
Abstract
Background
A century ago, pantheras were abundant across Asia. Illegal hunting and trading along with loss of habitat have resulted in the designation of Panthera as a genus of endangered species. In addition to the onslaught from humans, pantheras are also susceptible to outbreaks of several infectious diseases, including babesiosis. The latter is a hemoprotozoan disease whose causative agents are the eukaryotic parasites of the apicomplexan genus Babesia. Babesiosis affects a varied range of animals including humans (Homo sapiens), bovines (e.g. Bos taurus), pantheras (e.g. Panthera tigris, P. leo, P. pardus) and equines. Babesia spp. are transmitted by the tick vector Ixodes scapularis or ticks of domestic animals, namely Rhipicephalus (Boophilus) microplus and R. (B.) decoloratus. At the level of protein translation within these organisms, the conserved aminoacyl tRNA synthetase (aaRS) family offers an opportunity to identify the sequence and structural differences in the host (Panthera) and parasites (Babesia spp.) in order to exploit these for drug targeting Babesia spp.
Methods
Using computational tools we investigated the genomes of Babesia spp. and Panthera tigris so as to annotate their aaRSs. The sequences were analysed and their subcellular localizations were predicted using Target P1.1, SignalP 3.0, TMHMM v.2.0 and Deeploc 1.0 web servers. Structure-based analysis of the aaRSs from P. tigris and its protozoan pathogens Babesia spp. was performed using Phyre2 and chimera.
Results
We identified 33 (B. bovis), 34 (B. microti), 33 (B. bigemina) and 33 (P. tigris) aaRSs in these respective organisms. Poor sequence identity (~ 20–50%) between aaRSs from Babesia spp. and P. tigris was observed and this merits future experiments to validate new drug targets against Babesia spp.
Conclusions
Overall this work provides a foundation for experimental investigation of druggable aaRSs from Babesia sp. in an effort to control Babesiosis in Panthera.
Similar content being viewed by others

Background
Panthera is a genus within the family Felidae, comprising five species: P. leo, P. onca, P. uncia, P. pardus and P. tigris. Sadly, Panthera spp. are now endangered due to illegal hunting, loss of habitat and trading [1]. Furthermore, Panthera spp. have been subjected to outbreaks of babesiosis which is a zoonotic, hemoprotozoan disease caused by tick-borne piroplasmids of the genus Babesia [2]. Babesia spp. are the second most common haemoparasites of mammals after trypanosomes. They have a wide range of hosts including humans (Homo sapiens), bovines (e.g. Bos taurus), pantheras (e.g. P. tigris, P. leo, P. pardus), equines, and a number of other mammal and bird species [3,4,5]. To date, more than 100 species of Babesia have been identified, which may be attributed to the fact that Babesia is not strictly host-specific. While babesiosis infection is most commonly asymptomatic, the disease can cause fever, fatigue and haemolytic anaemia that at times can be fatal [5]. Babesiosis is believed to be a major cause of mortality for big cats held in captivity [6,7,8,9,10,11,12,13,14,15,16,17] (Table 1). Species of Babesia infecting Panthera spp. are usually Babesia leo or Babesia canis [12]. A number of effective therapeutics are currently in use which include imidocarb dipropionate, diminazene aceturate, clindamycin, azithromycin and antiparasitic drugs such as atovaquone-azithromycin [14, 18]. The current drug application is often accompanied by intravenous fluids and blood transfusion; these are difficult to administer in the wilderness [14, 18] and therefore developing new and effective therapeutics against babesiosis infection is very important for animal conservation.
Being a crucial part of the protein translation machinery, aminoacyl tRNA synthetases (aaRSs) are currently being studied as drug targets for several pathogens [19,20,21,22,23,24,25,26,27,28]. The aaRSs catalyse the addition of amino acids to respective tRNAs, and are usually multi-domain proteins with an anti-codon binding domain (ABD) as well as a conserved catalytic domain. Often aaRSs possess editing domains that remove incorrectly charged tRNAs. The 20 aaRSs fall into either Class I or Class II based on their modes of substrate binding and structural folds. Class I aaRSs contain a Rossmann fold which is characterized by KMSKS and HIGH motifs. Class II aaRSs have three motifs and another different β-sheet arrangement [29]. The aaRSs can localise to different subcellular compartments where they are responsible for protein synthesis [30]. More recently, aaRSs have been explored from several eukaryotic pathogens such as Plasmodium, Toxoplasma and Leishmania as potential druggable targets [19,20,21,22,23,24,25,26,27,28]. In the present study we used bioinformatics tools to investigate the genomes of Babesia spp. (B. bovis, B. microti and B. bigemina) and P. tigris and annotated their aaRSs. We identified 33 (B. bovis), 34 (B. microti), 33 (B. bigemina) and 33 (P. tigris) aaRSs in these organisms. We have analysed the aaRSs sequences and predicted their cellular localizations. Finally, we determined the percentage sequence identity in aaRSs from Babesia spp. with respect to P. tigris to identify divergent aaRSs. This work hence provides a resource for launching experimental investigations of druggable Babesia aaRSs in an effort to control babesiosis.
Methods
Open reading frames (ORFs) for the annotated B. bovis, B. microti, B. bigemina and P. tigris were downloaded from the National Center for Biotechnology Information (NCBI) database of genomes and computationally translated. All 20 aaRSs were individually used to perform BLAST searches against non-redundant databases available at the NCBI (Additional file 1: Dataset S1). For each, hidden Markov models (HMMs) [31] were generated using homologs identified from BLAST searches. Sequence alignment and modeling software system (SAM) was used to generate multiple sequence alignment (MSA), and the HMMER package was used for building profile HMM for each MSA. Significantly similar matches for each HMM profile were identified within the genomic sequences for the three Babesia spp. and for P. tigris. Genome sequences were analysed using blastx and four additional aaRSs were identified. Pfam domains were assigned to computationally translated sequences using the ‘Pfam web server for analysis of domains’ [32]. Details on domain functions were also obtained from the Pfam database. Subcellular localizations were predicted using Target P1.1 [33], SignalP 3.0 [34], TMHMM Server v.2.0 [35] and Deeploc 1.0 web server [36]. The presence of secretory signals was detected using Target P1.1 and SignalP, while transmembrane domains were detected using TMHMM. Furthermore, DeepLoc-1.0 was used to predict the subcellular localisations. If a protein was predicted to have a signal peptide and at least one transmembrane domain and predicted to be either mitochondrial or apicoplastic via DeepLoc-1.0 it was annotated as an organellar protein. Homology modelling for protein sequences was performed using Phyre2 server [37] and Chimera [38] was used for structure visualization and analyses.
Results
The Babesia spp. that infect Panthera spp. are usually B. leo or B. canis [12]. Since the genomes of B. leo and B. canis are currently unavailable, we used the open reading frames (ORFs) and genome sequences of B. bovis, B. microti and B. bigemina that are known to infect bovine species and at times Panthera spp. as well [39]. Genome sizes for B. bovis, B. microti and B. bigemina are ~ 8.2, ~ 6.3 and ~ 13.8 Mbp, respectively. The aaRSs in the genomes of B. bovis, B. microti, B. bigemina and P. tigris were identified via HMM-based searches [31]. We annotated aaRSs corresponding to each of the 20 amino acids in these four studied genomes (Tables 2, 3, 4, 5, Additional file 2: Tables S1–S4). Our analyses have annotated 33 aaRSs in B. bovis, 34 aaRSs in B. microti, 33 aaRSs in B. bigemina and 33 aaRSs in P. tigris (Fig. 1a).

Aminoacyl-tRNA synthetases (aaRSs) and their sites. Number of annotated aaRSs (a) and predicted subcellular localisations (b) for the studied Babesia spp. (B. bovis, B. microti and B. bigemina) and Panthera tigris
The predicted compartmentalisations of all aaRSs were analysed based on the presence of signal sequences using Target P1.1 [33], SignalP 3.0 [34], TMHMM v.2.0 [35] and Deeploc 1.0 web servers [36]. Our analyses predicted subcellular localisation for multiple aaRSs with gene copies in both cytoplasm and in an organelle (apicoplast/mitochondria) for Babesia spp. (Fig. 1b, Tables 2, 3, 4). For P. tigris, the predicted localisations cover both cytoplasm and mitochondria (Fig. 1b, Table 5). Hereafter, the aaRSs with a predicted subcellular localisation to apicoplast in Babesia spp. will be referred to as organellar. We emphasize the need for experimental validation to positively assess the localisations as aaRSs may co-reside in mitochondria [40,41,42]. In B. bovis, the predictions are for 16 aaRSs as cytoplasmic, 12 aaRSs as organellar and 5 aaRSs [cysteinyl-tRNA synthetase (CRS), alanyl-tRNA synthetase (ARS), aspartyl-tRNA synthetase (DRS), glycyl-tRNA synthetase (GRS), threonyl-tRNA synthetase (TRS)] as potential co-localisers between the cytoplasm and organelle (Fig. 1b, Table 2). In B. microti, 16 aaRSs are cytoplasmic, 14 aaRSs are organellar and 4 aaRSs (CRS, ARS, DRS, GRS) are possibly dual-localised (Fig. 1b, Table 3). In B. bigemina, 16 aaRSs are cytoplasmic, 12 aaRSs are organellar and 5 aaRSs (CRS, ARS, DRS, GRS, TRS) may reside in both the cytoplasm and organelle (Fig. 1b, Table 4). Our HMM profile-based analyses failed to predict a complete set of 20 organellar aaRSs for three aaRSs [glutaminyl-tRNA synthetase (QRS), valyl-tRNA synthetase (VRS), phenylalanyl-tRNA synthetase (FRS)] in B. bovis, three aaRSs (VRS, FRS, TRS) in B. microti and three aaRSs (QRS, VRS, FRS) in B. bigemina. In an attempt to predict the missing organellar aaRSs within the Babesia spp., we searched the non-redundant protein sequence databases. Our searches resulted in identification of tRNA synthetases (isoleucine, leucine, methionine and valine; I, L, M and V, respectively) in B. bovis, B. microti and B. bigemina with ~ 55%, 54% and 55% sequence identity, respectively, with, for example, predicted VRS from each of the three species (Tables 2, 3, 4). Searches using Blastx resulted in the identification of four additional aaRSs: methionyl-tRNA synthetase (MRS) (cytoplasmic) in B. bovis; QRS (organellar) and tryptophanyl-tRNA synthetase (WRS) (organellar) in B. microti; and tyrosyl-tRNA synthetase (YRS) (cytoplasmic) in B. bigemina. Based on previous reports, a non-discriminating organellar glutamyl-tRNA synthetase (ERS) is known to convert tRNAGln into a misacylated Glu-tRNAGln [43]. The misacylated Glu-tRNAGln is then converted into Gln-tRNAGln by an organellar glu-tRNAGln amidotransferase [43]. While this explains the absence of QRS in the organelles of the three Babesia spp., there is a need to identify the missing organellar TRS in B. microti to complete its set of 20 aaRSs. A plausible reason for failure to predict a complete set may be poor sequence identity with aaRS homologs that were used for building profile HMMs.
In contrast to Babesia spp., for P. tigris, 19 aaRSs are present within the cytoplasm including a bi-functional aaRS, namely glutamyl-prolyl-tRNA synthetase (EPRS). In addition, 14 aaRSs are mitochondrial in P. tigris (Fig. 1b, Table 5). These observations are supported by previous reports which show that despite the occurrence of protein synthesis in the three distinct compartments in Plasmodium falciparum, this apicomplexan does not possess three complete sets of 20 aaRSs for each compartment (i.e. not 60 but has 36 aaRSs) [42]. Furthermore, while cytoplasmic aaRSs supposedly can drive translation in that compartment, several studies have revealed that some aaRSs have the potential to be multi-localized [30]. When a complete set of 20 aaRSs is not available for organellar protein synthesis, as an example, the mitochondria are able to import charged tRNAs, as shown in yeast, Leishmania, Trypanosoma, Plasmodium and Toxoplasma [44,45,46,47,48]. The requirement for their own mitochondrial aaRSs in these organisms is therefore bypassed as charged tRNAs are transported between the cellular compartments, thereby compensating for the absence of any aaRS(s) [40,41,42].
One aaRS enzyme is particularly interesting as it can occur as heterodimers of two different genes: the FRSs. It is well established that FRSs can exist in (αβ)2 subunit organization, with exceptions of mitochondrial FRSs from yeast and Homo sapiens and possibly others that are monomeric [40]. Our analyses predicted cytoplasmic FRS α and FRS β subunit in all the four studied genomes. A third gene for FRS was predicted in B. bovis and B. bigemina to localise within the cytoplasm, while the gene for FRS in B. microti was predicted to localise within the organelle (Tables 2, 3, 4). Mitochondria of the apicomplexan P. falciparum import tRNAs along with an active FRS, and so we analysed the FRSs from B. bovis, B. microti and B. bigemina using Target P1.1 [33] and SignalP 3.0 [34] for the presence of signal peptide and transmembrane domain [35]. The encoded cytoplasmic FRSs from B. bovis and B. bigemina showed no identifiable organelle targeting sequence or transmembrane domains. However, a transmembrane domain was predicted within the FRS from B. microti (Table 3). These predicted FRSs need to be characterised experimentally for validation.
Domain architectures of aminoacyl tRNA synthetases in Babesia spp. and Panthera tigris
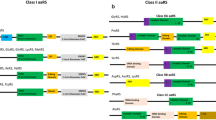
Based on HMM searches, we identified a total of 33 aaRSs in B. bovis, 34 aaRSs in B. microti, 33 aaRSs in B. bigemina and 33 aaRSs in P. tigris. Amongst these, 18 aaRSs belong to Class I and 15 aaRSs to Class II in B. bovis; 18 to Class I and 16 Class II in B. microti; 18 to Class I and 15 to Class II in B. bigemina; and 16 to Class I and 17 to Class II in P. tigris (Tables 2, 3, 4, 5). Pfam server predicted catalytic domains for all these aaRSs. Additionally, anticodon-binding domains (ABD), N-terminal domain (NTD) and C-terminal domain (CTD), DALR [aspartate (D), alanine (A), leucine (L), arginine (R)], DHHA [aspartate (D), histidine (H), histidine (H), alanine (A)], SAD (second additional domain), WHEP-TRS (Wh-T) and glutathione S-transferase (GST) domains were predicted.
Panthera tigris aaRSs are also predicted to contain DALR and DHHA domains (Fig. 2). DALR is an all alpha-helical anti-codon binding domain named after the characteristic conserved amino acids: aspartate (D), alanine (A), leucine (L) and arginine (R). The DHHA domain is named after conserved amino acids: aspartate (D), histidine (H), histidine (H), alanine (A) in the cytoplasmic version of ARS [49]. Furthermore, in P. tigris a GST-like domain was predicted at the N-termini of dual-localised MRS, EPRS and cytoplasmic VRS (Fig. 2). While the functional implications of the GST domains may vary, these seem to be involved in protein assembly and folding [50, 51]. Several studies have reported that GST or GST-like domains play an important role in the formation of complexes between aaRSs and multifunctional factors (p18, p38, p43) [52]. Additionally, Wh-T domain was also predicted in cytoplasmic MRS, WRS, GRS and in the dual-localised HRS/EPRSs (Fig. 2). Wh-T domain has a characteristic helix-turn-helix motif, with consensus lysine and arginine residues. These residues are critical for protein-protein interactions within the multi-synthetase complex [53,54,55,56].

NCBI reference sequence and pfam assigned domains in B. bovis. Abbreviations: aaRSs, aminoacyl-tRNA synthetase; CD, catalytic domain; ABD, anticodon binding domain; SAD, second additional domain; NTD, N-terminal domain; CTD, C-terminal domain; ATED, aminoacyl tRNA editing domain; TBA, tRNA binding arm; GST, glutathione S-transferase
We observed no distinct domains amongst the aaRSs from B. bovis, B. microti and B. bigemina (Figs. 3, 4, 5). In Babesia spp., RRS and SRS were found to have an additional NTD, while their PRSs have an additional CTD, with no predicted function for either of them (Figs. 3, 4, 5) [57, 58]. Interestingly, for B. microti, a ZnF domain was annotated for the organellar IRS. The C-terminal peptide (CP) and C-terminal enzyme-bound zinc participate in aminoacylation of tRNAIle (Fig. 4). These domains were not found for other two species, B. bovis and B. bigemina. The differences observed within the aaRSs of three Babesia spp. and P. tigris provide a unique opportunity to exploit some aaRSs as potential drug targets.

NCBI reference sequence and domain annotations of aaRSs in B. microti. Abbreviations: aaRSs, aminoacyl-tRNA synthetases; ABD, anticodon binding domain; SAD second additional domain; NTD, N-terminal domain; CTD, C-terminal domain; ATED, aminoacyl tRNA editing domain; GST, glutathione S-transferase domain

Domain annotations and NCBI reference sequence for aaRSs in B. bigemina. Abbreviations: aaRSs, aminoacyl-tRNA synthetases; ABD, anticodon binding domain; SAD, second additional domain; NTD, N-terminal domain; CTD, C-terminal domain; ATED, aminoacyl tRNA editing domain; TBA, tRNA binding arm, GST, glutathione S-transferase domain

NCBI reference sequence and Pfam assigned domains in P. tigris. Abbreviations: aaRSs, aminoacyl-tRNA synthetases; ABD, anticodon binding domain; TBA, tRNA-binding arm; NTD, N-terminal domain; CTD, C-terminal domain; ATED, aminoacyl tRNA editing domain; GST, glutathione S-transferase domain; WT, WHEP-TRS domain
We subsequently studied the sequence identities between Babesia spp. (B. bovis, B. microti and B. bigemina) and P. tigris (Tables 2, 3, 4, 5). While the aaRSs identified in the three Babesia spp. share similarities with their homologs in P. tigris, our analyses reveal poor identity in the range of ~ 20–50% (Tables 2, 3, 4, 5). The aaRSs with less than 30% identity with the corresponding protein in P. tigris include ERS, IRS, MRS, WRS, YRS, HRS, NRS and SRS (Tables 2, 3, 4, 5). Out of eight aaRSs with sequence identity less than 30% between Babesia spp. and P. tigris, ERS, MRS, YRS and NRS are cytoplasmic. Among the aaRSs with sequence identity less than 30%, IRS and LRS are the targets of drugs currently in use, i.e. IRS (bacterial: muciprocin, available as a 2% topical preparation) and LRS (fungal: tavaborole/AN2690, available as a 5% topical preparation) [19, 24, 59,60,61].
Structure-based analysis of cladosporin binding sites in Babesia and Panthera KRSs
The Plasmodium KRS (for cladosporin, CLD) and PRS (for halofuginone) are currently being studied as potential drug targets [19, 24, 59,60,61]. We analysed CLD which, along with its analogs, is being investigated as an inhibitor of P. falciparum-KRS (Pf-KRS) and of various other pathogen KRSs [21, 62]. Several amino acid residues in CLD binding pocket are highly conserved except at two positions near the ATP binding pocket and adjacent to CLD methyl moiety (Fig. 6a). The basis for CLD selectivity has been ascribed to these two key selectivity residues that show clear divergence across species. As proof of concept, we conducted a structural analysis of CLD bound Homo sapiens KRS (Hs-KRS) (PDB: 4YCU) and Plasmodium falciparum KRS (Pf-KRS) (PDB: 4PG3) in comparison with the three Babesia spp. and P. tigris (Fig. 6b). We built a three-dimensional structure model for KRSs using Phyre2 (Protein Homology/AnalogY Recognition Engine)-based protein structure prediction [37]. The two key residues bestowing selectivity to CLD were analysed in Babesia spp. and P. tigris. In Pf-KRS, valine and serine (VS) occupy the two selectivity residue positions thereby providing tight binding. Our analysis here reveals the presence of structurally smaller residues, cysteine and serine (CS), in B. bovis and B. bigemina, with an exception of B. microti that has a smaller valine and a bigger non-favourable threonine (VT) at the site (Fig. 6b). In Hs-KRS, these two positions are occupied by bulkier glutamine-threonine (QT) residues that likely hamper CLD binding. The corresponding position in P. tigris is also occupied by these bulkier QT residues (Fig. 6b). It is noteworthy that biochemical analysis of recombinant KRSs has previously shown that CLD displays a nanomolar range potency of inhibition (IC50 ~ 40–90 nM) against Pf-KRS, which is ~ 500-fold higher when compared with Hs-KRS [20, 62, 63]. This suggests potential poor selectivity for P. tigris KRS due to the same bulkier residues (QT) at the CLD binding pocket in comparison to the smaller, more favourable residues (CS/VT) in Babesia spp. This analysis suggests that small molecule targeting of KRS active site in Babesia spp. is an attractive avenue from the perspective of developing anti-infectives.

Analysis of CLD binding site in KRSs from Babesia spp. and P. tigris. a Sequence alignment of KRSs. The two key residues are highlighted in a black box and other conserved residues responsible for CLD binding are shown in blue boxes. b Structural superimposition of Hs-KRS (PDB: 4YCU), Pf-KRS (PDB: 4PG3), P. tigris and three Babesia spp. (Bb, B. bovis; Bm, B. microti and Bg, B. bigemina) (built structure model, this study). The smaller CLD selectivity residues of cysteine-serine (for Bb, Bg) and valine-threonine (for Bm) may accommodate CLD in the binding pocket of Babesia KRSs. The bulkier glutamine-threonine (for Hs, H. sapiens; Pt, P. tigris) potentially hinder high potency CLD binding. Abbreviations: CLD, cladosporin; KRS, lysyl-tRNA synthetase
Discussion
The treatment of babesiosis presents an emerging challenge. It is a hemoprotozoan disease whose causative agents are apicomplexan Babesia spp. Panthera spp. have been subjected to outbreaks of babesiosis, caused by Babesia spp., the second most common haemoparasites of mammals after trypanosomes, with a worldwide distribution. The current regime of therapy for targeting babesiosis requires the use of antibiotics and antiparasitic drugs. While atovaquone plus azithromycin is used to treat most cases, clindamycin plus quinine is used in more severe cases with chances of relapses [19, 24, 59,60,61]. Therefore, new drugs with high specificity and low toxicity are desirable. The recent availability of Babesia spp. (B. bovis, B. microti and B. bigemina) genomes has paved the way for screening of new chemotherapy targets. In this study, we focused on the housekeeping enzymes aminoacyl-tRNA synthetases that are essential for protein synthesis and cell viability. Lately, parasitic, microbial and fungal aaRSs have been explored for druggability [19,20,21,22,23,24,25,26,27]. A bacterial IRS inhibitor, mupirocin (marketed as Bactroban), and a fungal LRS inhibitor, 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690), have been developed for human use [19, 24, 59,60,61]. Recent studies on potential anti-malarial compounds like CLD (against KRS) and halofuginone (against PRS) are also promising [19, 20, 22, 24, 59, 64]. Considering this evidence, we have identified Babesia aaRSs with poor sequence identity to Panthera aaRSs as proteins of interest [65, 66]. As proof-of-concept, we performed a structure-based analysis of P. tigris and Babesia KRSs and have indicated a potential for selective drug targeting (Fig. 6). Hence, our work here lays a foundation for the future to further investigate and exploit Babesia aaRSs as potential targets.
Conclusions
In the present study, we provide data on genome-wide identification and annotation of aaRSs from Babesia spp. and P. tigris. Poor sequence identity (~ 20–50%) between pathogen/host aaRS pairs offers a window for specific studies to explore druggability. This detailed genomic cataloguing of aaRSs from pathogenic Babesia merits future experiments to validate new drug targets against Babesia spp.
Availability of data and materials
Aminoacyl-tRNA synthetase sequences used to generate HMM profiles are provided in Additional file 1: Dataset S1. The datasets analysed during the present study are available in UniProt repository (https://www.uniprot.org) and are included within the article and Additional file 2: Tables S1–S4. The HMM profiles generated are available from the corresponding author upon request.
Abbreviations
- ARS:
-
alanyl-tRNA synthetase
- aaRSs:
-
aminoacyl-tRNA synthetase
- ABD:
-
anticodon binding domain
- ATED:
-
aminoacyl-tRNA editing domain
- CRS:
-
cysteinyl-tRNA synthetase
- CTD:
-
c-terminal domain
- DRS:
-
aspartyl-tRNA synthetase
- DALR:
-
aspartate (D), alanine (A), leucine (L), arginine (R)
- DHHA:
-
aspartate (D), histidine (H), histidine (H), alanine (A)
- EPRS:
-
glutamyl-prolyl-tRNA synthetase
- ERS:
-
glutamyl-tRNA synthetase
- FRS:
-
phenylalanyl-tRNA synthetase
- GRS:
-
glycyl-tRNA synthetase
- GST:
-
glutathione S-transferase
- HRS:
-
histidyl-tRNA synthetase
- HMM:
-
hidden Markov model
- IRS:
-
isoleucyl-tRNA synthetase
- KRS:
-
lysyl-tRNA synthetase
- LRS:
-
leucyl-tRNA synthetase
- MRS:
-
methionyl-tRNA synthetase
- NRS:
-
asparaginyl-tRNA synthetase
- NCBI:
-
National Center for Biotechnology Information
- NTD:
-
n-terminal domain
- ORF:
-
open reading frame
- PRS:
-
prolyl-tRNA synthetase
- QRS:
-
glutaminyl-tRNA synthetase
- RRS:
-
arginyl-tRNA synthetase
- SRS:
-
seryl-tRNA synthetase
- SAD:
-
second additional domain
- TRS:
-
threonyl-tRNA synthetase
- TBA:
-
tRNA-binding domain
- VRS:
-
valyl-tRNA synthetase
- WRS:
-
tryptophanyl-tRNA synthetase
- Wh-T:
-
WHEP-TRS domain
- YRS:
-
tyrosyl-tRNA synthetase
References
Goodrich JM, Lyam A, Miquelle DG, Wibisono HT, Kawanishi K, Pattanavibool A, et al. Panthera tigris. The IUCN red list of threatened species. 2015; e.T15955A50659951.
Dantas-Torres F, Alves LC, Uilenberg G. Babesiosis. In: Marcondes C, editor. Arthropod Borne Diseases. Cham: Springer; 2016.
Healy G, Ristic M. Human babesiosis. In: Miodrag R, editor. Babesiosis Domest Anim Man. Florida: CRC Press; 2018.
Hunfeld KP, Hildebrandt A, Gray JS. Babesiosis: recent insights into an ancient disease. Int J Parasitol. 2008;38:1219–37.
Mosqueda J, Olvera-Ramirez A, Aguilar-Tipacamu G, Canto GJ. Current advances in detection and treatment of babesiosis. Curr Med Chem. 2012;19:1504–18.
André MR, Dumler JS, Scorpio DG, Teixeira RHF, Allegretti SM, Machado RZ. Molecular detection of tick-borne bacterial agents in Brazilian and exotic captive carnivores. Ticks Tick Borne Dis. 2012;3:247–53.
Baviskar BS, Gawande PJ, Maske DK, Jayraw AK, Bawaskar SS, Bhandarkar AG, et al. Occurrence of Babesia infection in leopard Panthera pardus at Nagpur. Zoos Print J. 2007;22:2736–7.
Githaka N, Konnai S, Kariuki E, Kanduma E, Murata S, Ohashi K. Molecular detection and characterization of potentially new Babesia and Theileria species/variants in wild felids from Kenya. Acta Trop. 2012;124:71–8.
Kinge YA, Sarode DB, Dakshinkar NP. Babesiosis in a lioness (Panthera leo). Vet World. 2010;3:133.
Lopez-Rebollar LM, Penzhorn BL, de Waal DT, Lewis BD. A possible new piroplasm in lions from the Republic of South Africa. J Wildl Dis. 2013;35:82–5.
McDermid KR, Carroll JP, Verreynne FJ, Penzhorn BL, Yabsley MJ, Snyman A. Surveillance for viral and parasitic pathogens in a vulnerable African lion (Panthera leo) population in the Northern Tuli Game Reserve, Botswana. J Wildl Dis. 2016;265:19208-1.
Munson L, Terio KA, Kock R, Mlengeya T, Roelke ME, Dubovi E, et al. Climate extremes promote fatal co-infections during canine distemper epidemics in African lions. PLoS ONE. 2008;3:e2545.
Nighot NK, Mote CS, Jadhav RV, More BK, Bansal N, Mathkari C. Clinico-therapeutic aspects of babesiosis in a white tigress (Panthera tigris tigris). Zoos Print J. 2012;27:19–20.
Rafiqi SI, Kumar S, Reena KK, Garg R, Ram H, Karikalan M, et al. Molecular characterization of Hepatozoon sp. and Babesia sp. isolated from endangered asiatic lion (Panthera leo persica). Indian J Anim Sci. 2018;88:662–6.
Sinha KP, Sinha M, Pankaj NK, Singh VK. Babesiosis in a tigress. Zoos Print J. 2000;15(327):1.
Upadhye SV, Dhoot VM. Sudden death of a leopard (Panthera pardus) due to babesiosis. Zoos Print J. 2013;15:327.
Williams BM, Berentsen A, Shock BC, Teixiera M, Dunbar MR, Becker MS, et al. Prevalence and diversity of Babesia, Hepatozoon, Ehrlichia, and Bartonella in wild and domestic carnivores from Zambia, Africa. Parasitol Res. 2014;113:911–8.
Ayoob AL, Hackner SG, Prittie J. Clinical management of canine babesiosis. J Vet Emerg Crit Care. 2010;20:77–89.
Jain V, Yogavel M, Kikuchi H, Oshima Y, Hariguchi N, Matsumoto M, et al. Targeting prolyl-tRNA synthetase to accelerate drug discovery against malaria, leishmaniasis, toxoplasmosis, cryptosporidiosis, and coccidiosis. Structure. 2017;25(1495–1505):e6.
Khan S, Sharma AA, Belrhali H, Yogavel M, Sharma AA. Structural basis of malaria parasite lysyl-tRNA synthetase inhibition by cladosporin. J Struct Funct Genomics. 2014;15:63–71.
Das P, Babbar P, Malhotra N, Sharma M, Jachak GR, Gonnade RG, et al. Specific stereoisomeric conformations determine the drug potency of cladosporin scaffold against malarial parasite. J Med Chem. 2018;61:5664–78.
Khan S, Garg A, Camacho N, Van Rooyen J, Kumar Pole A, Belrhali H, et al. Structural analysis of malaria-parasite lysyl-tRNA synthetase provides a platform for drug development. Acta Crystallogr Sect D Biol Crystallogr. 2013;69:785–95.
Yogavel M, Chaturvedi R, Babbar P, Malhotra N, Jain V, Sharma A. Drug targeting of one or more aminoacyl-tRNA synthetase in the malarial parasite Plasmodium falciparum. Drug Discov Today. 2018;23:26–33.
Sharma A, Sharma M, Yogavel M, Sharma A. Protein translation enzyme lysyl-tRNA synthetase presents a new target for drug development against causative agents of loiasis and schistosomiasis. PLoS Negl Trop Dis. 2016;10:e0005084.
Kato N, Comer E, Sakata-Kato T, Sharma A, Sharma M, Maetani M, et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature. 2016;538:344–9.
Hussain T, Yogavel M, Sharma A. Inhibition of protein synthesis and malaria parasite development by drug targeting of methionyl-tRNA synthetases. Antimicrob Agents Chemother. 2015;59:1856–67.
Jain V, Sharma A, Singh G, Yogavel M, Sharma A. Structure-based targeting of orthologous pathogen proteins accelerates antiparasitic drug discovery. ACS Infect Dis. 2017;3:281–92.
Jain V, Kikuchi H, Oshima Y, Sharma A, Yogavel M. Structural and functional analysis of the anti-malarial drug target prolyl-tRNA synthetase. J Struct Funct Genomics. 2014;15:181–90.
Chaliotis A, Vlastaridis P, Mossialos D, Ibba M, Becker HD, Stathopoulos C, et al. The complex evolutionary history of aminoacyl-tRNA synthetases. Nucleic Acids Res. 2017;45:1059–68.
Debard S, Bader G, De Craene JO, Enkler L, Bär S, Laporte D, et al. Nonconventional localizations of cytosolic aminoacyl-tRNA synthetases in yeast and human cells. Methods. 2017;113:91–104.
Wu J. The beauty of mathematics in computer science. Boca Raton: CRC Press; 2019.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30.
Emanuelsson O, Nielsen H, Brunak S, Von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–16.
Bendtsen JD, Nielsen H, Von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–95.
Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H, Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017;33:4049.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat Protoc. 2015;10:845–58.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Góes TS, Góes VS, Ribeiro MFB, Gontijo CM. Bovine babesiosis: anti-erythrocyte antibodies purification from the sera of naturally infected cattle. Vet Immunol Immunopathol. 2008;116:215–8.
Sharma A, Sharma A. Plasmodium falciparum mitochondria import tRNAs along with an active phenylalanyl-tRNA synthetase. Biochem J. 2015;465:459–69.
Nabholz C, Horn E, Schneider A. tRNAs and proteins are imported into mitochondria of Trypanosoma brucei by two distinct mechanisms. Mol Biol Cell. 1999;10:2547–57.
Chaubey S, Kumar A, Singh D, Habib S. The apicoplast of Plasmodium falciparum is translationally active. Mol Microbiol. 2005;56:81–9.
Mailu BM, Ramasamay G, Mudeppa DG, Li L, Lindner SE, Peterson MJ, et al. A nondiscriminating glutamyl-tRNA synthetase in the Plasmodium apicoplast: the first enzyme in an indirect aminoacylation pathway. J Biol Chem. 2013;288:32539–52.
Khan S, Sharma A, Jamwal A, Sharma V, Pole AK, Thakur KK, et al. Uneven spread of cis- and trans-editing aminoacyl-tRNA synthetase domains within translational compartments of P. falciparum. Sci Rep. 2011;1:188.
Jackson KE, Habib S, Frugier M, Hoen R, Khan S, Pham JS, et al. Protein translation in Plasmodium parasites. Trends Parasitol. 2011;27:467–76.
Esseiva AC, Naguleswaran A, Hemphill A, Schneider A. Mitochondrial tRNA import in Toxoplasma gondii. J Biol Chem. 2004;279:42363–8.
Hancock K, Jahduk SL. The mitochondrial tRNAs of Trypanosoma brucei are nuclear encoded. J Biol Chem. 1990;265:19208–15.
Schneider A. Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu Rev Biochem. 2011;80:1033–53.
Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-tRNA synthetases-analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710.
Lee SW. Aminoacyl-tRNA synthetase complexes: beyond translation. J Cell Sci. 2004;117:3725–34.
Simader H, Hothorn M, Suck D. Structures of the interacting domains from yeast glutamyl-tRNA synthetase and tRNA-aminoacylation and nuclear-export cofactor Arc1p reveal a novel function for an old fold. Acta Crystallogr Sect D Biol Crystallogr. 2006;62:1510–9.
Ko Y-G, Park H, Kim HJ, Lee J-W, Kim S, Kim JY, et al. p38 is essential for the assembly and stability of macromolecular tRNA synthetase complex: implications for its physiological significance. Proc Natl Acad Sci. 2002;99:7912–6.
Rho SB, Lee KH, Kim JW, Jo YJ, Shiba K, Kim S. Interaction between human tRNA synthetases involves repeated sequence elements. Proc Natl Acad Sci USA. 2002;93:10128–33.
Rho SB, Lee JS, Jeong EJ, Kim KS, Kim YG, Kim S. A multifunctional repeated motif is present in human bifunctional tRNA synthetase. J Biol Chem. 1998;273:11267–73.
Cahuzac B. A recurrent RNA-binding domain is appended to eukaryotic aminoacyl-tRNA synthetases. EMBO J. 2002;19:445–52.
Otero FJ, Skene RJ, Yang X-L, Schimmel P, McRee DE, Ribas de Pouplana L. Crystal structures that suggest late development of genetic code components for differentiating aromatic side chains. Proc Natl Acad Sci USA. 2003;100:15376–80.
Sati SP, Singh SK, Kumar N, Sharma A. Extra terminal residues have a profound effect on the folding and solubility of a Plasmodium falciparum sexual stage-specific protein over-expressed in Escherichia coli. Eur J Biochem. 2002;269:5259–63.
Gowri VS, Ghosh I, Sharma A, Madhubala R. Unusual domain architecture of aminoacyl tRNA synthetases and their paralogs from Leishmania major. BMC Genomics. 2012;13:621.
Jain V, Yogavel M, Oshima Y, Kikuchi H, Touquet B, Hakimi MASA. Structure of prolyl-tRNA synthetase-halofuginone complex provides basis for development of drugs against malaria and toxoplasmosis. Structure. 2015;23:819–29.
Pines M, Spector I. Halofuginone—the multifaceted molecule. Molecules. 2015;20:573–94.
Yogavel M, Chaturvedi R, Babbar P, Malhotra N, Jain V, Sharma A, et al. Drug targeting of one or more aminoacyl-tRNA synthetase in the malaria parasite Plasmodium falciparum. Drug Discov Today. 2018;23:1233–40.
Chhibber-Goel J, Sharma A. Side chain rotameric changes and backbone dynamics enable specific cladosporin binding in Plasmodium falciparum lysyl-tRNA synthetase. Proteins. 2019;87:730–7.
Fang P, Han H, Wang J, Chen K, Chen X, Guo M. Structural basis for specific inhibition of tRNA synthetase by an ATP competitive inhibitor. Chem Biol. 2015;22:734–44.
Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, Aust T, et al. Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–63.
Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25:451–7.
Bull SC, Doig AJ. Properties of protein drug target classes. PLoS ONE. 2015;10:e0117955.
Acknowledgements
We thank Dr Alex Rai for constant encouragement, inspiration and insights. We also thank Dr D. L. S. Armstrong for enabling access to the wilderness.
Funding
AS is funded by Department of Science and Technology (DST) JC Bose fellowship (JCB-41) and JCG is funded under the BioCARe scheme and AS is funded by Department of Biotechnology (DBT), (BT/PR30603/BIC/101/1104/2018). Funding was provided by Medicines for Malaria Venture (MMV), Global Health Innovative Technology (GHIT).
Author information
Authors and Affiliations
Contributions
AS designed the study. JCG, SJ and AS collected the genomic data. JCG and AS wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Dataset S1.
Aminoacyl-tRNA synthetase sequences used to generate the HMM profiles.
Additional file 2: Table S1.
Location of aaRS domains in the B. bovis genome. Table S2. Location of aaRS domains in the B. microti genome. Table S3. Location of aaRS domains in the B. bigemina genome. Table S4. Location of aaRS domains in the P. tigris genome.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chhibber-Goel, J., Joshi, S. & Sharma, A. Aminoacyl tRNA synthetases as potential drug targets of the Panthera pathogen Babesia. Parasites Vectors 12, 482 (2019). https://doi.org/10.1186/s13071-019-3717-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3717-z