
Abstract
Background
Anaplasma phagocytophilum is a Gram-negative obligate intracellular bacterium that replicates in neutrophil granulocytes. It is transmitted by ticks of the Ixodes ricinus complex and causes febrile illness in humans and animals. The geographical distribution of A. phagocytophilum spans the Americas, Europe, Africa and Asia. However, human disease predominantly occurs in North America but is infrequently reported from Europe and Asia. In North American strains, the absence of the drhm gene has been proposed as marker for pathogenicity in humans whereas no information on the presence or absence of the drhm gene was available for A. phagocytophilum strains circulating in Europe. Therefore, we tested 511 European and 21 North American strains for the presence of drhm and compared the results to two other typing methods: multilocus sequence typing (MLST) and ankA-based typing.
Results
Altogether, 99% (478/484) of the analyzable European and 19% (4/21) of the North American samples from different hosts were drhm-positive. Regarding the strains from human granulocytic anaplasmosis cases, 100% (35/35) of European origin were drhm-positive and 100% (14/14) of North American origin were drhm-negative. Human strains from North America and Europe were both part of MLST cluster 1. North American strains from humans belonged to ankA gene clusters 11 and 12 whereas European strains from humans were found in ankA gene cluster 1. However, the North American ankA gene clusters 11 and 12 were highly identical at the nucleotide level to the European cluster 1 with 97.4% and 95.2% of identity, respectively.
Conclusions
The absence of the drhm gene in A. phagocytophilum does not seem to be associated with pathogenicity for humans per se, because all 35 European strains of human origin were drhm-positive. The epidemiological differences between North America and Europe concerning the incidence of human A. phagocytophilum infection are not explained by strain divergence based on MLST and ankA gene-based typing.

Similar content being viewed by others


Background
Anaplasma phagocytophilum is a Gram-negative obligate intracellular bacterium that replicates in neutrophil granulocytes [1]. It causes febrile illness in humans and animals and is transmitted by ticks of the Ixodes ricinus complex [2, 3]. The main vectors of A. phagocytophilum are I. ricinus in much of Europe, I. persulcatus in north-eastern Europe and East Asia and I. scapularis and I. pacificus in North America [2]. Anaplasma phagocytophilum has a wide geographical distribution that spans the Americas, Europe, Africa and Asia [2]. However, human disease predominantly occurs in North America with 4008 anaplasmosis cases in 2018 in the USA (https://wonder.cdc.gov/nndss/nndss_annual_tables_menu.asp?mmwr_year=2018). In contrast, human granulocytic anaplasmosis is infrequently reported from Europe [4] and Asia [5,6,7,8,9]. Of note, most patients from China initially described as to be infected by A. phagocytophilum suffered from a bunyavirus infection called severe fever with thrombocytopenia syndrome (SFTS) [5, 10,11,12].
Anaplasma phagocytophilum is not only geographically widely distributed, but also has a broad host range. Clinically apparent disease is mainly observed in humans [3], dogs [13], horses [14], cats [15] as well as in sheep and cattle [16]. Symptomatic granulocytic anaplasmosis in domestic ruminants has been observed in Europe [17], Africa [18, 19], the Near [20] and Far East [21], whereas it has not been reported from North America so far [22].
Transovarial transmission of A. phagocytophilum is inefficient at least in Ixodes ticks [23, 24]. It is therefore thought to depend on reservoir hosts to complete its life-cycle. The white-footed mouse (Peromyscus leucopus) is probably the main reservoir for human infection in the USA [25, 26], whereas the situation is less clear for Europe. Several species including wild ruminants, small mammals and wild boar have been considered in the past [22].
A variety of single and multilocus sequence typing schemes have been used (i) to elucidate the epidemiological differences mentioned above; (ii) to find markers for human pathogenicity; (iii) to study host adaptation of distinct A. phagocytophilum strains; and (iv) to determine reservoir hosts for human and animal infection [22]. The absence of the drhm gene has been proposed as a marker for pathogenicity in humans and dogs when seven whole genome sequences from five North American and two European strains from different hosts were compared [27]. However, this could not be verified on a larger series of 117 samples from the USA because 25% (4/16) of dog strains were drhm positive [28]. No information on the presence or absence of the drhm gene has been available so far for A. phagocytophilum circulating in Europe.
Therefore, we here tested 511 European and 21 North American A. phagocytophilum strains for the presence of drhm and compared the results to two other typing methods that we found in the past to have high discriminatory power: multilocus sequence typing (MLST) and ankA-based typing [29, 30].
Methods
Samples
In total, 686 A. phagocytophilum strains were included. Of them, 98 were from this study and originated from 3 humans, 35 domestic animals (18 horses, 14 dogs, 2 cats and 1 cow), 57 wild animals (19 red deer, 9 roe deer, 9 sika deer, 6 wild boars, 5 mouflons, 4 fallow deer, 2 ibexes, 2 red foxes and 1 bird) and 3 ticks (2 I. ricinus and 1 I. frontalis). The two nymphs and one adult female were engorged and removed from blackbirds (Turdus merula). A total of 577 strains were reported previously. All human strains originated from patients with human granulocytic anaplasmosis. Reference, host species, country of origin, year of sampling and disease state of the host are shown in Additional file 1: Table S1.
Presence or absence of the drhm gene
The drhm sequences available at GenBank were aligned by ClustalW. Four primers, drhm 1f (5′-CGT CAT GTG CAC TAA TAG CC-3′), drhm 2r (5′-CTC TCA TGA AAA CTA GAC GAT-3′), drhm 3f (5′-GCT ATT GCA ACA GTA ATG ACT-3′) and drhm 4r (5′-GGT ATG GTT CCA TTC TCC TG-3′) were designed for nested amplification of a 392-bp fragment. Primers drhm 1f and drhm 2r were used in the first and drhm 3f and drhm 4r in the second PCR round.
The genomic region, which contains the drhm gene, was partially amplified as well to prove its presence. It is flanked by the inversely duplicated genes APH_0919 and APH_0922 [27]. APH_0919 and APH_0922 sequences were extracted from the A. phagocytophilum whole genome sequences available on GenBank and aligned by ClustalW. Degenerated primers APH_0919 1f (5′-ATA TCC CTG CCR TTT RTK CTG), APH_0919 2r (5′-GTG GTA YAT TAG ATG TAT CAA AA-3′), APH_0919 3f (5′-CTG CTT CAY GCA ACS CTR TTA-3′) and AHP_0919 4r (5′-TTA GAT GTA TCA AAA CAY ATT GC-3′) were designed to amplify a 482-bp fragment of APH_0919 and/or APH_0922. Primers APH_0919 1f and APH_0919 2r were applied in the first and APH_0919 3f and APH_0919 4r in the second PCR round.
Two μl of DNA were used as a template in a 50 μl reaction mixture containing 50 mM KCl, 20 mM Tris-HCl (pH 8.4), 2 mM MgCl2, 0.2 mM deoxynucleoside triphosphates, 0.4 μM concentrations of each primer and 0.2 μl (1 U) of Taq DNA polymerase (Invitrogen, Karlsruhe, Germany). The PCR were performed by a 2720 GeneAmp thermal cycler (Applied Biosystems, Darmstadt, Germany) under the following conditions: an initial denaturation at 94 °C for 3 min; followed by 40 cycles of denaturation at 94 °C for 30 s, annealing at 54 °C for 30 s, extension at 72 °C for 30 s; and a final extension step at 72 °C for 10 min. Individual drhm and APH_0919/APH_0922 amplicons were bidirectionally sequenced to prove PCR specificity.
MLST and ankA gene
Seven housekeeping genes (pheS, glyA, fumC, mdh, sucA, dnaN and atpA) were amplified for MLST and sequenced bidirectionally as reported previously [29]. Clonal complexes (CC) were defined by sharing identical alleles at five of the seven loci with at least one other member of the group. The ankA gene was partially amplified and bidirectionally sequenced as described [29]. Full length ankA sequences were obtained in tick_CM20 and tick_CS2 (cluster 6) as detailed in Additional file 2: Text S1, and in horse_S1523_07 (cluster 7) as described previously [31].
Phylogenetic analysis
Sequences were codon-aligned by ClustalW applying the PAM (Dayhoff) matrix. Trees were constructed using the neighbor-joining (NJ) method with the Jukes-Cantor model and the complete deletion option in the program MEGA X version 10.0.5 [32]. Bootstrap analysis was conducted with 1000 replicates. Net average distances between nucleotide sequences of MLST and ankA gene clusters were computed using the Jukes-Cantor matrix and applying the complete deletion option. Net average distances between protein sequences of ankA gene clusters were calculated using the PAM (Dayhoff) matrix and applying the complete deletion option.
Comparison of typing methods
To test for the concordance between different typing methods, adjusted Wallace coefficients [33] were calculated using the online tool available at: http://www.comparingpartitions.info/index.php?link=Tool. For example, the Wallace coefficient host → MLST cluster is the probability that two strains are found in the same MLST cluster, if they are from the same host.
Results
Presence or absence of the drhm gene
The presence or absence of the drhm gene was determined in 532 A. phagocytophilum strains, 511 from Europe and 21 from North America (Additional file 1: Table S1). The information was extracted from GenBank in 13 cases (3 from Europe, 10 from the USA). The DNA was used up or information on drhm was not available on GenBank in 154 of the 668 strains in total.
The absence of a gene is difficult to prove due to methodical reasons. Therefore, the amplification of the flanking genes APH_0919/APH_0922 was undertaken in 519 samples in order to prove that the genomic region containing the drhm gene was present (Additional file 1: Table S1). This information was extracted from GenBank in 13 cases. 95% (505/532) of the A. phagocytophilum strains investigated contained the APH_0919 and/or APH_0922 gene (Table 1). The 27 APH_0919/APH_0922-negative samples were exclusively of European origin and from 22 voles, 3 shrews and 2 I. ricinus ticks removed from blackbirds (T. merula) (Table 2). All APH_0919/APH_0922-negative samples were also negative for drhm (Table 1).
Of the APH_0919/APH_0922-positive samples, 95% (482/505) were also drhm-positive. This was true for 99% (478/484) of the European and 19% (4/21) of the North American A. phagocytophilum strains (Table 1). The drhm-positive samples from the USA originated from 1 horse and 3 I. scapularis ticks (Table 2). Six European strains (from 3 dogs, 2 roe deer and 1 red deer) were APH_0919/APH_0922-positive, but drhm-negative (Table 2). The 17 APH_0919/APH_0922-positive, but drhm-negative samples from the USA originated from 14 humans, 1 dog, 1 jumping meadow mouse (Zapus hudsonicus) and 1 chipmunk (Tamias striatus).
Regarding the human strains, 100% (35/35) of the strains with European origin were drhm-positive and 100% (14/14) of those with North American origin were drhm-negative.
MLST
In general, different sequences of a given locus (pheS, glyA, fumC, mdh, sucA, dnaN and atpA) were ascribed a unique, but arbitrary allele number and each unique combination of alleles was assigned a sequence type (ST). Full profiles were obtained for 653 A. phagocytophilum strains. Of these, 93 were from this study, 491 were reported previously by our group and in 69 cases the information was extracted from GenBank or PubMLST (Additional file 1: Table S1). Housekeeping gene sequences with double peaks in the chromatograms were regarded as non-typeable. Therefore, a ST could not be ascribed in 139 A. phagocytophilum strains revealing a typeability of 79% (514/653). Clonal complexes (CC) were defined by sharing identical alleles at five of the seven loci with at least one other member of the group. CC 18 and CC 19 are newly described and contained two roe deer samples each. Allele numbers, ST, CC and MLST cluster for each A. phagocytophilum strain are shown in Additional file 1: Table S1.
A total of 520 A. phagocytophilum strains without ambiguous nucleotides were included in the phylogenetic analysis. The sequences segregated into 8 clusters (Fig. 1, Additional file 3: Fig. S1). Clusters 1 to 3 [29] and 4 to 6 [34, 35] have been described before. Cluster 1 contained strains from humans, domestic animals (dogs, horses and cats), farm animals (cattle, sheep and goats), wild animals (red deer, sika deer, fallow deer, European bison, mouflon, chamois, ibex, wild boar and red foxes), small mammals (hedgehogs, jumping meadow mouse and chipmunk) and I. ricinus ticks. Cluster 2 harbored mainly samples from roe deer and I. ricinus ticks, but also sporadically sequences from domestic ruminants. Cluster 3 was restricted to strains from voles and shrews from Europe. Cluster 4 was small and constituted by samples from 2 roe deer and 1 red deer. Cluster 5 and cluster 6 contained strains from I. persulcatus, I. pavlovskyi and their hybrids from the Asian part of Russia as described previously [34, 35]. Samples from Asian voles were found exclusively in cluster 6. Cluster 7 harbored 2 strains from I. ricinus ticks removed from blackbirds (Turdus merula). Cluster 8 contained 3 isolates from I. scapularis ticks from the USA that have been classified as non-human pathogenic A. phagocytophilum Ap-variant 1 [36]. However, the other North American samples from 11 humans, 1 dog, 1 horse, 1 chipmunk and 1 jumping meadow mouse were part of cluster 1 (Fig. 1, Additional file 3: Fig. S1).

Phylogenetic tree calculated from the concatenated housekeeping gene sequences of 520 samples without ambiguous nucleotides. Tree construction was achieved by the NJ method using the Jukes-Cantor matrix with the complete deletion option. Bootstrap values ≥ 64% are shown next to the branches. The scale-bar indicates the number of nucleotide substitutions per site. The final data set contained 2877 positions. Identical ST are displayed only once per species. The number in parenthesis indicates the frequency with which the respective ST was found. Key: red circles, sequences from humans, dogs, horses and cats; dark blue diamonds, sequences from domestic ruminants (cattle, sheep, goats and water buffalo); light blue diamonds, sequences from wild ruminants (roe deer, red deer, sika deer, fallow deer, European bison, mouflon, chamois and ibex); green triangles, sequences from small mammals (hedgehogs, voles, shrews, chipmunk and jumping meadow mouse); yellow squares, sequences from wild boars; purple triangles, sequences from red foxes; white triangles, sequences from ticks
The largest cluster was cluster 1. It contained samples from Europe and North America. However, all other clusters were restricted to samples from either Europe or North America or Asia (Fig. 1, Additional file 3: Fig. S1). Net average distances between the MLST clusters are shown in Table 3. The highest identity of 98.6% was observed between clusters 1 and 8 and the lowest identity of 88.3% between clusters 6 and 8.
ankA
Partial ankA sequences were available for 637 A. phagocytophilum strains. Of these, 94 were from this study, 491 were reported previously by us and in 52 cases the information was extracted from GenBank (Additional file 1: Table S1). A total of 17 samples from cattle, red deer, roe deer, sika deer and I. ricinus were regarded as non-typeable, because they contained ankA variants belonging to different clusters (Additional file 1: Table S1). In 15 cases two ankA gene variants and in two cases three ankA gene variants were present. The typeability regarding the ankA gene cluster was 97% (620/637). The ankA gene cluster for each A. phagocytophilum strain is shown in Additional file 1: Table S1.
A total of 623 ankA sequences without ambiguous nucleotides were included in the phylogenetic analysis. The sequences segregated into 12 clusters (Fig. 2, Additional file 4: Fig. S2). Clusters 1 to 5 [29] as well as 8 and 10 [34] have been described before. Cluster 1 contained strains from humans, domestic animals (dogs, horses and cats), farm animals (cattle, sheep and goats), wild animals (red deer, sika deer, fallow deer, European bison, chamois, wild boar and red foxes), small mammals (hedgehogs) and I. ricinus ticks. Cluster 2 harbored mainly samples from roe deer and I. ricinus ticks, but also sporadically sequences from domestic ruminants. Cluster 3 comprised mostly strains from roe deer, but also single sequences from red deer and sika deer. Cluster 4 contained samples from one horse, farm animals (cattle, sheep and goats), wild animals (red deer, sika deer, fallow deer, roe deer, European bison, mouflon, chamois and ibex) and I. ricinus ticks. Cluster 5 was restricted to strains from voles and shrews from Europe. Cluster 6 harbored one sample from a blackbird (T. merula) and two strains from I. ricinus ticks removed from blackbirds. Cluster 7 contained one strain from a horse and two strains from I. ricinus ticks. Cluster 8 comprised samples from I. persulcatus and I. pavlovskyi ticks from the Asian part of Russia as described previously [34]. Cluster 9 was restricted to one sequence [37] from an Ixodes sp. tick removed from a woodchat shrike (Lanius senator senator). Cluster 10 contained only samples from voles as well as from I. persulcatus, I. pavlovskyi and their hybrids from the Asian part of Russia as described previously [34]. Clusters 11 and 12 harbored solely strains from North America. Cluster 11 comprised samples from humans, one dog, one horse and small mammals (jumping meadow mouse and chipmunk). Cluster 12 contained strains from humans and I. scapularis ticks. One of the tick strains (USG3) has been described as human pathogenic A. phagocytophilum Ap-ha strain [38], whereas the other three have been classified as non-human pathogenic A. phagocytophilum Ap-variant 1 [36].

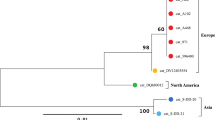
Phylogenetic tree calculated from the ankA sequences of 623 samples without ambiguous nucleotides. Tree construction was achieved by the NJ method using the Jukes-Cantor matrix with the complete deletion option. Bootstrap values ≥ 69% are shown next to the branches. The scale-bar indicates the number of nucleotide substitutions per site. The final data set contained 510 positions. Identical ankA sequences are displayed only once per species. The number in parenthesis indicates the frequency with which the respective sequence was found. Key: red circles, sequences from humans, dogs, horses and cats; dark blue diamonds, sequences from domestic ruminants (cattle, sheep, goats and water buffalo); light blue diamonds, sequences from wild ruminants (roe deer, red deer, sika deer, fallow deer, European bison, mouflon, chamois and ibex); green triangles, sequences from small mammals (hedgehogs, voles, shrews, chipmunk and jumping meadow mouse); yellow squares, sequences from wild boars; purple triangles, sequences from red foxes; pink square, sequence from a bird, white triangles, sequences from ticks
Samples from Europe were found in clusters 1–7 and 9, those from Asia in clusters 8 and 10 and those from North America in clusters 11 and 12. Net average distances between the ankA gene clusters are shown in Table 4. At the nucleotide level, the highest identity of 97.4% was observed between clusters 1 and 11 and the lowest identity of 47.8% between cluster 1 and cluster 10.
Full length ankA sequences of the A. phagocytophilum strains horse_S1523_07 (present study) and tick_W271 [31] which both belonged to cluster 7 were compared to all other complete ankA sequences available so far [30, 31, 39]; the highest identities were observed to cluster 1 (83.9%) and to cluster 4 (86.3%) sequences. However, the identity of cluster 7 sequences was 99.4% to cluster 1 sequences when nucleotides 1–1639 were considered and 98.2% to cluster 4 sequences when nucleotides 1604–3720 were taken into account. This means that cluster 7 ankA sequences probably arose by recombination of cluster 1 and cluster 4 sequences.
Concordance between typing methods
To test for the concordance between different typing methods, adjusted Wallace coefficients [33] were calculated. The information regarding drhm presence was obtained in 392 A. phagocytophilum strains that were typeable by MLST and ankA. The concordance between drhm status and continent was 71% (Table 5) which reflects the fact that 94% (478/511) of the European samples, but only 19% (4/21) of those from North America were drhm-positive. The association between drhm presence and the other partitions was low (host, ST, CC, MLST cluster, ankA cluster and country). However, the concordance between ST, CC and ankA gene cluster on one hand and drhm status on the other hand was high (> 75%), because CC 5, CC 12 and ankA gene clusters 11 and 12 were restricted to North American strains (Table 5). The concordance between host and MLST cluster was 88% which indicates a host association of certain A. phagocytophilum variants (Table 5). The association between ankA cluster and MLST cluster was 96%.
The information regarding drhm presence was unavailable for all Asian strains. Therefore, adjusted Wallace coefficients were calculated in 467 A. phagocytophilum strains that were typeable by MLST and ankA, but lacked the partition drhm presence. Then, the concordance between MLST cluster and continent was 68% and between ankA gene cluster and continent 100% (Table 6).
Discussion
Presence or absence of the drhm gene
The absence of the drhm gene in A. phagocytophilum strains has been proposed as a marker for pathogenicity in humans and dogs [27]. Eight human strains from North America investigated so far have been drhm-negative [27]. Here, we included further human samples from the USA that did not possess drhm either. However, all 35 human strains from Europe were drhm-positive. Thus, the absence of the drhm gene in A. phagocytophilum does not seem to be associated with pathogenicity for humans per se. On the other hand, drhm negativity could indicate that those strains are of higher virulence, because human granulocytic anaplasmosis is infrequently reported from Europe compared to the USA [4].
In contrast to human disease, canine [13] and equine [14] granulocytic anaplasmosis equally occurs in North America and Europe. In a previous study, 25% (4/16) of dog strains and 53% (11/21) of horse strains from the USA were positive for drhm [28]. Here, 96% (67/70) of canine and 100% (44/44) of equine samples from Europe possessed the drhm gene. Thus, the drhm status seems not to be associated with pathogenicity or virulence in dogs and horses. The same is probably true in humans because A. phagocytophilum strains from humans, dogs and horses have been reported to be homologous [29, 30, 40] and dogs and horses have been reported to be susceptible to infection with human isolates [41,42,43].
The concordance between drhm status and host and vice versa was low. A similar finding has been reported previously [28]. Thus, presence or absence of drhm is probably not associated with certain hosts. However, a tendency has been observed for A. phagocytophilum strains from the Northeast of the USA to be drhm-negative, in contrast to samples from the Southeast, Midwest and West [28]. In the present study, the concordance between drhm status and country of origin and vice versa was low. Therefore, presence or absence of drhm is not geographically informative in Europe. However, 94% (478/511) of the European and 19% (4/21) of the North American A. phagocytophilum strains were drhm-positive yielding a concordance between drhm status and continent of 71%. Thus, drhm positivity seems to be associated with European origin, although the concordance was not very strong (< 75%).
Twenty-seven European A. phagocytophilum strains were APH_0919/APH_0922- and drhm-negative. They originated from voles, shrews and I. ricinus ticks removed from blackbirds (T. merula) and belonged to MLST clusters 3 and 7. Strains from these clusters were more distantly related to the MLST clusters 1, 2, 4 and 8 for which information on APH_0919/APH_0922 and drhm was available. Thus, the most likely reasons for negativity in APH_0919/APH_0922 and drhm are primer mismatches or a different genomic organization.
MLST and ankA-based typing
A ST could not be ascribed in 139 A. phagocytophilum strains because of double peaks in the chromatograms. This phenomenon has been observed before, most prominently in wild [29] and domestic ruminants [44, 45], probably reflecting their co- or superinfection with different A. phagocytophilum variants.
Among others, wild boar and small mammals have been considered in the past as reservoir hosts for human infection in Europe [22]. Here, concatenated housekeeping and ankA gene sequences from human strains from Europe clustered most closely together with hedgehogs and wild boar indicating that they might harbor human-pathogenic A. phagocytophilum variants. In contrast, samples from voles and shrews from Europe and Asia were only distantly related. Asian A. phagocytophilum strains from humans were not available for analysis. Thus, it is unclear whether Asian voles might harbor human-pathogenic variants. At least in Europe, voles and shrews are unlikely to serve as reservoir hosts for human infection.
In the USA, two major 16S rRNA gene variants of A. phagocytophilum have been described: the A. phagocytophilum Ap-ha and the A. phagocytophilum Ap-variant 1 strain. Both were defined by a two-base pair difference in the 16S rRNA gene [46] and it has been claimed that A. phagocytophilum Ap-ha is pathogenic for humans whereas A. phagocytophilum Ap-variant 1 is not [22]. However, single locus 16S rRNA gene-based typing of A. phagocytophilum has been proven to not reliably define A. phagocytophilum genotypes [29,30,31, 47,48,49]. Here, the I. scapularis strains CRT35 (ST 217), CRT38 (ST 216) and CRT53-1 (ST 218) that have been classified as non-human pathogenic A. phagocytophilum Ap-variant 1 [36] were found in MLST cluster 8, whereas the human strains from the USA were part of cluster 1. However, concerning the ankA-based typing, seven North American strains from humans were part of cluster 12 together with the three A. phagocytophilum Ap-variant 1 isolates from I. scapularis. In our opinion, this finding questions the classification of A. phagocytophilum Ap-variant 1 as non-human pathogenic using the 16S rRNA gene as a marker.
The epidemiological differences between North America and Europe regarding the incidence of human infection are not explained when MLST and ankA-based typing are considered, because human strains from North America and Europe were both part of MLST cluster 1. Further, the North American ankA gene clusters 11 and 12 were highly identical at the nucleotide level to the European cluster 1 with 97.4% and 95.2%, respectively.
In contrast to the drhm status, the concordance between host and MLST cluster was 88% indicating a host association of certain A. phagocytophilum variants. Bird-related MLST cluster 7 and avian ankA clusters 6 and 9 are newly described here. Bird-specific A. phagocytophilum strains have been reported before because groEL ecotype IV was restricted to samples from a blackbird and from five ticks feeding on blackbirds [50]. Recently, a bird-associated groEL cluster 7 has been characterized as well [51].
Ixodes persulcatus, I. pavlovskyi ticks and their hybrids from the Asian part of Russia were restricted to MLST clusters 5 and 6 and ankA clusters 8 and 10. Ixodes persulcatus from its European distribution area was not investigated. It is therefore unclear whether the clustering is reflected by tick species or geography.
The ankA sequences from cluster 7 found in a horse and two I. ricinus ticks were probably recombinants of cluster 1 and 4 sequences. It has been shown before that the ankA gene might undergo recombination [39]. Here, the infection of a horse with an A. phagocytophilum strain of ankA cluster 4 was observed for the first time. However, all 44 equine samples of European origin were part of cluster 1. A double or triple infection with A. phagocytophilum variants belonging to different ankA clusters was detected in 17 cases in cattle, deer and a tick. In roe deer, this phenomenon has been observed before [52]. Thus, multiple infections as a prerequisite for recombination occur.
In general, the genetic diversity was higher in Europe than in North America and Asia because European samples belonged to five MLST and eight ankA clusters, whereas North American and Asian strains were part of two MLST and ankA clusters. However, a considerable sampling bias must be taken into account, as 585, 72 and 29 strains were of European, Asian and North American origin, respectively.
The concordance between MLST cluster and continent was 68%, and between ankA gene cluster and continent 100%. Thus, both typing methods were geographically informative. A broader host spectrum especially from North America and Asia should be typed by MLST and ankA-gene-based typing to further elucidate host association and geographical distribution of distinct A. phagocytophilum strains.
Conclusions
The absence of the drhm gene in A. phagocytophilum does not seem to be associated with pathogenicity for humans per se, because all 35 European strains from human granulocytic anaplasmosis cases were drhm positive. The epidemiological differences between North America and Europe concerning the incidence of human A. phagocytophilum infection are not explained by strain divergence based on MLST and ankA gene-based typing. The concordance between host and MLST cluster was 88% which indicates a host association of certain A. phagocytophilum strains. Human strains from Europe clustered most closely together with hedgehogs and wild boars indicating that they might serve as reservoir hosts for human infection.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its additional files. All nucleotide sequences are available at GenBank. GenBank accession numbers are shown in Additional file 1: Table S1. The MLST profiles for samples without ambiguous nucleotides were submitted to the A. phagocytophilum isolates database hosted on PubMLST (https://pubmlst.org/aphagocytophilum/).
Change history
30 September 2020
An amendment to this paper has been published and can be accessed via the original article.
Abbreviations
- CC:
-
Clonal complex
- MLST:
-
Multilocus sequence typing
- NJ:
-
Neighbor-joining
- SNP:
-
Single nucleotide polymorphism
- ST:
-
Sequence type
References
Dumler JS, Barbet AF, Bekker CPJ, Dasch GA, Palmer GH, Ray SC, et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51:2145–65.
Stuen S, Granquist EG, Silaghi C. Anaplasma phagocytophilum - a widespread multi-host pathogen with highly adaptive strategies. Front Cell Infect Microbiol. 2013;3:31.
Ismail N, McBride JW. Tick-borne emerging infections: ehrlichiosis and anaplasmosis. Clin Lab Med. 2017;37:317–40.
Matei IA, Estrada-Peña A, Cutler SJ, Vayssier-Taussat M, Varela-Castro L, Potkonjak A, et al. A review on the eco-epidemiology and clinical management of human granulocytic anaplasmosis and its agent in Europe. Parasit Vectors. 2019;12:599.
Xu B, Liu L, Hunang X, Ma H, Zhang Y, Du Y, et al. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: discovery of a new bunyavirus. PLoS Pathog. 2011;7:e1002369.
Zhang L, Wang G, Liu Q, Chen C, Li J, Long B, et al. Molecular analysis of Anaplasma phagocytophilum isolated from patients with febrile diseases of unknown origin. PLoS ONE. 2013;8:e57155.
Ohashi N, Gaowa W, Kawamori F, Wu D, Yoshikawa Y, et al. Human granulocytic anaplasmosis, Japan. Emerg Infect Dis. 2013;19:289–92.
Kim KH, Yi J, Oh WS, Kim NH, Choi SJ, Choe PG, et al. Human granulocytic anaplasmosis, South Korea, 2013. Emerg Infect Dis. 2014;20:1708–11.
Tsai KH, Chung LH, Chien CH, Tung YJ, Wei HY, Yen TY, et al. Human granulocytic anaplasmosis in Kinmen, an offshore island of Taiwan. PLoS Negl Trop Dis. 2019;13:e0007728.
Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364:1523–32.
Cui F, Cao HX, Wang L, Zhang SF, Ding SJ, Yu XJ, et al. Clinical and epidemiological study on severe fever with thrombocytopenia syndrome in Yiyuan County, Shandong Province, China. Am J Trop Med Hyg. 2013;88:510–2.
Wormser GP. Accuracy of diagnosis of human granulocytic anaplasmosis in China. Emerg Infect Dis. 2016;22:1728–31.
Carrade DD, Foley JE, Borjesson DL, Sykes JE. Canine granulocytic anaplasmosis: a review. J Vet Intern Med. 2009;23:1129–41.
Saleem S, Ijaz M, Farooqi SH, Ghaffar A, Ali A, Iqbal K, et al. Equine granulocytic anaplasmosis 28 years later. Microb Pathog. 2018;119:1–8.
Lappin MR. Update on flea and tick associated diseases of cats. Vet Parasitol. 2018;254:26–9.
Atif FA. Anaplasma marginale and Anaplasma phagocytophilum: rickettsiales pathogens of veterinary and public health significance. Parasitol Res. 2015;114:3941–57.
Woldehiwet Z. The natural history of Anaplasma phagocytophilum. Vet Parasitol. 2010;167:108–22.
Ben Said M, Belkahia H, Messadi L. Anaplasma spp. in North Africa: a review on molecular epidemiology, associated risk factors and genetic characteristics. Ticks Tick Borne Dis. 2018;9:543–55.
Djiba ML, Mediannikov O, Mbengue M, Thiongane Y, Molez JF, Seck MT, et al. Survey of Anaplasmataceae bacteria in sheep from Senegal. Trop Anim Health Prod. 2013;45:1557–61.
Aktas M, Ozubek S. Bovine anaplasmosis in Turkey: first laboratory confirmed clinical cases caused by Anaplasma phagocytophilum. Vet Microbiol. 2015;178:246–51.
Zhan L, Cao WC, Jiang JF, Zhang XA, Wu XM, Zhang WY, et al. Anaplasma phagocytophilum in livestock and small rodents. Vet Microbiol. 2010;144:405–8.
Dugat T, Lagreé AC, Maillard R, Boulouis HJ, Haddad N. Opening the black box of Anaplasma phagocytophilum diversity: current situation and future perspectives. Front Cell Infect Microbiol. 2015;5:61.
Ogden NH, Bown K, Horrocks BK, Woldehiwet Z, Bennett M. Granulocytic Ehrlichia infection in ixodid ticks and mammals in woodlands and uplands of the U.K. Med Vet Entomol. 1998;12:423–9.
Krücken J, Schreiber C, Maaz D, Kohn M, Demeler J, Beck S, et al. A novel high-resolution melt PCR assay discriminates Anaplasma phagocytophilum and “Candidatus Neoehrlichia mikurensis”. J Clin Microbiol. 2013;51:1958–61.
Telford SRI, Dawson JE, Katavolos P, Warner CK, Kolbert CP, Persing DH. Perpetuation of the agent of human granulocytic ehrlichiosis in a deer tick-rodent cycle. Proc Natl Acad Sci USA. 1996;93:6209–14.
Stafford KCI, Massung RF, Magnarelli LA, IJdo JW, Anderson JF. Infection with agents of human granulocytic ehrlichiosis, Lyme disease, and babesiosis in wild white-footed mice (Peromyscus leucopus) in Connecticut. J Clin Microbiol. 1999;37:2887–92.
Al-Khedery B, Barbet AF. Comparative genomics identifies a potential marker of human-virulent Anaplasma phagocytophilum. Pathogens. 2014;3:25–35.
Foley J, Stephenson N, Cubilla MP, Qurollo B, Breitschwerdt EB. A putative marker for human pathogenic strains of Anaplasma phagocytophilum correlates with geography and host, but not human tropism. Ticks Tick Borne Dis. 2016;7:390–3.
Huhn C, Winter C, Wolfsperger T, Wüppenhorst N, Strašek Smrdel K, Skuballa J, et al. Analysis of the population structure of Anaplasma phagocytophilum using multilocus sequence typing. PLoS One. 2014;9:e93725.
Scharf W, Schauer S, Freyburger F, Petrovec M, Schaarschmidt-Kiener D, Liebisch G, et al. Distinct host species correlate with Anaplasma phagocytophilum ankA gene clusters. J Clin Microbiol. 2011;49:790–6.
von Loewenich FD, Baumgarten BU, Schröppel K, Geißdörfer W, Röllinghoff M, Bogdan C. High diversity of ankA sequences of Anaplasma phagocytophilum among Ixodes ricinus ticks in Germany. J Clin Microbiol. 2003;41:5033–40.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Severiano A, Pinto FR, Ramirez M, Carriço JA. Adjusted Wallace coefficient as a measure of congruence between typing methods. J Clin Microbiol. 2011;49:3997–4000.
Mukhacheva TA, Shaikhova DR, Kovalev SY, von Loewenich FD. Phylogeographical diversity of Anaplasma phagocytophilum in the Asian part of Russia based on multilocus sequence typing and analysis of the ankA gene. Infect Genet Evol. 2020;80:104234.
Langenwalder DB, Silaghi C, Nieder M, Pfeffer M, von Loewenich FD. Co-infection, reinfection and superinfection with Anaplasma phagocytophilum strains in a cattle herd based on ankA gene and multilocus sequence typing. Parasit Vectors. 2020;13:157.
Massung RF, Levin ML, Munderloh UG, Silverman DJ, Lynch MJ, Gaywee JK, et al. Isolation and propagation of the Ap-Variant 1 strain of Anaplasma phagocytophilum in a tick cell line. J Clin Microbiol. 2007;45:2138–43.
Hoffman T, Wilhelmsson P, Barboutis C, Fransson T, Jaenson TGT, Lindgren PE, et al. A divergent Anaplasma phagocytophilum variant in an Ixodes tick from a migratory bird; Mediterranean basin. Infect Ecol Epidemiol. 2020;10:1729653.
Storey JR, Doros-Richert LA, Gingrich-Baker C, Munroe K, Mather TN, Coughlin RT, et al. Molecular cloning and sequencing of three granulocytic Ehrlichia genes encoding high-molecular-weight immunoreactive proteins. Infect Immun. 1998;66:1356–63.
Majazki J, Wüppenhorst N, Hartelt K, Birtles R, von Loewenich FD. Anaplasma phagocytophilum strains from small mammals exhibit specific ankA gene sequences. BMC Vet Res. 2013;9:235.
Foley JE, Nieto NC, Barbet A, Foley P. Antigen diversity in the parasitic bacterium Anaplasma phagocytophilum arises from selectively-represented, spatially clustered functional pseudogenes. PLoS One. 2009;4:e8265.
Barlough JE, Madigan JE, DeRock E, Dumler JS, Bakken JS. Protection against Ehrlichia equi is conferred by prior infection with the human granulocytotropic Ehrlichia (HGE agent). J Clin Microbiol. 1995;33:3333–4.
Pusterla N, Pusterla JB, Braun U, Lutz H. Experimental cross-infections with Ehrlichia phagocytophila and human graunlocytic ehrlichia-like agent in cows and horses. Vet Rec. 1999;145:311–4.
Scorpio DG, Dumler JS, Barat NC, Cook JA, Barat CE, Stillman BA, et al. Comparative strain analysis of Anaplasma phagocytophilum infection and clinical outcomes in a canine model of granulocytic anaplasmosis. Vector Borne Zoonotic Dis. 2011;11:223–9.
Lagrée AC, Rouxel C, Kevin M, Dugat T, Girault G, Durand B, et al. Co-circulation of different A phagocytophilum variants within cattle herds and possible reservoir role for cattle. Parasit Vectors. 2018;11:163.
Tegtmeyer P, Ganter M, von Loewenich FD. Simultaneous infection of cattle with different Anaplasma phagocytophilum variants. Ticks Tick Borne Dis. 2019;10:1051–6.
Courtney JW, Dryden RL, Montgomery J, Schneider BS, Smith G, Massung RF. Molecular characterization of Anaplasma phagocytophilum and Borrelia burgdorferi in Ixodes scapularis ticks from Pennsylvania. J Clin Microbiol. 2003;41:1569–73.
Casey AN, Birtles RJ, Radford AD, Bown KJ, French NP, Woldehiwet Z, et al. Groupings of highly similar major surface protein (p44)-encoding paralogues: a potential index of genetic diversity amongst isolates of Anaplasma phagocytophilum. Microbiology. 2004;150:727–34.
Bown KJ, Lambin X, Ogden NH, Petrovec M, Shaw SE, Woldehiwet Z, et al. High-resolution genetic fingerprinting of European strains of Anaplasma phagocytophilum by use of multilocus variable-number tandem-repeat analysis. J Clin Microbiol. 2007;45:1771–6.
Bown KJ, Lambin X, Ogden NH, Begon M, Telford G, Woldehiwet Z, et al. Delineating Anaplasma phagocytophilum ecotypes in coexisting, discrete enzootic cycles. Emerg Infect Dis. 2009;15:1948–54.
Jahfari S, Coipan EC, Fonville M, van Leeuwen AD, Hengeveld P, Heylen D, et al. Circulation of four Anaplasma phagocytophilum ecotypes in Europe. Parasit Vectors. 2014;7:365.
Jaarsma RI, Sprong H, Takumi K, Kazimirova M, Silaghi C, Mysterud A, et al. Anaplasma phagocytophilum evolves in geographical and biotic niches of vertebrates and ticks. Parasit Vectors. 2019;12:328.
Jouglin M, Chagneau S, Faille F, Verheyden H, Bastian S, Malandrin L. Detecting and characterizing mixed infections with genetic variants of Anaplasma phagocytophilum in roe deer (Capreolus capreolus) by developing an ankA cluster-specific nested PCR. Parasit Vectors. 2017;10:377.
Acknowledgements
The work presented here is part of the MD thesis of Denis B. Langenwalder. We are grateful to Eva Olsson (National Veterinary Institute, Stockholm, Sweden) for her generous supply of blood infected with the A. phagocytophilum rosa isolate.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
DL and FvL designed the study. DL, SS and FvL performed the genetic characterization. FvL undertook the phylogenetic analyses. CS, JS, NP, IM, AM, UG, JZ, MG, TH, ES and MP contributed materials. FvL wrote the manuscript. All authors critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The samples from humans, domestic and farm animals were obtained as part of routine diagnostic evaluations. Part of the ovine samples were collected for the Q-fever surveillance program of the federal state of Lower-Saxony. The red foxes were killed for the official rabies monitoring program of the federal state of Brandenburg. All other wild animals were shot by professional hunters during the annual hunting seasons. Hedgehogs from a captive population in the federal state of Baden-Württemberg and from hedgehog caretaking stations in various areas of Germany were bled from the saphenous vein or died naturally. None of the hedgehogs were sacrificed for this study. For Germany, permission to trap rodents was given by the District Government Stuttgart, Germany. For the UK, protocols for the handling and sampling of wild small mammals were approved by the University of Liverpool Committee on Research Ethics. For Romania, samples were collected based on permits issued by ARBDD. For Russia, rodent samples were kindly provided by the Institute of Plant and Animal Ecology (Yekaterinburg, Russia).
Consent for publication
Written informed consent was obtained from the patients and owners, respectively.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1
. Reference, host species, ST, CC, MLST cluster, allele numbers, ankA gene cluster, drhm status, APH_0919/APH_0922 status, country of origin, year of sampling, disease state of the host and GenBank accession numbers for the 686 A. phagocytophilum strains.
Additional file 2: Text S1.
Conditions for amplification by nested PCR and sequencing of the complete open reading frame of ankA gene cluster 6.
Additional file 3: Figure S1.
Phylogenetic tree calculated from the concatenated housekeeping gene sequences of 520 samples without ambiguous nucleotides. Tree construction was achieved by the NJ method using the Jukes-Cantor matrix with the complete deletion option. Bootstrap values ≥ 64% are shown next to the branches. The scale-bar indicates the number of nucleotide substitutions per site. The final data set contained 2877 positions. Identical ST are displayed only once per species. The number in parenthesis indicates the frequency with which the respective ST was found. Key: red circles, sequences from humans, dogs, horses and cats; dark blue diamonds, sequences from domestic ruminants; light blue diamonds, sequences from wild ruminants; green triangles, sequences from small mammals; yellow squares, sequences from wild boars; purple triangles, sequences from red foxes; white triangles, sequences from ticks.
Additional file 4: Figure S2.
Phylogenetic tree calculated from the ankA sequences of 623 samples without ambiguous nucleotides. Tree construction was achieved by the NJ method using the Jukes-Cantor matrix with the complete deletion option. Bootstrap values ≥ 69% are shown next to the branches. The scale-bar indicates the number of nucleotide substitutions per site. The final data set contained 510 positions. Identical ankA sequences are displayed only once per species. The number in parenthesis indicates the frequency with which the respective sequence was found. Key: red circles, sequences from humans, dogs, horses and cats; dark blue diamonds, sequences from domestic ruminants; light blue diamonds, sequences from wild ruminants; green triangles, sequences from small mammals; yellow squares, sequences from wild boars; purple triangles, sequences from red foxes; pink square, sequence from a bird, white triangles, sequences from ticks.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Langenwalder, D.B., Schmidt, S., Silaghi, C. et al. The absence of the drhm gene is not a marker for human-pathogenicity in European Anaplasma phagocytophilum strains. Parasites Vectors 13, 238 (2020). https://doi.org/10.1186/s13071-020-04116-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04116-z