP18-120) to collect specimens and the National Environment Agency for the licence (NEA/PH/CLB/19-00003) to collect and rear mosquitoes.
Funding
This research is supported by the National University of Singapore and the Ministry of Education, Singapore through a startup grant and AcRF Tier 1 grants (R15400A56133; R154000A75114).
Author information
Authors and Affiliations
Contributions
HY and NP designed the research. HD and HY collected the mosquitoes from the field. HY identified the mosquito samples. HD performed the DNA extraction and PCR. HD and HY carried out the sequence analyses. HD, HY, and NP interpreted the results and wrote the manuscript. All the authors read and approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Polymerase chain reaction (PCR) screening of Cardinium, Rickettsia, and Spiroplasma in wild mosquitoes from Singapore.
Additional file 2: Figure S1.
Weighted reproductive tissue length across various mosquito species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ding, H., Yeo, H. & Puniamoorthy, N. Wolbachia infection in wild mosquitoes (Diptera: Culicidae): implications for transmission modes and host-endosymbiont associations in Singapore. Parasites Vectors 13, 612 (2020). https://doi.org/10.1186/s13071-020-04466-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04466-8