
Abstract
Background
Pediculus humanus, which includes two ecotypes (body and head lice), is an obligate bloodsucking parasite that co-evolved with their human hosts over thousands of years, thus providing a valuable source of information to reconstruct the human migration. Pediculosis due to head lice occurred each year throughout the world and several pathogenic bacteria, which are usually associated with body lice, are increasingly detected in them. In Gabon, where this pediculosis is still widespread, there is a lack of data on genetic diversity of head lice and their associated bacteria.
Methods
This study aimed to investigate the phylogeny of head lice collected in Gabon and their associated bacteria, using molecular tools. Between 26 March and 11 April 2018, 691 head lice were collected from 86 women in Franceville. We studied the genetic diversity of these lice based on the cytochrome b gene, then we screened them for DNA of Bartonella quintana, Borrelia spp., Acinetobacter spp., Yersinia pestis, Rickettsia spp., R. prowazekii, Anaplasma spp. and C. burnetii, using real time or standard PCR and sequencing.
Results
Overall 74.6% of studied lice belonged to Clade A, 25.3% to Clade C and 0.1% to Clade E. The phylogenetic analysis of 344 head lice yielded 45 variable positions defining 13 different haplotypes from which 8 were novel. Bacterial screening revealed the presence of Borrelia spp. DNA in 3 (0.4%) of 691 head lice belonging to Clade A and infesting one individual. This Borrelia is close to B. theileri (GenBank: MN621894). Acinetobacter spp. DNA has been detected in 39 (25%) of the 156 screened lice; of these 13 (8.3%) corresponded to A. baumannii. Acinetobacter nosocomialis (n = 2) and A. pittii (n = 1) were also recorded.
Conclusions
To of our knowledge, this study is the first to investigate the genetic diversity of head lice from Gabon. It appears that Clade C is the second most important clade in Gabon, after Clade A which is known to have a global distribution. The detection of Borrelia spp. DNA in these lice highlight the potential circulation of these bacteria in Gabon.

Similar content being viewed by others


Background
Archaeological studies have shown that lice are very old human parasites [1]. Indeed, lice and their nits had been found on hair remains from an archaeological site in Brazil dating back to 8000 years BC, on the hair of an individual living in the cave of Nahal Hemar in Israel, dating back to 9000 years BC [2]. More recently, lice and louse nits had been found in Hatzeva in the Judean desert, in Moa, and around the Dead Sea (Arava), on combs dating back 2000 years [3]. These findings provide ample evidence that lice have parasitized humans for a very long time and have probably completed their migration “Out of Africa” through the migration of their human host [1]. Therefore, lice could be a good biological marker to understand human migration and evolution [4]. Lice are host specific; only two genera of lice infest humans: the genus Pthirus with a unique representative Pthirus pubis (crab lice) and the genus Pediculus [5]. The latter is of concern and contains two ecotypes: Pediculus humanus capitis (head lice) which lives and thrive on scalp and hair and P. h. humanus (body lice) which lives and thrive on skin and clothes [6].
Phylogenetic studies based on mitochondrial genes, mainly cytochrome b (cytb) and cytochrome c oxidase subunit 1 (cox1), have permitted to split lice into six divergent clades, five of which (A to E) are well known and one named clade F, which has recently been discovered [7]. Head lice encompass the full genetic diversity of clades, while body lice belong only to clades A and D [8]. Clade A has a global distribution and is the most prevalent [9, 10]. Clade B is frequently found in Americas, Europe and Australia, but has recently been detected in South Africa, Saudi Arabia and Algeria [8, 11]. Clade C was found in Ethiopia, Congo-Brazzaville, Nepal, Pakistan and Thailand [7, 12]. Clade D was found in the Democratic Republic of the Congo (DR Congo), the Republic of the Congo (Congo-Brazzaville), Ethiopia and Zimbabwe [6, 7, 12, 13]. Clade E has been described in Senegal, Mali, DR Congo and found among Nigerian refugees in Algeria and migrant communities living in Bobigny, France [14,15,16,17].
In addition to their role as markers of human migration, lice can be a significant health hazard. Body lice are able to transmit pathogenic bacteria such as Bartonella quintana (the agent of trench fever, bacillary angiomatosis, chronic lymphadenopathies and endocarditis) [18], Borrelia recurrentis (the causative agent of louse-borne relapsing fever) [19] and Rickettsia prowazekii (responsible for epidemic typhus) [20]. The role of head lice as vector of these bacteria is still suggestive. Head lice have been shown to have a stronger immune response than body lice, resulting in the rapid elimination of ingested bacteria [21]. However, studies on lice collected worldwide indicate the presence of a large number of pathogenic bacteria, including, Acinetobacter spp. in head lice collected in France, Algeria [22], Republic of the Congo and DR Congo [15, 16, 22, 23]. Bartonella quintana had been found in head lice collected in Madagascar, Senegal [24], in Nepal [25], the USA [26], and on head lice nits of a homeless person in Marseille [27]. Coxiella burnetii had been detected in head lice collected in Mali, Algeria and France [17, 28, 29]. Borrelia recurrentis and B. theileri had been found on head lice of pygmies from the Republic of the Congo [12]. Yersinia pestis was detected in head lice collected from persons living in a highly plague-endemic area in DR Congo [13]. Rickettsia aeschlimannii, as well as the DNA of potential new species from the genera Anaplasma and Ehrlichia were also detected in head lice [17]. Moreover, under experimental conditions head lice are able to acquire, maintain and transmit R. prowazekii and B. quintana [21]. Since head lice are able to carry bacteria, they could thus passively transmit them to their host, by excreting them on the injured skin of the host. Since pediculosis due to head lice is widespread throughout the world [30], controlling their associated pathogens is important to prevent the potential outbreaks related to these pathogens.
In Gabon, pediculosis caused by head lice is very prominent but poorly reported, because its leads to stigmatization of the carriers. To the best of our knowledge, this is the first study investigating the genetic diversity of head lice from Gabon and their associated bacteria.
Methods
Lice collection and DNA extraction
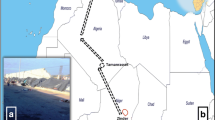
Between March 26th and April 11th, 2018, the staff of the Centre International de Recherche Médicales de Franceville (CIRMF) harvested lice from inhabitants of Franceville town (Fig. 1), in the department of M’Passa. Eighty-eight apparently healthy women were recruited in 11 very close neighborhoods of Franceville city. These women were thoroughly examined for head lice and a total of 691 lice were collected. For each individual, lice were collected in a Falcon tube containing 70% ethanol which was then stored at -20 °C at CIRMF, before being sent to IHU Mediterranée-Infection (Marseille, France) for further analysis.

Map of Africa with location of Gabon and study area in Gabon
Once in Marseille, each louse was analyzed individually. To overcome any false positives due to environmental contamination, the lice were disinfected by immersion in ethanol for 5 min and rinsed twice by immersion in sterile distilled water. Then, the lice were dried and cut in half, if possible; small individuals were treated whole. A prelysis of lice was performed in 200 μl of buffer G2 and 10 μl Proteinase K supplied in the Qiagen DNA Tissue kit (Qiagen, Courtaboeuf, France), after being crushed with a scalpel blade. DNA extraction was automatically performed in the BioRobot EZ1 Advanced XL instrument (Qiagen); the elution volume was 100 μl. DNA quantification was carried out using a NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA, USA). DNA was stored at − 20 °C until the start of analysis.
Genotyping lice by real-time quantitative PCR (qPCR)
Lice clades were initially determined by qPCR using the A-D and B-C/E duplex primers (Additional file 1: Table S1) [15, 17]. For each qPCR run, the final volume of 20 μl was composed of 10 μl of the Roche master mix (Roche Applied Science, Mannheim, Germany), 3 μl of water, 0.5 μl of each primer and probe, 0.5 μl of UDG (uracil DNA glycosylase) and 5 μl lice genomic DNA. Amplification was performed in a CFX96 Real-Time PCR detection system (Bio-Rad Laboratories, Foster City, CA, USA) according to the following amplification parameters: one step at 50 °C for 2 min, an initial denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s for annealing extension.
Determination of clades by standard PCR coupled with sequencing
For better characterization and identification of possible new haplotypes, we amplified and sequenced a 347-bp fragment of the cytochrome b gene for 344 randomly selected samples and for lice where clade determination by qPCR was not successful. The final reaction volume of 25 µl consisted of 12.5 µl of Amplitaq gold master mix, 0.75 µl of each primer (20 µM), 6 µl of water and 5 µl of genomic DNA. Amplification was performed in a Peltier PTC-200 model thermal cycler (MJ Research Inc, Watertown, MA, USA) according to the following parameters: one step of incubation at 95 °C for 15 min, 40 cycles of 1 min at 95 °C, 30 s at 56 °C and 1 min at 72 °C, followed by a final extension step at 72 °C for 5 min. PCR results were visualized on a 1.5% agarose gel and stained with SYBR Safe (Invitrogen, San Diego, CA, USA) under transilluminator UV light. The PCR amplicons were then purified using the Macherey Nagel NucleoFast 96 PCR plate (Macherey-Nagel, Hoerdt, France), following the manufacturer’s instructions. Once purified, PCR products were sequenced using the big Dye Terminator Sequencing kit (Perkin Elmer Applied Biosystems, Foster City, CA, USA) using a 3500XL Genetic Analyzer (Thermo Fisher Scientific). The sequences obtained were analyzed with ChromasPro software (ChromasPro 1.7, Technelysium Pty Ltd., Tewantin, Australia).
Screening of bacterial DNA in head lice
In order to screen louse-borne pathogens, lice DNA was pooled in 68 pools of 10 lice (10 µl of each louse) and 1 pool of 11 head lice. A total of 69 DNA pools were screened for the presence of DNA from B. quintana, Borrelia spp., Acinetobacter spp., Y. pestis, Rickettsia spp., R. prowazekii, Anaplasma spp. and C. burnetii. The DNA was screened using qPCRs as previously described [15]. We created a DNA pool with 10 samples of 10 µl each. In the pools, each sample was therefore in a proportion of 1 Log of its initial concentration. We therefore raised the number of cycles from 40 Cq to 45 Cq to ensure we did not miss any potentially positive samples in the pools. A pool consisted of 10 consecutive samples in our database, and the 11 remaining samples were pooled together. The ready to use Master mix Light Cycler 480 Probes (Roche Applied Science) was used in the PCR reactions carried out in a CFX96 Real-Time PCR detection system. Genomic DNA of targeted bacteria was used as positive controls and the master mix was used as a negative control in each PCR run. The amplification of DNA was performed according to the following parameters: one step of incubation at 50 °C for 2 min (for UDG activation), a step of 95 °C for 5 min for initial denaturation and 45 cycles of 5 s at 95 °C and 30 s at 60 °C. When a pool was found positive in a qPCR with a quantification cycle (Cq) ≤ 38, lice of this pool were tested individually. A cycle threshold for an individual louse was considered at Cq ≤ 35. Samples positive by qPCR for Acinetobacter spp. were subjected to qPCR specific for A. baumannii, as previously reported [31]. For those negative for A. baumannii but positive for Acinetobacter spp., we sequenced the rpoB gene, as previously described [32]. Samples positive by Borrelia spp. qPCR were studied in a standard PCR targeting the flaB gene using primers designed for nested PCR [33].
Data analyses
The electropherograms obtained were assembled and edited using ChromasPro 1.7 software (Technelysium Pty Ltd, City, Country). The corrected sequences were then analyzed using BLAST (www.ncbi.nlm.nih.gov/blast/Blast.cgi) and compared with sequences in the GenBank database. ClustalW alignments were performed in MEGA6 [34]. Haplotypes were identified using DnaSP v5.10 software [35]. The phylogenetic tree was inferred using the Maximum Likelihood method based on the Tamura 3-parameter model for nucleotide sequences under 500 bootstrap replicates.
Results
Genotyping of lice clades
A total of 86 women reported to have head lice were recruited in Franceville from which 691 head lice were collected. Phylogenetic molecular clade identification (qPCR and cytb amplification/sequencing) analysis showed that 515 of 691 (74.6%) analyzed lice belonged to clade A, 175 (25.3%) belonged to clade C and 1 (0.1%) belonged to clade E (Table 1).
For the phylogenetic study, a partial cytb gene (347 bp) of 344 head lice was sequenced. cytb sequence analysis yielded 45 variable positions defining 13 different haplotypes (Fig. 2) from which 8 were novel (Table 2). Of these, 325 (94.5% of 344) belonged to clade A, from which 306 sequences corresponded to haplotype A17 and 11 corresponded to haplotype A5. The 8 remaining clade A sequences corresponded to new haplotypes which are named here A72 (1 sequence), A73 (1 sequence), A74 (1 sequence), A75 (1 sequence), A76 (1 sequence), A77 (1 sequence) and A78 (2 sequences). Eighteen sequences (20.9% of 344) were clade C, from which 13 sequences corresponded to haplotype C74, 4 to haplotype C75 and one was a novel haplotype named here C80. One louse showing a very low amplitude melting curve with a clade C probe was identified as a member of clade E by sequencing and corresponded to haplotype E46. All the new haplotypes found in this study were deposited in the GenBank database under the accession numbers MN515370-MN515373 and MN515375-MN515380.

Molecular phylogenetic analysis of cytb sequences of lice from Gabon. The evolutionary history was inferred using the Maximum Likelihood method based on the Tamura 3-parameter model [34]. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 27 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + noncoding. There was a total of 272 positions in the final dataset. Evolutionary analyses were conducted in MEGA7. Haplotypes marked by a black circle are those identified in this study
Single and multiple infections with lice of different clades
Among the 86 women sampled, 73 (84.9%) were infested with lice belonging to clade A only, and 8 (9.3%) were infested with lice of clade C only. Four (4.6%) of the 86 individuals were concomitantly infested with lice belonging to clade A + C, and one (1.2%) was infested with lice belonging to three clades (A + C + E). Only one clade E louse was identified in our study. Thus, no co-infection with clades A + E or C + E was observed.
Identification of head lice pathogens
We searched the DNA of 7 pathogenic bacteria out of a total of 691 head lice collected. Among all screened bacterial genera, Acinetobacter spp. were found in 29 of the 69 DNA pools. Of the 290 lice belonging to these 29 pools, we screened 156 head lice for which DNA was available. Thirty-nine of the 156 (25%) screened lice were positive for Acinetobacter spp. Among them, 13 out of 39 (33.3%) lice infesting 8 patients were positive for A. baumannii. The number of lice infesting the 8 individuals, and the number of lice infested with A. baumannii in these individuals, are recorded in Additional file 2: Table S2. Nine lice belonged to clade A and the 4 belonged to clade C. Of the remaining 26 head lice that were positive for the Acinetobacter spp. qPCR and negative for A. baumannii, only 3 were successfully sequenced. Two of the 3 sequences shared 99.7% and 100% of similarity, respectively, with A. nosocomialis (GenBank: CP020588) and the last sequence shared 99% similarity with A. pittii (GenBank: CP040911) (Fig. 3). The two lice positive for A. nosocomialis infested the same patient and belonged to clade A. The louse harboring A. pittii also belonged to clade A.

Molecular phylogenetic analysis of rpoB sequences of Acinetobacter spp. The rpoB sequences of Acinetobacter spp. were aligned using the CLUSTALW method and evolutionary analyses were conducted in MEGA7 software using the Maximum Likelihood method based on the Tamura-Nei model. There was a total of 262 positions in the final dataset. Species in bold are those identified in this study
We also detected DNA of Borrelia spp. in one of the 69 DNA pools. After testing the 10 samples of this pool, 3 samples were positive using qPCR. Sequencing of a 148-bp fragment of the qPCR product targeting Borrelia 16S rRNA sequence, allowed us to confirm that these lice were undoubtedly infected with Borrelia spp. (Fig. 4). The 3 sequences obtained were 100% identical to each other and the BLAST search of these sequences in GenBank showed that the amplified sequence shared 100% identity with B. theileri (GenBank: MN621894). All these 3 positive head lice infested the same individual and belonged to clade A.

Alignment of the 148-bp fragment of Borrelia recurrentis 16S rRNA with those of the two lice positive for this gene using BioEdit software (version 7.2)
No samples were positive for B. quintana, Y. pestis, Rickettsia spp., R. prowazekii, Anaplasma spp. or C. burnetii.
Discussion
We studied the genetic diversity and associated pathogens of head lice from Gabon. To the best of our knowledge, this is the first study of this kind in Gabon, which investigates both phylogeny of head lice and their associated pathogens. Overall, 691 head lice were collected from 86 female individuals. We found that the studied lice belonged to the three mitochondrial clades, A (74.6%), C (25.3%) and E (0.1%). The presence of clade A in Gabon is not surprising, because this clade is found in all studies investigating the genetic diversity of human lice worldwide, thus proving its worldwide distribution [36]. However, we observed a very low interhaplogroup genetic diversity in the lice studied. Indeed, 84.9% of individuals were infested with head lice belonging to clade A only, while 9.3% of individuals were infested with lice belonging to clade C only. Clade C has already been described in head lice of pygmies (37.3% of 86 screened individuals) from the Republic of the Congo [12], which borders Gabon.
There is a significant migratory flow between Gabon and the Republic of the Congo, so the exchange between both countries is highly possible. Clade C has also been described in head lice from Ethiopia, Nepal, Pakistan, and recently in France [8, 23, 25, 37].
Four of the 86 individuals were concomitantly infested with lice belonging to clade A and C as described elsewhere [12]. One individual was infested with lice belonging to clades A and C, and only one louse of an individual belonged to clade E. This clade is more prevalent in West Africa and haplotype E46 has already been reported among head lice collected from Mali [17]. This individual could have been in close contact with a person returning from Mali and infested with head lice. Clade E had been recently described in head lice of women from DR Congo [15], but it is frequently identified in West African countries (Mali and Senegal) [17].
Among all screened bacteria, we found A. baumannii in 13 head lice infesting seven individuals. Acinetobacter baumannii has recently emerged as an important agent of nosocomial infection [38]. Its ability to acquire resistance to the main antibiotics, makes it a potentially fatal pathogen and was classified by the WHO in 2017 in the list of priority pathogens for research of new antibiotics [38, 39]. Carbapenem-resistant A. baumannii might be responsible for almost 60% of mortality in hospital-acquired pneumoniae and bloodstream infections [40]. Two other species of the Acinetobacter calcoaceticus-A. baumannii complex were found, A. nosocomialis in two head lice and A. pittii in one louse. Studies have shown that these species have a low prevalence rate and are less resistant and virulent than A. baumannii [41]. Finally, several species of the genus Acinetobacter have been reported in head lice collected worldwide, particularly in Africa [15, 22, 28].
We found that three of 691 lice infesting one individual were positive for a species of Borrelia close to B. theileri based on analysis of the newly generated 148-bp sequence of the 16S gene. Our attempts to amplify the almost entire flaB gene of these samples failed. The low number of DNA copies of Borrelia spp. could explain this failure. Indeed, a study performed in Malaysia on the detection of Borrelia in the tick Haemaphysalis hystricis showed that only one of the 12 ticks detected positive by high-throughput sequencing was found to be positive by conventional PCR targeting the 16S rRNA sequence and flaB gene of Borrelia spp. [42]. The 11 other samples positive by high-throughput sequencing showed a low relative abundance, and thus a low number of DNA copies, compared to the sample which was positive by conventional PCR. Furthermore, a previous study performed in the part of Gabon studied here found Borrelia spp. in blood samples of two afebrile children by qPCR targeting 16S rRNA sequences. However, the authors failed to amplify and sequence the flaB gene of Borrelia spp., because of the low amount of DNA copies, as shown by a cycle threshold of the qPCR [43].
The presence of Borrelia spp. in head lice is surprising, as only B. recurrentis is known to be associated with louse transmission. BLAST analysis revealed that this spirochete is closest to B. theileri, which is known as the causative agent of cattle borreliosis [44] and has never been reported in humans. However, this species has recently been detected in head lice from Congo-Brazzaville [12]. Amanzougaghene et al. [12] argued that, since head lice are specific to humans, their infection with this bacterium may only occur during a blood meal on a human host with B. theileri bacteremia. This hypothesis has also been advocated in a previous study reporting the presence of B. theileri in head lice of pygmies, as a result of ‘accidental spill-over’ of infection in animal hosts, like those described for B. duttoni found in chickens and swine [12, 45].
Conclusions
To the best of our knowledge, our study is the first to investigate simultaneously the genetic diversity of head lice and associated pathogens in Gabon. Overall, we revealed the predominance of clade A in the head lice studied. This study presents certain limits concerning the detection of pathogens, such as limited quantities of DNA for certain samples. In addition, the method of detection of Borrelia spp. should be optimized for small organisms such as lice, given the low sensitivity of standard PCR coupled with sequencing of the flagellin gene (for samples with low quantity of genomic DNA). Further investigations of the genetic diversity and bacteria associated with head lice from Gabon are needed, because data on this topic are scarce.
Availability of data and materials
All data generated as well as material used during this study are included in this published article. The newly generated sequences were deposited in the GenBank database under the accession numbers: MN515370-MN515373 and MN515375-MN515380.
Abbreviations
- cox1:
-
Cytochrome c oxidase subunit 1
- cytb :
-
Cytochrome b
- DR Congo:
-
Democratic Republic of the Congo
- UDG:
-
Uracil DNA glycosylase
- BLAST:
-
Basic Local Alignment Search Tool
References
Ascunce MS, Toups MA, Kassu G, Fane J, Scholl K, Reed DL. Nuclear genetic diversity in human lice (Pediculus humanus) reveals continental differences and high inbreeding among worldwide populations. PLoS ONE. 2013;8:e57619.
Ewing HE. Lice from human mummies. Science. 1924;60:389–90.
Amanzougaghene N, Mumcuoglu KY, Fenollar F, Alfi S, Yesilyurt G, Raoult D, et al. High ancient genetic diversity of human lice, Pediculus humanus, from Israel reveals new insights into the origin of Clade B lice. PLoS ONE. 2016;11:e0164659.
Demastes JW, Spradling TA, Hafner MS, Spies GR, Hafner DJ, Light JE. Cophylogeny on a fine scale: Geomydoecus chewing lice and their pocket gopher hosts, Pappogeomys bulleri. J Parasitol. 2012;98:262–70.
Reed DL, Light JE, Allen JM, Kirchman JJ. Pair of lice lost or parasites regained: the evolutionary history of anthropoid primate lice. BMC Biol. 2007;5:7.
Light JE, Toups MA, Reed DL. What’s in a name: the taxonomic status of human head and body lice. Mol Phylogenet Evol. 2008;47:1203–16.
Amanzougaghene N, Fenollar F, Davoust B, Djossou F, Ashfaq M, Bitam I, et al. Mitochondrial diversity and phylogeographic analysis of Pediculus humanus reveals a new Amazonian clade “F”. Infect Genet Evol. 2019;70:1–8.
Ashfaq M, Prosser S, Nasir S, Masood M, Ratnasingham S, Hebert PDN. High diversity and rapid diversification in the head louse, Pediculus humanus (Pediculidae: Phthiraptera). Sci Rep. 2015;5:14188.
Ascunce MS, Fane J, Kassu G, Toloza AC, Picollo MI, González-Oliver A, et al. Mitochondrial diversity in human head louse populations across the Americas. Am J Phys Anthropol. 2013;152:118–29.
Light JE, Allen JM, Long LM, Carter TE, Barrow L, Suren G, et al. Geographic distributions and origins of human head lice (Pediculus humanus capitis) based on mitochondrial data. J Parasitol. 2008;94:1275–81.
Al-Shahrani SA, Alajmi RA, Ayaad TH, Al-Shahrani MA, Shaurub ESH. Genetic diversity of the human head lice, Pediculus humanus capitis, among primary school girls in Saudi Arabia, with reference to their prevalence. Parasitol Res. 2017;116:2637–43.
Amanzougaghene N, Akiana J, Mongo Ndombe G, Davoust B, Nsana NS, Parra HJ, et al. Head lice of pygmies reveal the presence of relapsing fever borreliae in the Republic of Congo. PLoS Negl Trop Dis. 2016;10:e0005142.
Drali R, Davoust B, Shako JC, Raoult D, Diatta G. A new clade of African body and head lice infected by Bartonella quintana and Yersinia pestis - Democratic Republic of the Congo. Am J Trop Med Hyg. 2015;93:990–3.
Louni M, Amanzougaghene N, Mana N, Fenollar F, Raoult D, Bitam I, et al. Detection of bacterial pathogens in clade E head lice collected from Niger’s refugees in Algeria. Parasit Vectors. 2018;11:348.
Boumbanda Koyo CS, Amanzougaghene N, Davoust B, Tshilolo L, Lekana-Douki JB, Raoult D, et al. Genetic diversity of human head lice and molecular detection of associated bacterial pathogens in Democratic Republic of Congo. Parasit Vectors. 2019;12:290.
Candy K, Amanzougaghene N, Izri A, Brun S, Durand R, Louni M, et al. Molecular survey of head and body lice, Pediculus humanus, in France. Vector Borne Zoonotic Dis. 2018;18:243–51.
Amanzougaghene N, Fenollar F, Sangaré AK, Sissoko MS, Doumbo OK, Raoult D, et al. Detection of bacterial pathogens including potential new species in human head lice from Mali. PLoS ONE. 2017;12:e0184621.
Foucault C, Brouqui P, Raoult D. Bartonella quintana characteristics and clinical management. Emerg Infect Dis. 2006;12:217–23.
Bryceson AD, Parry EH, Perine PL, Warrell DA, Vukotich D, Leithead CS. Louse-borne relapsing fever. Q J Med. 1970;39:129–70.
Zinsser H, Grob GN. Rats, lice and history. Transaction Publishers; 2011.
Kim JH, Previte DJ, Yoon KS, Murenzi E, Koehler JE, Pittendrigh BR, et al. Comparison of the proliferation and excretion of Bartonella quintana between body and head lice following oral challenge: immune response of human lice to B. quintana. Insect Mol Biol. 2017;26:266–76.
Mana N, Louni M, Parola P, Bitam I. Human head lice and pubic lice reveal the presence of several Acinetobacter species in Algiers, Algeria. Comp Immunol Microbiol Infect Dis. 2017;53:33–9.
Bouvresse S, Socolovschi C, Berdjane Z, Durand R, Izri A, Raoult D, et al. No evidence of Bartonella quintana but detection of Acinetobacter baumannii in head lice from elementary schoolchildren in Paris. Comp Immunol Microbiol Infect Dis. 2011;34:475–7.
Socolovschi C, Olive M-M, Doumbo OK, Drali R, Rogier C, Raoult D, et al. Detection of Bartonella quintana in African body and head lice. Am J Trop Med Hyg. 2014;91:294–301.
Sasaki T, Poudel SKS, Isawa H, Hayashi T, Seki N, Tomita T, et al. First molecular evidence of Bartonella quintana in Pediculus humanus capitis (Phthiraptera: Pediculidae), collected from Nepalese Children. J Med Entomol. 2006;43:110–2.
Bonilla DL, Kabeya H, Henn J, Kramer VL, Kosoy MY. Bartonella quintana in body lice and head lice from homeless persons, San Francisco, California, USA. Emerg Infect Dis. 2009;15:912–5.
Angelakis E, Rolain J-M, Raoult D, Brouqui P. Bartonella quintana in head louse nits. FEMS Immunol Med Microbiol. 2011;62:244–6.
Louni M, Mana N, Bitam I, Dahmani M, Parola P, Fenollar F, et al. Body lice of homeless people reveal the presence of several emerging bacterial pathogens in northern Algeria. PLoS Negl Trop Dis. 2018;12:e0006397.
Amanzougaghene N, Mediannikov O, Ly TDA, Gautret P, Davoust B, Fenollar F, et al. Molecular investigation and genetic diversity of Pediculus and Pthirus lice in France. Parasit Vectors. 2020;13:177.
Falagas ME, Matthaiou DK, Rafailidis PI, Panos G, Pappas G. Worldwide prevalence of head lice. Emerg Infect Dis. 2008;14:1493–4.
Ly TDA, Kerbaj J, Hoang VT, Louni M, Dao TL, Badiaga S, et al. The presence of Acinetobacter baumannii DNA on the skin of homeless people and its relationship with body lice infestation. Preliminary results. Front Cell Infect Microbiol. 2019;9:86.
La Scola B, Gundi VA, Khamis A, Raoult D. Sequencing of the rpoB gene and flanking spacers for molecular identification of Acinetobacter species. J Clin Microbiol. 2006;44:827–32.
Johnson BJB, Happ CM, Mayer LW, Piesman J. Detection of Borrelia burgdorferi in ticks by species-specific amplification of the flagellin gene. Am J Trop Med Hyg. 1992;47:730–41.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinforma Oxf Engl. 2009;25:1451–2.
Reed DL, Smith VS, Hammond SL, Rogers AR, Clayton DH. Genetic analysis of lice supports direct contact between modern and archaic humans. PLoS Biol. 2004;2:e340.
Angelakis E, Diatta G, Abdissa A, Trape JF, Mediannikov O, Richet H, et al. Altitude-dependent Bartonella quintana genotype C in head lice, Ethiopia. Emerg Infect Dis. 2011;17:2357–9.
Towner KJ. Acinetobacter: an old friend, but a new enemy. J Hosp Infect. 2009;73:355–63.
Tacconelli E, Magrini N, Kahlmeter G, Singh N. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. WHO. 2017;27:318–27.
Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, Spellberg B. Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin Microbiol Rev. 2017;30:409–47.
Chusri S, Chongsuvivatwong V, Rivera JI, Silpapojakul K, Singkhamanan K, McNeil E, et al. Clinical outcomes of hospital-acquired infection with Acinetobacter nosocomialis and Acinetobacter pittii. Antimicrob Agents Chemother. 2014;58:4172–9.
Khoo JJ, Lim FS, Tan KK, Chen FS, Phoon WH, Khor CS, et al. Detection in Malaysia of a Borrelia sp. From Haemaphysalis hystricis (Ixodida: Ixodidae). J Med Entomol. 2017;54:1444–8.
Mourembou G, Fenollar F, Socolovschi C, Lemamy GJ, Nzoughe H, Kouna LC, et al. Molecular detection of fastidious and common bacteria as well as Plasmodium spp. in febrile and afebrile children in Franceville, Gabon. Am J Trop Med Hyg. 2015;92:926–32.
McCoy BN, Maïga O, Schwan TG. Detection of Borrelia theileri in Rhipicephalus geigyi from Mali. Ticks Tick Borne Dis. 2014;5:401–3.
Cutler SJ, Abdissa A, Trape J-F. New concepts for the old challenge of African relapsing fever borreliosis. Clin Microbiol Infect. 2009;15:400–6.
Acknowledgements
We are very grateful to the Agence Nationale des Bourses du Gabon (ANBG) and Infectiopôle Sud foundation for the thesis grant of CSB-K. We are grateful to Philippe Engandja, Alain Prince Okouga who helped us in sample collection.
Funding
This study was supported by the Institut Hospitalo-Universitaire (IHU) Méditerranée Infection, the National Research Agency under the program “Investissements d’avenir”, reference ANR-10-IAHU-03, the Région Provence-Alpes-Côte d’Azur, and European funding FEDER PRIMI. Funding sources had no role in the design and conduct of the study, collection, management, analysis and interpretation of the data, and preparation, review, or approval of the manuscript. CIRMF is supported by Gabonese Government and Total. UNEEREP is a member of the CANTAM network funded by EDTCP.
Author information
Authors and Affiliations
Contributions
FF, OM, JBLD and DR performed the conception or design of the work and revision of the manuscript. CSB-K performed the analysis, carried out data interpretation and drafted the manuscript. NA contributed to the interpretation of the data and substantial manuscript revisions. SLOL and RKIL carried out sample collection. OM, FF, JBLD and DR performed substantial revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the National Ethic Committee for Research (CNER) of Gabon (N°0020/2015/SG/CNE). All required written consents were obtained from each participant or their legal guardian in the case of children.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Sequences of primers and probes used in the current study.
Additional file 2: Table S2.
Number of lice carried by patients with lice positive for Acinetobacter baumannii and a breakdown by patient.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Boumbanda-Koyo, C.S., Mediannikov, O., Amanzougaghene, N. et al. Molecular identification of head lice collected in Franceville (Gabon) and their associated bacteria. Parasites Vectors 13, 410 (2020). https://doi.org/10.1186/s13071-020-04293-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04293-x