
Abstract
Background
The tick vector Rhipicephalus microplus which transmits Babesia spp. and rickettsial pathogens has not been reported in Kenya since 1998. More recently, the pathogenic Babesia bovis has been detected in cattle blood DNA. The status of R. microplus in Kenya remains unknown. This study employed morphological and molecular tools to characterize R. microplus originating from Kenya and assess the genetic relationships between Kenyan and other African R. microplus genotypes.
Methods
Ticks were collected in south-eastern Kenya (Kwale County) from cattle and characterized to investigate the existence of R. microplus. Genetic and phylogenetic relationships between the Kenyan and other annotated R. microplus reference sequences was investigated by analysis of the cytochrome c oxidase subunit 1 (cox1) gene. To further characterize Kenyan ticks, we generated low coverage whole genome sequences of two R. microplus, one R. decoloratus and R. appendiculatus. A B. bovis specific TaqMan probe qPCR assay was used to detect B. bovis in gDNA from R. microplus ticks.
Results
Occurrence of R. microplus was confirmed in Kwale County, Kenya. The Kenyan R. microplus cox1 sequences showed very high pairwise identities (> 99%) and clustered very closely with reference African R. microplus sequences. We found a low genetic variation and lack of geographical sub-structuring among the African cox1 sequences of R. microplus. Four complete mitochondrial (mt) genomes for two R. microplus, one R. decoloratus and one R. appendiculatus were assembled from next generation sequence data. The mitochondrial genome sequences of the two Kenyan R. microplus ticks clustered closely with reference genome sequences from Brazil, USA, Cambodia and India forming R. microplus Clade A. No B. bovis was detected in the Kwale R. microplus DNA.
Conclusions
These findings confirm the presence of R. microplus in Kenya and suggest that R. microplus Clade A is prevalent in cattle in sub-Saharan Africa. These and other recent findings of widespread occurrence of R. microplus in Africa provide a strong justification for urgent surveillance to determine and monitor the spread of R. microplus and vector competence of Boophilus ticks for B. bovis in Africa, with the ultimate goal of strategic control.

Similar content being viewed by others


Background
The Asian blue tick, Rhipicephalus microplus, has a widespread distribution in many tropical and subtropical areas of Asia, north-eastern Australia, South and Central America, southern and eastern Africa [1]. Historically, R. microplus has been known to occur in East and South Africa since the second wave of cattle introduction into Africa following the rinderpest epidemic in 1896 when R. microplus-infested cattle were imported from southern Asia via Madagascar [2]. In the last decade it has been reported in many West African countries [3,4,5,6,7,8] and more recently in Angola [9], Cameroon [10] and Uganda [11].
Rhipicephalus microplus is considered more economically important than other African ticks including R. decoloratus and R. annulatus because of its invasive capabilities and higher competence for Babesia bovis, the causative agent for a fatal form of bovine babesiosis [12]. In addition to pathogen transmission, the tick also causes direct economic losses to productivity and through hide damage [13]. The spread of R. microplus to new areas in Africa is a big threat to livestock industries and livelihoods of rural populations, who often depend on livestock for their survival. Annual economic losses associated with babesiosis and anaplasmosis in Kenya was estimated at about $6.9 million per year [14] and 70% of cattle are assumed to be at risk. The economic importance of R. microplus is also compounded by its propensity to displace other tick species, its higher vectoral capacity and ability to accumulate resistance to acaricides [15]. Already, widespread acaricide resistance has been reported in West Africa, where the tick is spreading [3, 5].
Rhipicephalus microplus was first recorded in Kenya in 1974 [16] and then later in 1998 [17] within a very limited area around Kwale County along the Kenyan Coast. However, more recently, B. bovis has been detected in cattle blood in central and western Kenya [18, 19], strongly suggesting the presence of its vector, R. microplus. In order to prevent the spread of B. bovis, validated methods to identify R. microplus are required, discriminating between this species and the endemic R. decoloratus and other Boophilus ticks. Based on cytochrome c oxidase subunit 1 (cox1) and mitochondrial genome phylogenetics, five distinct geographical clusters of the R. microplus species complex have been reported [20, 21]. They include Clade A (Africa, Asia and South America) and Clade B (southern China and northern India) of Burger et al. [20]; Clade C (Malaysia and India) of Low et al. [21], R. australis and R. annulatus. The Clade B lineage which was found to be more closely related to R. annulatus than to ticks in Clade A or C is thought to constitute a cryptic species restricted to China and parts of India [20].
Therefore, to determine if R. microplus does exist in Kenya, this study employed morphological and molecular tools to characterize and genotype tick specimens collected from Kwale County. Ticks were genetically and morphologically characterized to deliver reference material for the region. The reference material was characterised using scanning electron microscopy, digital microscopy, sequencing of cox1 as well as analysis of complete mtDNA from whole genome sequence data. DNA isolated from ticks was also tested for the presence of the pathogenic B. bovis DNA.
Methods
Study site
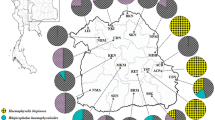
Tick samples were collected in May 2019 from cattle herds in several sites in Kwale County, Kenya (Fig. 1, Additional file 1: Table S1). Kwale County has an area of 8270.3 km2 and borders Indian Ocean to the East and South-East and Tanzania to the South-West. It lies between latitudes 30.05° to 40.75° South and longitudes 38.52° to 39.51° East. The County has a coastal plain that lies 30 m above sea level after which there is a foot plateau at an altitude of between 60–135 m above sea level. A coastal range characterized by hills rises steeply from the foot plateau to an altitude between 150–462 m above sea level. The final zone is a semi-arid plateau that stands at an altitude of about 180–300 m above sea level on the western boundary of the County. The County has a tropical type of climate influenced by monsoon seasons. The average temperature is about 23 °C with a maximum temperature of 25 °C being experienced in March and minimum temperature of 21 °C experienced in July. On average, annual precipitation in the County is less than 800 mm. Rainfall is bi-modal with a short rainy season from October to December and a long rainy season from April to July. There is a strong east to west gradient of decreasing precipitation with eastern (coastal) parts of the County receiving greater than 1000 mm of precipitation per year, while a majority of the central to west areas receive around 500–750 mm. Some areas along the western side of the County receive less than 500 mm of precipitation per year.

Map of Kenya showing Kwale County. Localities shown on the map were sampled for ticks. The tick samples analysed in this study were mainly collected from Matuga Tangini, Shimoni, Shimoni Kidimu and several other unspecified sites in the County
Tick samples
Live adult ticks were plucked directly from cattle by use of steel forceps and placed in tubes. A total of 35 ticks, all from localities in Kwale County [Kwale Kidimu (n = 6), Matuga Tangini (n = 6), Shimoni Kidimu (n = 5) and unspecified locations in Kwale County (n = 18)] (Additional file 1: Table S1) were subjected to morphological and molecular analyses. In Kenya, R. decoloratus is endemic and widely distributed and is likely to be confused with R. microplus. Thus, reference R. microplus and R. decoloratus ticks were compared to the Kwale ticks. The 11 reference specimens included three R. australis from the Tick Fever Centre, Queensland, Australia, two R. microplus and two R. decoloratus from Cameroon, two R. microplus from Laos and two R. decoloratus from a laboratory colony maintained at the International Livestock Research Institute (ILRI), Nairobi (Additional file 1: Table S1). Since most of the ticks were semi- or fully engorged and some had missing mouth parts, only those specimens that were intact could be tentatively identified based on morphological features described in the identifications guides and keys used.
Morphological identifications
Key morphological features of the tick specimens were observed under a compound microscope following standard keys and identification guides in Walker et al. [22] and Barker & Walker [23]. Images of key characteristic features were captured using a digital microscope (VHX-6000, KEYENCE Inc., Japan). Scanning electron microscopy (SEM) analysis of some reference specimens was done. Whole tick specimen was adhered to a mounting SEM stub (Ted Pella Inc., USA) with a double-sided carbon tape. The stub with the tick was then placed on a rotary planetary specimen stage within a K550X Sputter Coater unit (Quorum Technologies, Kent, UK) and coated with gold (Au) using the following parameters: current 25 mA, time 2:00 min and coating 15 nm. Once coated, the specimen was placed in a JEOL Neoscope, JCM-6000 (JEOL Inc., Nikon Inc., Japan) for imaging.
DNA isolation, PCR amplification and sequencing of the cox1 gene
Using sterile single-use scalpel blades, 3–4 cuts were made on the body of the tick sparing key features important in morphological identifications. All semi- and fully engorged ticks were cut into two halves along the abdomen. DNA was extracted using the ISOLATE II Genomic DNA Kit (Bioline, Sydney, Australia) following the manufacturer’s protocol. Total DNA was eluted into 70 µl of elution buffer (Tris buffer, pH = 8.5, preheated to 70 °C). A 604 nucleotide 5′-fragment of the cytochrome c oxidase subunit 1 (cox1) gene was amplified in a conventional PCR using the forward primer S0725 (F1) (5′-TAC TCT ACT AAT CAT AAA GAC ATT GG-3′) and reverse primer S0726 (R1) (5′-CCT CCT CCT GAA GGG TCA AAA AAT GA-3′) [24]. MyTaq™ Red Mix (Bioline, Sydney, Australia) was used for cox1 amplification in 25 μl reactions using 1 μl of each of the two primers (10 pmol) and 2 μl template DNA. The PCR run conditions were: initial denaturation at 95 °C for 5 min, followed by 34 cycles of 94 °C for 10 s, 55 °C for 10 s and 72 °C for 15 s, and a final extension step at 72 °C for 7 min. A positive control and a negative no-template water control were included in all the reactions which were performed in a T100™ Thermal Cycler (Bio-Rad, Australia). PCR products were purified and sequenced at Macrogen Ltd (Seoul, South Korea).
Sequencing and assembly of mitochondrial DNA from whole genome sequencing data
Isolated genomic DNA was used for NEBNext® DNA Library preparation following manufacturer’s recommendations. Indices were added to each of the four samples sequenced followed by the next-generation sequencing using 150-bp paired-end Illumina HiSeq 2500 sequencing systems utilizing a depth of 1 Gb of raw sequence data (Novogene, Singapore). The complete mitochondrial genome (mtDNA) of the four specimens comprising of two R. microplus, one R. decoloratus and one R. appendiculatus was assembled from FastQ data using the MITObim pipeline available at https://github.com/chrishah/MITObim with the complete mtDNA sequence of R. microplus (KC503260) as bait. The assembly was repeated three times with varying percentage of the raw FastQ sequence data used (10–50%), keeping mtDNA coverage at 60–100×. The obtained mtDNA was annotated with the aid of MITOS Web Server available at http://mitos.bioinf.uni-leipzig.de/ and aligned with available Rhipicephalus species genomes.
TaqMan qPCR assay for detection of mammalian and Babesia bovis DNA
A TaqMan qPCR assay targeting a mammalian housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [25] was conducted to confirm the presence of mammalian DNA in the Kwale R. microplus DNA samples. To detect B. bovis DNA, an assay targeting two different B. bovis-specific genes was performed. One set targeted the nuclear 18S rDNA [26] and the other targeted the mitochondrial cytochrome b gene [27]. The primer and probe sequences used for detection of both mammalian and B. bovis DNA are listed in Table 1. The qPCR was conducted in a CFX96 Touch™ Real-Time PCR detection system (Bio-Rad, Australia). The 20 µl reactions included 10 µl of SensiFAST 2× Probe Mix (Bioline, Sydney, Australia), 0.8 µl of each oligonucleotide primer, 0.2 µl of the FAM or HEX labelled probe, and 2 µl of genomic DNA template. Temperature cycling conditions were: 95 °C for 3 min followed by 39 cycles of 95 °C for 10 s, 54 °C for 15 s and 72 °C for 30 s. The sensitivity and efficiency of the B. bovis assay was determined by using serial 10-fold dilutions of B. bovis control DNA (48 ng/µl) ranging from 1:10–1:107. Positive and negative controls (no-template PCR grade water) were included in each PCR run. The threshold was set to 100 relative fluorescence units (RFUs) for the three assays and the cycle quantification (Cq) scores corresponding to the PCR cycle number at which the amplification curve of each sample intersected the threshold line were recorded for each sample.
Data analysis
Cox1 sequence chromatograms were visually inspected and resulting sequences edited manually using CLC Main Workbench 20 software (CLC bio, Qiagen GmbH, Hilden, Germany). Sequences were trimmed to remove low quality reads at the 5′- and 3′-ends and consensus sequences generated from the sequenced fragments. Molecular identity of the study ticks was confirmed via BLASTN [28] searches of the cox1 against the GenBank’s non-redundant nucleotide sequence database. Multiple sequence alignments of the cox1 gene and mtDNA genomes were performed using ClustalW2 in CLC Main Workbench. Cox1 sequences of the 23 R. microplus ticks were collapsed into haplotypes, using DnaSP v5.10.01 [29]. Percent identity analyses was performed using Clustal Omega multiple sequence analyses tool [30] (https://www.ebi.ac.uk/Tools/msa/clustalo/). A cox1 phylogenetic tree was constructed by employing the Maximum Likelihood (ML) algorithm implemented in MEGA X [31] using a total of 42 nucleotide sequences which included three Kwale R. microplus cox1 haplotypes and one R. appendiculatus sequence, 11 sequences from reference ticks and 28 GenBank reference sequences. The best nucleotide substitution model which gave the lowest Bayesian Information Criterion (BIC) score (3833.705) was Tamura 3-parameter (T92+I) as determined using MEGA X. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 69.54% sites). All positions with less than 95% site coverage were eliminated (partial deletion option). Clade support was assessed via 1000 bootstrap replications. Bootstrap values below 70% were collapsed. There were a total of 403 positions in the final cox1 dataset. A mtDNA phylogenetic tree was constructed using complete mtDNA genome sequences of two R. microplus, one R. decoloratus and one R. appendiculatus from this study and 13 reference tick mtDNA sequences available in GenBank. The best substitution model for the mtDNA tree which gave the lowest BIC score (148759.37) was General Time Reversible (GTR) [32]. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.5523)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 30.59% sites). The analysis involved 17 nucleotide sequences. There were a total of 18,435 positions in the final mtDNA dataset. Evolutionary analyses were conducted in MEGA X.
Results
Morphological identification of tick species
Of the 35 ticks collected from Kwale County, Kenya, 16 specimens were semi-engorged, 11 were fully engorged, 3 were partially fed and 5 were slightly fed (Additional file 1: Table S1). Morphologically, all but two ticks belonged to the genus Rhipicephalus (n = 33). Two ticks represented Amblyomma variegatum. On microscopic examination, 12 Rhipicephalus specimens were found to have damaged or missing mouth parts while 21 specimens were intact. Further examination revealed presence of 10 R. appendiculatus specimens. The remaining 23 specimens were compared to reference R. microplus, R. australis and R. decoloratus. Female reference R. microplus and R. australis had four rows of denticles on each side of the hypostome and a concavity with no setae on the medial aspect of article one of the palpal segment (Fig. 2). Reference R. decoloratus had three rows of denticles on each side of the hypostome, females had a convex protuberance with setae on the first palpal segments while males had long distinct adanal plates with long spurs which extended beyond the posterior body margin and were clearly visible outside the scutum (Figs. 3, 4). Twelve ticks were found to have morphological features of female R. microplus (Fig. 5). The remaining ticks (n = 11) had damaged mouth parts but spiracular plate consistent with that of subgenus Boophilus (Figs. 2, 3, 4).

Images of three key morphological features of reference R. australis (a) and R. microplus (b, c). Left: mouth parts. The characteristic 4 + 4 rows of dentition are shown by black (left row) and clear (right) arrows. Centre: spiracular plate. Right: anal aperture. The three specimens are: a R. australis (Australia); b R. microplus (Laos); and c R. microplus (Cameroon)

SEM images (ventral view) of male reference R. decoloratus and R. australis showing some characteristic morphological features a Upper anterior body of R. decoloratus (Kenya). Block pointed arrow shows coxa I anterior spur. Clear arrows show coxa I posterior spurs which are usually distinct and short in males. b Posterior body of R. decoloratus. White arrows show long distinct adenal plate spurs that are normally visible dorsally. The clear white arrow shows the caudal appendage which is long and visible dorsally. c Complete body of R. australis (Australia). Block pointed arrow shows coxa I anterior spur which is usually long and visible dorsally. Clear arrows show coxa I posterior spurs which are distinct and long in males. Black arrows show short indistinct adenal plate spurs which are not visible dorsally. The clear white arrow shows the caudal appendage which is usually short and narrow

Images of key morphological features of reference R. decoloratus from Kenya (a) and Cameroon (b). Left: mouth parts and spiracular plate images of Kenyan specimens KBF6 (upper plate) and KBF7 (lower plate). Big clear arrows show the characteristic pectinate setae on palp article I. Black and clear-pointed arrows show the characteristic 3 + 3 dentition rows. Right: mouth parts and spiracular plate images of Cameroonian specimens CdF6 (upper plate) and CdF1 (lower plate)

Mouth parts images of six female R. microplus specimens from Kwale County. Upper plate: Left: KF13; Middle: KSF2; Right: KFK6. Lower plate: Left: KF23; Middle: KF14; Right: KFK4. Most of the ticks were semi- or fully engorged
Molecular identification and genetic characterization based on cox1
Molecular analysis involving partial amplification and sequencing of the cox1 gene was performed for 32 ticks from Kwale, Kenya (12/12 R. microplus; 11/11 Rhipicephalus sp.; 8/10 R. appendiculatus; 1/2 A. variegatum). In addition, cox1 was generated for 11 reference tick specimens of R. australis (Australia, n = 3), R. microplus (Laos, n = 2), R. microplus (Cameroon, n = 2), R. decoloratus (Cameroon, n = 2) and R. decoloratus (Kenya, n = 2).
The cox1 DNA sequence of all R. microplus (n = 12) and Rhipicephalus sp. (n = 11) was > 99.5% identical to each other representing R. microplus. Presence of R. microplus was demonstrated in all sites where ticks were collected; Kwale Kidimu (6/6), Matuga Tangini (3/6) and Shimoni Kidimu (4/5), as well in across the unspecified location in Kwale County (10/18). Only two informative polymorphic sites were found, resulting in three haplotype sequences differing in two variable polymorphic sites at position 51 and 483 with a haplotype diversity (Hd) of 0.5692. Haplotype 1 consisted of six sequences which had the bases TG while 14 sequences formed haplotype 2 with bases CG at the two variable positions. Haplotype 3 consisted of 3 sequences with bases CA at the two polymorphic sites. The distribution of the three haplotypes across the sampled sites is shown in Additional file 2: Table S2. The cox1 sequences of the 23 Kwale R. microplus and 11 sequences of reference ticks were deposited in the GenBank database under the accession numbers MT181192–MT181227 (Additional file 1: Table S1), while the three haplotype sequences were deposited under the accession numbers MT181228–MT181230. Three R. microplus haplotypes from Kenya were > 99% identical to each of the 10 R. microplus available sequences from Africa; Cameroon (CF4, CF5, MK648412, MG983832, MG983831), Democratic Republic of Congo (MF45873), South Africa (KY678117), Benin (KY678120), Madagascar (KY678118) as well as a cox1 sequence from a tick collected on a zebra from Kenya (KX228549) (Additional file 3: Table S3). The Kwale R. microplus cox1 sequences had an identity of 94.5% to R. australis sequences. The cox1 sequences from reference R. microplus specimens from Laos and GenBank reference sequences from China matched with a lower identity of 92%.
cox1 phylogeny and genetic relationships
Phylogenetic analysis based on cox1 sequences was undertaken to determine the genetic relationships between the Kwale R. microplus haplotype sequences and reference R. microplus, R. australis and R. decoloratus (Fig. 6). The Kenyan R. microplus clustered in a major clade (R. microplus Clade A) together with sequences from Cameroon (CF4 and CF5). The cluster was strongly separated (100%) from a GenBank R. microplus sequence from Kenya (KX228549). Clade A was strongly separated (92%) from a R. australis sister clade. Two sequences of R. microplus specimens from Laos analysed in this study clustered with GenBank reference sequences from China (R. microplus Clade B). The Kenyan R. decoloratus sequences analysed in this study (KBF6, KBF7) clustered closely in one clade with those from Cameroon (CdF1 and CdF6).

Maximum Likelihood (ML) cox1 tree showing the phylogenetic relationships between ticks analysed in this study and 28 reference sequences. The tree was constructed based on Tamura 3-parameter (T92) model [51]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 69.54% sites). This analysis involved 42 nucleotide sequences. Tick samples analysed in this study are highlighted. The scale represents 0.10 nucleotide substitutions per site. There was a total of 403 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [31]. Bootstrap values (1000 replications) above 70% are shown
Mitochondrial genomes characterization and phylogeny
Four new complete mitochondrial genomes (mtDNA) were assembled from Illumina reads from two specimens of R. microplus from Kwale (KF13, KSF2), one R. apendiculatus from Kwale (KF10) and from a specimen representing R. decoloratus colony at ILRI Kenya (KBF6) (MT430985–MT430988; Additional file 1: Table S1). MITOS, a web server for automatic annotation of metazoan mtDNA was used to annotate protein coding genes, tRNAs and non-coding RNAs in the four complete mtDNA genomes. Multiple sequence (MS) analysis was performed on the four mtDNA sequences and 13 reference genomes from GenBank. These included six R. microplus, one R. annulatus, one unverified R. decoloratus, one R. geigyi, a partial R. appendiculatus, one R. sanguineus, one R. turanicus and one H. longicornis reference genome sequences. There were 13 protein-coding genes, 22 tRNAs and two rRNAs annotated (Fig. 7).

Graphical overview of the 13 mitochondrial proteins, 22 tRNAs and 2 rRNAs annotated by MITOS in the four mitochondrial genomes sequenced in this study. Genes located on the plus strand are drawn in the upper part. Genes annotated on the minus strand are shown in the lower region. A small vertical line is drawn every 1000 nucleotides. KF13 and KSF2 refer to mtDNA genomes of the two R. microplus specimens sequenced while KF10 and KBF6 are genomes of R. appendiculatus and R. decoloratus respectively
From the MITOS prediction, the arrangement and length of the annotated mitochondrial features in the two R. microplus (KF13 and KSF2) genomes was very similar (Fig. 7). The program predicted the presence of two trnE genes (65 bp) in the two genomes. The first occurred in the plus strand in position 4614–4678 while the second was in position 4744–4808 for both KF13 and KSF2. Both predictions had a similar e-value of 3.313e × 10−5. In the MS alignment, this region was part of control region I which occurs within the tandem repeat region annotated between trnE and nad1 in other reference R. microplus genomes. The program also predicted pseudo-copies of tRNA genes for trnA (gca), trnR (cga), trnN (aac), trnS1(aga) and trnE (gaa) (Fig. 7) in position 4854–5184 of the R. decoloratus (KBF6) mt genome. The MS alignment showed the first prediction in position 4368–4610 to be conserved and common to the reference mtDNAs. The pseudo-prediction from position 4854–5184 was a 331 bp long AT rich sequence lacking in the unverified R. decoloratus reference sequence and in other reference genomes. It occurs as part of the tandem repeat region annotated between trnE and nad1 in other reference R. microplus genomes. MITOS could not locate the gene for trnS1 in the R. apendiculatus (KF10) genome. In the reference mtDNA annotations, the trnS1 gene is reasonably conserved located upstream of the trnN gene in position 4557–4612 for KF13 and KSF2 and 4555–4610 for KBF6. In the R. appendiculatus KF10 sequence, the region (4543–4599) is predicted by MITOS to be a pseudo non-standard tRNA feature (trnX) (Fig. 7). In the MS alignment, two nucleotides are missing from the 5′-start of the gene in KF10 and the partial R. appendiculatus Zimbabwe reference genome when compared to the predicted trnS1 gene of the R. microplus reference genomes. MITOS also predicted a short nad4 gene and two nad5 genes in the minus strand in KF10 (Fig. 7). The first nad5 (1500 bp long) at position 8251–9750 with a quality value of 2.05 × 108 and a second tiny pseudo-fragment (45 bp) at position 11081–11125 with a value of 375.9. With such a lower value, the second prediction is highly unlikely. In the MS alignment, this fragment is part of nad4 which appears to be conserved across the other reference genomes.
The two R. microplus mtDNA (KF13 and KSF2) had high nucleotide similarity (> 99%) and matched with an identity of greater than > 98% to the R. microplus reference genome sequences from India, Cambodia, Brazil and Texas (USA) (Additional file 4: Table S4). They matched to the R. australis reference genome (KC503255) with a similarity of 96% and to the China sequence (KC503259) with an identity of 94%. Their similarity to the R. decoloratus sequence (KBF6) was 87% and 83% to the R. appendiculatus sequence (KF10). In the mtDNA phylogenetic tree, four major clusters were observed with all the nodes strongly supported by 100% bootstrap value (Fig. 8). The two R. microplus genome sequences (KF13 and KSF2) clustered closely with R. microplus reference sequences from Brazil (KC503261), USA (KP143546), Cambodia (KC503260) and India (MK234703).

Maximum Likelihood (ML) mtDNA tree inferred from 17 nucleotide sequences. The analysed sequences included four mitochondrial genomes sequenced in this study (highlighted) and annotated reference genomes available on GenBank. The four mtDNA genomes analysed in this study were: R. microplus KF13 and KSF2, R. appendiculatus KF10 and R. decoloratus KBF6. Six R. microplus genomes and one genome each for R. annulatus, R. geigyi, R. sanguineus, R. turanicus and H. longicornis from GenBank were included. A partial mtDNA genome of R. appendiculatus (KC503257) and an unverified R. decoloratus genome (KY457525) available on GenBank were used to compare the genome sequences of R. appendiculatus and R. decoloratus. The tree was reconstructed using the General Time Reversible (GTR) (Nei & Kumar [32]) in MEGA X. There was a total of 18,435 positions in the final dataset
Molecular detection of B. bovis in R. microplus ticks
Genomic DNA samples from 23 specimens confirmed by molecular analysis to be R. microplus were subjected to sensitive molecular quantitative qPCR assays to detect presence of bovine blood and consequently B. bovis parasites. Presence of cattle blood DNA was tested in undiluted (neat) DNA by amplifying mammalian glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The cycle quantification (Cq) values ranged from a high of 39.45 in KSF3 to a low of 23.40 in KF16. Bovine DNA was not detected in 2 samples (KF-K6 and KSF5), analysis of a 1:10 diluted DNA of these two samples resulted in the detection of bovine DNA (Cq value of 37.26) in KSF5, but no DNA was detected in the diluted KF-K6 sample.
Having confirmed the presence of bovine DNA in the extracted tick DNA, B. bovis-specific primers were used to amplify specific regions of cytochrome b (cytb) and 18S rDNA genes to detect presence of B. bovis parasites. At a threshold of 100 RFU, all material was considered negative for B. bovis DNA using either assay (Cq values ≥ 40.00).
Discussion
The invasive R. microplus, a highly efficient vector of tick fevers caused by B. bovis, B. bigemina and A. marginale has rapidly spread into many African countries over the last decade [3,4,5,6,7,8,9,10,11]. In many areas, it has been reported to progressively displace the indigenous R. decoloratus and R. annulatus [3, 5, 11, 33,34,35,36] sometimes establishing itself in areas predicted to be climatically unsuitable for its survival [7, 33, 36]. The latest findings of widespread occurrence of R. microplus in eastern Uganda [11] and recent introduction and spread of the tick in Cameroon [10] have been attributed to uncontrolled live animal movements from neighbouring countries and cross-border animal trade. To date, the widely accepted genetic characterizations and phylogenetic clustering of R. microplus are based on the cox1 gene. Therefore, we applied the same marker to characterize and genotype Kenyan Boophilus ticks.
In Kenya, R. microplus was first recorded in 1974 in a limited area along the Kenyan coast [16]. This study confirms that R. microplus still occurs in the region, although B. bovis was not detected in the limited number of ticks analysed. The coastal plain in Kwale is usually hot and humid with an average temperature above 25 °C and precipitation greater than 1000 mm annually [37]. In southern Africa, R. microplus has been shown to prefer warm and humid conditions [38]. Warm temperatures of at least 15–20 °C are required for egg-laying and larval hatching to occur. Thus, the eco-climatic conditions in Kwale are suitable for the survival of R. microplus. The tick is widespread over most of the neighbouring Tanzanian mainland [36]. The cross-border animal market at Tanga and transhumance at Shimoni port could be contributing to the re-introduction and spread of R. microplus from mainland Tanzania and the Indian ocean Islands into Kenya.
The cox1 gene has previously been shown to have greater intraspecies resolution within the R. microplus complex compared to 12S, 16S genes or ITS2 region [20, 21, 39, 40]. Based on cox1 phylogenetics, five distinct generic clusters have been shown to occur within the R. microplus species complex [20, 21, 40]. These include Clade A (ticks from Africa, Asia and South America) and Clade B (southern China and northern India) of Burger et al. [20]; Clade C (ticks from Malaysia and India) of Low et al. [21]. Rhipicephalus australis and R. annulatus have been found to be closely related to ticks in Clade B sensu Burger et al. [20]. The Kenyan R. microplus sequences analysed in this study clustered with other African R. microplus reference sequences into Clade A sensu Burger et al. [20].
Previous studies on genetic diversity and population structure of R. microplus have reported a low genetic variation and differentiation. Low et al. [21] reported a relatively low but significant genetic differentiation of R. microplus in Malaysia while in South African R. microplus a low variation within species was observed with cox1 and 16S rDNA genes [39]. A similar low divergence was also recently observed in Cameroonian R. microplus ticks by Silatsa et al. [10] using the two mitochondrial genes. Low levels of genetic differentiation and lack of population structuring amongst R. microplus populations based on microsatellite markers has also been observed [39, 41, 42]. In this study, we found a low divergence and lack of geographical sub-structuring among the African cox1 sequences analysed. The same low divergence has been observed with the 16S rRNA gene [10, 21].
A one-host tick, R. microplus, completes its entire parasitic life-cycle on a single host and is considered a cattle specialist [43]. Cattle are thought to be the only effective maintenance vertebrate hosts of R. microplus and although it can infest sheep, goats and other wild bovids, infestations of other hosts only occur when a population of ticks is maintained on cattle [22]. De Meeûs et al. [42] reported the divergence of R. microplus into two differentiated host-specific genetic groups with little or no genetic exchange within a short period of time after the tick was introduced in New Caledonia. One group was specific to cattle and another specific to the rusa deer which often co-grazes with cattle. This strict host specialization and the observed sympatric isolation can limit genetic exchange and flow resulting in very low genetic variation. All cox1 sequences analysed in this study were derived from ticks collected from cattle except for one Kenyan sequence (GenBank: KX228549) that had been collected from a migratory zebra [44]. Although R. microplus has been found to infest other livestock and some African wild ungulates including the African Cape buffalo, the role of African wildlife as reservoirs of R. microplus is currently not clear. A genetic drift caused by tick population bottlenecks such as a small effective population size or a founder event where a new population arises from few individuals such as was observed in New Caledonia can result in a decrease in allele frequencies leading to the low genetic variation and differentiation observed with R. microplus [42]. Analysis of a larger set of ticks from geographically separated areas in Africa using a combination of markers may reveal the gene flow patterns and population structure of R. microplus as it adapts into new areas.
Although R. microplus was first recorded in Kenya in the mid-1970s, it had been reported in Tanzania in the late 1960s [45]. Though the tick is thought to have been introduced into East and South Africa through cattle importations from Asia following the rinderpest epidemic in 1896 [2], the occurrences in eastern Africa were attributed to the documented spread of the tick from South Africa to the rest of southern and eastern African regions [22, 36]. Despite the different historical occurrences of R. microplus in Asia, Africa and South America, we observed the grouping of R. microplus from Africa, Asia and South America into the same cluster (Clade A). Although the use of markers with low discrimination power may contribute to the low genetic variation observed, the cox1 gene used has previously been shown to have high intraspecies resolution within the R. microplus complex resulting in the formation of the five known genetic groups [20, 21, 40]. Genetic variation within ticks has been shown to be influenced by several factors including the mammalian host range (specialist vs generalist), eco-climatic factors as well as geographical barriers to gene flow [43, 46, 47]. Although the numbers analysed in this study are few and from one general area, when combined with available data on other African genotypes, the sequence identities and cox1 clustering observed suggest a lack of phylogeographic structuring of R. microplus in Africa. It was anticipated that isolation-by-distance would result in the eastern Africa ticks being genetically divergent from those from Central and West Africa. However, based on cox1, our findings reveal that there is very low divergence of R. microplus populations from East, South, West and Central Africa.
Analysis of whole mitochondrial (mt) genomes of two Kenyan R. microplus confirmed the high conservation of mtDNA genome within the R. microplus complex as observed by Burger et al. [20]. The arrangement and length of the 13 mitochondrial proteins, 22 tRNAs (trn) and two rRNAs (rrn) in the two Kwale R. microplus genomes is similar to the reference genomes reported by Burger et al. [20]. The pseudo-trnE gene annotated by MITOS in the two Kenyan R. microplus genomes appears to be part of the tandem repeat region found to be present in all members of the R. microplus complex [20, 48]. This 150-bp tandem repeat consists of the 3′-end of trnS, trnE, the Tick-Box motif and the 3′-end of nad1. The Tick-Box is a 17-bp motif which intersperses the 3′-ends of nad1 and 16S rRNA during transcription in tick mt genomes [49]. Pseudo-copies of tRNA genes for trnA (gca), trnR (cga), trnN (aac), trnS1(aga) and trnE (gaa) were predicted in R. decoloratus (KBF6) mt genome (Fig. 7). This 331-bp insert occurred within the variable tandem repeat region annotated in other R. microplus reference genomes. It is lacking in the unverified R. decoloratus reference sequence and in other reference genomes. Two nucleotides were found to be missing from the 5′-start of the trnS1 gene in the annotated R. appendiculatus mtDNA and the partial R. appendiculatus Zimbabwe reference genome when compared to the predicted trnS1 gene of R. microplus reference genomes.
In Kenya and in many other African countries, diagnosis of tick-borne diseases including babesiosis is mostly achieved through observation of clinical signs and microscopic examination of blood smears, methods which are rapid and inexpensive but not specific or sensitive especially in immune carrier animals. The use of sensitive and specific molecular assays may increase and improve detection of tick-borne hemoparasites as indicated by the detection of B. bovis in Kenya [18, 19]. Highly sensitive quantitative PCR (qPCR) assays based on SYBR green [50] and TaqMan probes [26, 27] able to detect, quantify and differentiate B. bovis from B. bigemina are available. Validated sensitive diagnostic and epidemiological assays should be employed to screen and confirm the status and distribution of B. bovis in Kenya and other Africa countries where R. microplus has recently spread.
Conclusions
Our study has confirmed the occurrence of R. microplus in Kenya. The Kenyan R. microplus cox1 sequences clustered very closely with reference African genotypes. We found very low levels of genetic variation and lack of geographical sub-structuring among the African cox1 sequences belonging to R. microplus Clade A. With current invasion and rapid spread of R. microplus in many African countries, it is important to determine whether its occurrence is accompanied by the presence of the pathogenic B. bovis. Our findings and the recent reports of widespread occurrence of R. microplus in Africa provide the fundamental basis and rationale to implement diagnosis, urgent strategic control and surveillance to determine and monitor the spread of R. microplus and B. bovis in Kenya and in the region.
Availability of data and materials
The nucleotide dataset(s) supporting the conclusions of this article are available in the GenBank repository (http: //www.ncbi.nlm.nih.gov/genbank/). The 23 Kenyan R. microplus, haplotype and reference ticks cox1 sequences are under the accession numbers MT181192–MT181230. The whole mitochondrial genome DNA datasets appear in GenBank under the accession numbers MT430985–MT430988. The whole genome sequence data are available at SRA NCBI BioProject: PRJNA611067 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA611067). The exoskeletons of voucher specimens of the 35 Kenyan and 11 reference ticks characterized in this study have been deposited with the Australian National Insect Collection (ANIC) Museum, Canberra.
Abbreviations
- BLASTN:
-
basic local alignment search tool
- BIC:
-
Bayesian information criterion
- cox1:
-
cytochrome c oxidase subunit 1
- Cq:
-
cycle quantification
- DNA:
-
deoxyribonucleic acid
- dNTPs:
-
deoxyribonucleotide triphosphates
- ILRI:
-
International Livestock Research Institute
- GTR:
-
general time reversible
- mt:
-
mitochondrial
- mtDNA:
-
mitochondrial DNA
- MS:
-
multiple sequence
- rDNA:
-
ribosomal deoxyribonucleic acid
- qPCR:
-
quantitative PCR
- SEM:
-
scanning electron microscopy
- trn:
-
transfer RNA
- rrn:
-
ribosomal RNA
References
Estrada-Peña A, Bouattour A, Camicas J-L, Guglielmone A, Horak I, Jongejan F, et al. The known distribution and ecological preferences of the tick subgenus Boophilus (Acari: Ixodidae) in Africa and Latin America. Exp Appl Acarol. 2006;38:219–35.
Hoogstraal H. African Ixodoidea. VoI. I. Ticks of the Sudan (with special reference to Equatoria Province and with preliminary reviews of the genera Boophilus, Margaropus, and Hyalomma). Cairo: United States Naval Medical Research Unit No. 3; 1956.
Adakal H, Biguezoton A, Zoungrana S, Courtin F, de Clercq EM, Madder M. Alarming spread of the Asian cattle tick Rhipicephalus microplus in West Africa-another three countries are affected: Burkina Faso, Mali and Togo. Exp Appl Acarol. 2013;61:383–6.
Madder M, Thys E, Geysen D, Baudoux C, Horak I. Boophilus microplus ticks found in West Africa. Exp Appl Acarol. 2007;43:233–4.
Madder M, Thys E, Achi L, Touré A, De Deken R. Rhipicephalus (Boophilus) microplus: a most successful invasive tick species in West-Africa. Exp Appl Acarol. 2011;53:139–45.
Madder M, Adehan S, De Deken R, Adehan R, Lokossou R. New foci of Rhipicephalus microplus in West Africa. Exp Appl Acarol. 2012;56:385–90.
De Clercq EM, Vanwambeke SO, Sungirai M, Adehan S, Lokossou R, Madder M. Geographic distribution of the invasive cattle tick Rhipicephalus microplus, a country-wide survey in Benin. Exp Appl Acarol. 2012;58:441–52.
Kamani J, Apanaskevich DA, Gutiérrez R, Nachum-Biala Y, Baneth G, Harrus S. Morphological and molecular identification of Rhipicephalus (Boophilus) microplus in Nigeria, West Africa: a threat to livestock health. Exp Appl Acarol. 2017;73:283–96.
Gomes AF, Neves L. Rhipicephalus microplus (Acarina, Ixodidae) in Angola: evidence of its establishment and expansion. Exp Appl Acarol. 2018;74:117–22.
Silatsa BA, Kuiate JR, Njiokou F, Simo G, Feussom JMK, Tunrayo A, et al. A countrywide molecular survey leads to a seminal identification of the invasive cattle tick Rhipicephalus (Boophilus) microplus in Cameroon, a decade after it was reported in Cote d’Ivoire. Ticks Tick Borne Dis. 2019;10:585–93.
Muhanguzi D, Byaruhanga J, Amanyire W, Ndekezi C, Ochwo S, Nkamwesiga J, et al. Invasive cattle ticks in East Africa: morphological and molecular confirmation of the presence of Rhipicephalus microplus in south-eastern Uganda. Parasit Vectors. 2020;13:165.
Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(S1):S3.
Sutherst R, Maywald G, Kerr J, Stegeman D. The effect of cattle tick (Boophilus microplus) on the growth of Bos indicus × B taurus steers. Aust J Agric Res. 1983;34:317.
McLeod A, Kristjanson R. Impact of ticks and associated diseases on cattle in Asia, Australia and Africa ILRI and eSYS Report to ACIAR. Nairobi: International Livestock Research Institute; 1999.
Reck J, Klafke GM, Webster A, DallAgnol B, Scheffer R, Souza UA, et al. First report of fluazuron resistance in Rhipicephalus microplus: a field tick population resistant to six classes of acaricides. Vet Parasitol. 2014;201:128–36.
Walker JB. The ixodid ticks of Kenya. A review of present knowledge of their hosts and distribution. London and Reading, UK: Commonwealth Institute of Entomology, Eastern Press; 1974.
Zulu FP, Okello-Onen J, Punyua D, Essuman S, Malonza MM. A note on the ticks of domestic animals in Coast Province, Kenya. Int J Trop Insect Sci. 1998;18:163–5.
Njiiri NE, Bronsvoort BMC, Collins NE, Steyn HC, Troskie M, Vorster I, et al. The epidemiology of tick-borne haemoparasites as determined by the reverse line blot hybridization assay in an intensively studied cohort of calves in western Kenya. Vet Parasitol. 2015;210:69–76.
Adjou Moumouni PF, Aboge GO, Terkawi MA, Masatani T, Cao S, Kamyingkird K, et al. Molecular detection and characterization of Babesia bovis, Babesia bigemina, Theileria species and Anaplasma marginale isolated from cattle in Kenya. Parasit Vectors. 2015;8:496.
Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol. 2014;76:241–53.
Low VL, Tay ST, Kho KL, Koh FX, Tan TK, Lim YAL, et al. Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasit Vectors. 2015;8:341.
Walker A, Bouattour A, Camicas J, Estrada-peña A, Horak I, Latif A, et al. Ticks of domestic animals in Africa: a guide to identification of species. Edinburgh: Biosci Rep; 2003.
Barker SC, Walker AR. Ticks of Australia The species that infest domestic animals and humans. Zootaxa. 2014;3816:1–144.
Kushimo O. The tick genus Amblyomma in Africa: phylogeny and mutilocus DNA barcoding. Electron. Theses Diss. Pap. 835. Georgia Southern University, Statesboro, USA; 2013.
Nijhof AM, Balk JA, Postigo M, Jongejan F. Selection of reference genes for quantitative RT-PCR studies in Rhipicephalus (Boophilus) microplus and Rhipicephalus appendiculatus ticks and determination of the expression profile of Bm86. BMC Mol Biol. 2009;10:112.
Kim C, Iseki H, Herbas MS, Yokoyama N, Suzuki H, Xuan X, et al. Development of TaqMan-based real-time PCR assays for diagnostic detection of Babesia bovis and Babesia bigemina. Am J Trop Med Hyg. 2007;77:837–41.
Zhang B, Sambono JL, Morgan JAT, Venus B, Rolls P, Lew-Tabor AE. An evaluation of quantitative PCR assays (TaqMan® and SYBR Green) for the detection of Babesia bigemina and Babesia bovis, and a novel fluorescent-ITS1-PCR capillary electrophoresis method for genotyping B. bovis isolates. Vet Sci. 2016;3:23.
Altschul S, Gish W, Miller W, Myers E. Basic local alignment search tool. J Mol. 1990;215:403–10.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Madeira F, Park Y, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(1):W636–41.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford: Oxford University Press; 2000.
Berkvens DL, Geysen DM, Chaka G, Madder M, Brandt JRA. A survey of the ixodid ticks parasitising cattle in the Eastern province of Zambia. Med Vet Entomol. 1998;12:234–40.
Tønnesen MH, Penzhorn BL, Bryson NR, Stoltsz WH, Masibigiri T. Displacement of Boophilus decoloratus by Boophilus microplus in the Soutpansberg region, Limpopo Province, South Africa. Exp Appl Acarol. 2004;32:199–208.
Nyangiwe N, Harrison A, Horak IG. Displacement of Rhipicephalus decoloratus by Rhipicephalus microplus (Acari: Ixodidae) in the Eastern Cape Province, South Africa. Exp Appl Acarol. 2013;61:371–82.
Lynen G, Zeman P, Bakuname C, Di Giulio G, Mtui P, Sanka P, et al. Shifts in the distributional ranges of Boophilus ticks in Tanzania: evidence that a parapatric boundary between Boophilus microplus and B decoloratus follows climate gradients. Exp Appl Acarol. 2008;44:147–64.
The Ministry of Agriculture, Livestock and Fisheries (MoALF), Kenya. Climate Risk Profile for Kwale County. Kenya County climate risk profile series. 2016. https://ccafs.cgiar.org/publications/climate-risk-profile-kwale-county-kenya-county-climate-risk-profile-series#.XxaQV_gzZQI. Accessed 23 Jun 2020.
Gothe R. Investigations into the cold resistance of the eggs and larvae of Boophilus decoloratus (Koch, 1844), Boophilus microplus (Canestrini, 1888) and Margaropus winthemi Karsch, 1879. Onderstepoort J Vet Res. 1967;34:109–27.
Baron S, van der Merwe NA, Maritz-Olivier C. The genetic relationship between R. microplus and R. decoloratus ticks in South Africa and their population structure. Mol Phylogenet Evol. 2018;129:60–9.
Roy BC, Estrada-Peña A, Krücken J, Rehman A, Nijhof AM. Morphological and phylogenetic analyses of Rhipicephalus microplus ticks from Bangladesh, Pakistan and Myanmar. Ticks Tick Borne Dis. 2018;9:1069–79.
Sungirai M, Baron S, Van der Merwe NA, Moyo DZ, De Clercq P, Maritz-Olivier C, et al. Population structure and genetic diversity of Rhipicephalus microplus in Zimbabwe. Acta Trop. 2018;180:42–6.
De Meeûs T, Koffi BB, Barré N, de Garine-Wichatitsky M, Chevillon C. Swift sympatric adaptation of a species of cattle tick to a new deer host in New Caledonia. Infect Genet Evol. 2010;10:976–83.
McCoy KD, Léger E, Dietrich M. Host specialization in ticks and transmission of tick-borne diseases: a review. Front Cell Infect Microbiol. 2013;4:57.
Goolsby JA, Guerrero FD, Gaskin J, Bendele KG, Azhahianambi P, Amalin D, et al. Molecular comparison of cattle fever ticks from native and introduced ranges, with insights into optimal search areas for classical biological control agents. Southwest Entomol. 2016;41:595–604.
Yeoman GH, Walker JB, Ross JPJ, Docker TM. The ixodid ticks of Tanzania. A study of the zoogeography of the Ixodidae of an East African country. London: Commonwealth Institute of Entomology; 1967.
McCoy KD, Boulinier T, Tirard C, Michalakis Y. Host-dependent genetic structure of parasite populations: differential dispersal of seabird tick host races. Evolution. 2003;57:288.
McCoy KD. The population genetic structure of vectors and our understanding of disease epidemiology. Parasite. 2008;15:444–8.
Campbell NJH, Sturm RA, Barker SC. Large mitochondrial repeats multiplied during the polymerase chain reaction. Mol Ecol Notes. 2001;1:336–40.
Montagna M, Sassera D, Griggio F, Epis S, Bandi C, Gissi C. Tick-box for 3’-end formation of mitochondrial transcripts in Ixodida, basal chelicerates and Drosophila. PLoS ONE. 2012;7:e47538.
Buling A, Criado-Fornelio A, Asenzo G, Benitez D, Barba-Carretero JC, Florin-Christensen M. A quantitative PCR assay for the detection and quantification of Babesia bovis and B. bigemina. Vet Parasitol. 2007;147:16–25.
Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol. 1992;9:678–87.
Acknowledgements
We are very grateful for the permission, support, and facilitation of the Director of Veterinary Services (Dr Mwalonya Hamisi) and Sub County Veterinary Officers; Drs Mburu, Umlai M. A. and Nyamweya-Department of Agriculture, Livestock and Fisheries, Kwale County, Kenya, for their assistance with field work. We acknowledge the assistance offered by Thomas Njoroge, ILRI, Tiberius Marete, ICIPE and Gerald, Emmanuel Nuamawi and Patience Salama, Animal Health Assistants in Kwale who assisted with tick and blood sampling. We thank Silatsa Assongo Barberine, Molecular Parasitology & Entomology Subunit (MPES), Department of Biochemistry, Faculty of Science, University of Dschang, Cameroon for provision of Cameroonian ticks and Nichola Calvani, Sydney School of Veterinary Sciences, who collected the Lao ticks. Tick material from Queensland Australia were courtesy of the State of Queensland, Australia, through the Biosecurity Sciences Laboratory, the Department of Agriculture and Fisheries. We also acknowledge the facilities, scientific and technical assistance of Microscopy Australia at the Australian Centre for Microscopy & Microanalysis and Artemis HPC provided by the Sydney Informatics Hub, Core Research Facilities of the University of Sydney.
Funding
This work was financially supported by Australia Awards-Africa through a postdoctoral fellowship to EGK. The laboratory activities were supported by a research grant to DE. The field sampling and collection of ticks was supported by the CGIAR research program on Livestock (https://livestock.cgiar.org/).
Author information
Authors and Affiliations
Contributions
EGK, NG, EKN and RPB conceived the research idea. EGK, NG, EKN and DE developed the research concept. DE provided funding for the laboratory consumables. EGK and NG carried out field work. DE and JŠ designed, coordinated and supervised the morphological and molecular studies, data analysis and results presentation. EGK, DE and JŠ drafted the manuscript. All authors reviewed the manuscript to its current form. All authors read and approved the final manuscript.
Authors’ information
EGK is a Lecturer, Department of Biochemistry, School of Medicine, University of Nairobi, Kenya, and formerly a visiting postdoctoral fellow, Sydney School of Veterinary Science (SSVS), University of Sydney, Australia. DE is a Professor Veterinary Parasitology and JŠ (Orcid https://orcid.org/0000-0003-1459-9117) is a Professor of Veterinary and Molecular Parasitology, Sydney School of Veterinary Science (SSVS), University of Sydney, Australia. NWG is a Scientist & Tick Unit Manager, International Livestock Research Institute (ILRI), Nairobi, Kenya. EKN is a Professor, Department of Biochemistry, School of Medicine, University of Nairobi, Kenya. RPB is an Adjunct Professor, Department of Veterinary Microbiology and Pathology, Washington State University, Pullman, USA.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Detailed descriptions of the ticks analysed in the current study.
Additional file 2: Table S2.
Distribution of three cox1 haplotype sequences obtained in this study across sampled sites.
Additional file 3: Table S3.
cox1 sequences percent (%) identity matrix.
Additional file 4: Table S4.
Mitochondrial DNA (mtDNA) sequences percent (%) identity matrix.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kanduma, E.G., Emery, D., Githaka, N.W. et al. Molecular evidence confirms occurrence of Rhipicephalus microplus Clade A in Kenya and sub-Saharan Africa. Parasites Vectors 13, 432 (2020). https://doi.org/10.1186/s13071-020-04266-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04266-0