
Abstract
Background
The gut microbiota is known to play a role in a mosquito vector’s life history, a subject of increasing research. Laboratory experiments are essential for such studies and require laboratory colonies. In this study, the conservation of field-obtained midgut microbiota was evaluated in laboratory-reared Anopheles gambiae (s.l.) mosquitoes continuously hatched in water from field breeding habitats.
Methods
Pupae and late instars were obtained from the field and reared, and the emerged adults were blood-fed. The eggs obtained from them were hatched in either water from the field or in dechlorinated tap water. The mosquito colonies were maintained for 10 generations. Midguts of female adults from unfed F0 (emerging from field-caught pupae and larvae), F5 and F10 were dissected out and genomic DNA was extracted for 16S metagenomic sequencing. The sequences were compared to investigate the diversity and bacterial compositional differences using ANCOM and correlation clustering methods.
Results
Less than 10% of the bacterial families identified had differential relative abundances between generational groups and accounted for 46% of the variation observed. Although diversity reduced in F10 mosquitoes during laboratory colonization (Shannon-Weaver; P-value < 0.05), 50% of bacterial genera were conserved in those bred continuously in field-water compared to 38% in those bred in dechlorinated tap water.
Conclusions
To our knowledge, this study is the first report on the assessment of gut bacterial community of mosquitoes during laboratory colonization and recommends the use of water from the natural breeding habitats if they are intended for microbiota research.
Similar content being viewed by others


Background
Mosquito vector-borne diseases are major health concerns, causing significant morbidity and mortality worldwide [1]. Although chemotherapy and the search for vaccines for these diseases have improved over the years, vector control remains a very important strategy. Faced with the challenges posed by resistance to insecticides, there have been increased efforts to find innovative methods of control through gaining a better understanding of factors that also influence both vector competence and capacity. One promising strategy involves exploring the use of midgut microbiota for transmission-blocking [2].
Bacteria inhabiting the midgut of mosquitoes contribute significantly to reducing the developmental capabilities of parasites that are ingested during a blood meal [3,4,5]. To date, a few bacterial species isolated from the midgut of wild-caught mosquitoes have been characterised for their roles in the life history of the mosquito (reviewed in [6]). Although such bacteria are isolated from natural populations, studies to investigate their functions make use of laboratory populations. Laboratory colonies of mosquitoes are usually well-adapted to laboratory conditions and are useful in performing experiments involving large numbers of mosquitoes. However, continuous laboratory maintenance of field-derived populations over several generations results in the loss of the native microbiota [7, 8], most likely due to changes in larval breeding water and other laboratory procedures. Therefore, results obtained by studies on microbiota might not be an accurate representation of what occurs in the wild [9].
In this study, we investigated the potential for breeding a population of ‘field’ mosquitoes in the laboratory with the aim of maintaining the midgut microbiota composition and diversity over generations. This could help establish large numbers of mosquitoes under laboratory conditions while conserving the natural midgut microbiota profile for further studies.
Results
Alpha diversity indices indicate similarity between treatment replicates
Taxon similarity and alpha diversity were compared between replicates for all treatment groups. This evaluation was done to assess possible differences between replicates, as sampling of mosquitoes and water samples from the field were performed over several days during the experimental period. Following rarefaction of sequences, some replicates were lost due to low sequence counts and resulted in only one representing the experimental treatment. Replicate comparisons were therefore impossible for such groups, i.e. Lab_F5 and Lab_F10. Shannon-Weaver and Faith’s phylogenetic diversity (PD) indices demonstrated similarity between replicates of the same experimental treatment (P-values > 0.05) (Table 1), although variations in means were observed (Fig. 1a, b). The evenness index gave no indication of a dominant species (index ≠ 0) (Fig. 1c), although an index of 0.36 (Fig. 1c; Field_F10_2) may suggest a slight shift in taxon evenness in this replicate.

Shannon-Weaver (a), Faith’s phylogenetic piversity (PD) (b) and Pielou’s evenness indices (c) for samples. Black dots indicate index measure for each sample and error bars show standard error of the mean for each treatment replicate. Red dotted lines are the total average for all replicates for an experimental group
Number of observed features differed across generations
The diversity indices also gave a first-hand indication of differences between some experimental groups (Table 2). These were explored further by comparing features, operational taxonomic units (OTUs) and taxa classifications between treatments. The average number of OTUs ranged from as low as 6 in Lab_F10 to 35 in Field_F5 (Fig. 2). Interestingly, the average number of OTUs did not differ between baseline (F0) samples and any of the other experimental groups, likely due to the wide range of recorded points in some of the experimental groups and the reduced number of replicates. Field_F5 samples differed from all other groups except for the baseline (Fig. 2).

Comparison of observed OTUs between experimental treatments. Points are OTUs from samples (pool of 5 midguts) of each treatment. Error bars represent standard error of the mean
To be able to understand the source of the differences detected, the sequences were analysed based on bacterial families. In total, 99 families were taxonomically identified ranging from 1 to 31 per sample. The average number of bacterial families in mosquitoes reared in dechlorinated water was significantly lower than in field-water-reared (5 vs 12; Mann-Whitney U-test: U(30) = 45.5, P = 0.026) and baseline mosquitoes (5 vs 11; Mann-Whitney U-test: U(26) = 27 P = 0.0052) (Fig. 3a).

Comparison of 99 taxonomically identified bacterial families between experimental groups. Each point represents a sample (pool of 5 midguts). a Comparison between the baseline and all field and lab water bred samples. b Comparison of baseline and generational groups of field-water-bred mosquitoes. Error bars represent standard error of the mean
Differential compositional analyses
We looked closer at the field water-bred mosquitoes to determine whether the observation made for Field_F5 during the OTU analyses persisted when analysed at the bacterial family level. Both groups of field-water-bred generations (F5 and F10) differed in family number from the baseline, the F5 showing a higher number than F10 in comparison to the baseline (Fig. 3b). However, about 91% (90 out of 99) of the families identified each represented < 1% of the total number of analysed sequences. Re-analysis based on the 9 families with relative abundance ≥ 1% revealed that Field_F5 remained higher than the baseline (Mann-Whitney U-test: U(20) = 14, P < 0.0001), but Field_F10 and the dechlorinated water-reared mosquitoes were both similar to the baseline (P > 0.05) (Fig. 4).

Comparison of 9 bacterial families (relative abundance ≥ 1% of sequences) between experimental groups. Points represent samples. Error bars represent standard error of the mean
The ANCOM results, which were based on bacterial genera, confirmed the contribution of a few taxa to the observed differences between groups of midguts (Fig. 5; Additional file 1: Table S1) and to 46% of the variance (Additional file 2: Table S3). Five (out of 8) of these significant taxa, Thorsellia, Mesorhizobium, Microbacterium, Shingomonas and some unspecified Proteobacteria, were more likely to cluster or co-occur (Fig. 6). The balance formed by these genera (y0_numerator) was more pronounced in samples bred in dechlorinated water for 5 generations (Lab_F5). The remaining 3 differential bacterial groups separately joined balances with other low relative abundance bacteria (Fig. 6). Four balances (y1, y2, y5 and y9) significantly demonstrated which bacterial taxa contributed to the differences in Field_F5 and other samples bred in field water (Field_F0 and F10) (Additional file 2: Table S4). Notable among these are Micrococcaceae (genus Arthrobacter), Xanthomonadaceae (genus Stenotrophomonas), Enterobacteriaceae (genus Thorsellia) and Phyllobacteriaceae (genus Mesorhizobium) which significantly increased, while Acetobacteraceae (genus Acetobacter) and Pseudomonadaceae (genus Pseudomonas) decreased in Field_F5 compared to Field_F0 (baseline) (Fig. 6). Following 10 generations of laboratory maintenance, 50 and 38% of bacterial genera were conserved (maintained as present or absent) in field-water and dechlorinated tap water-bred mosquitoes, respectively (Additional file 1: Table S2).

Volcano plot for the analysis of composition of microbiomes (ANCOM) test. Only significant bacterial taxa are labelled. Taxa on the top-left corner are distinct species but with small proportions, i.e. low f-score. Truly different taxa are depicted as one moves towards the far right (high W-statistic)

Dendogram of bacterial families resulting from unsupervised correlation clustering. Balances (y0-y9) are shown by pink (numerator) and red (denominator) vertical bars on the left side of the map, and are not related to the scale
Discussion
Laboratory colonization of mosquitoes is useful for producing large numbers of samples for experiments on various aspects of mosquito life history, and selection of specific traits. However, for microbiota studies, the bacterial communities found in wild mosquitoes could be lost after a few generations, thus creating a discordance which limits the usefulness of laboratory colonies in efforts to understand the roles of microbiota [9]. The present study demonstrates that conservation of midgut microbiota can be achieved using water from breeding sites in the laboratory maintenance of mosquitoes. Relative bacterial abundances varied between generations of colonies continuously bred in field-water, while still maintaining significant field-derived taxa. However, dechlorinated water, as used in standard insectary procedures for egg hatching, resulted in a significant decrease in the relative abundance of many bacterial taxa while selectively keeping a few at high abundance.
Bacteria in the larval environment form a major part of the mosquito midgut through its aquatic developmental stages to emerged adults [10,11,12]. Breeding water is, however, dynamic with its bacterial community differing at different surface layers [13], and with abiotic and biotic effects such as contamination [14] and seasonal variations [15]. The laboratory environment is more controlled, presenting with less effects from natural external sources. Nevertheless, various factors in the laboratory may also cause changes in larval breeding water during mosquito maintenance. Such environmental variations could result in same mosquito species maintained in different insectaries having different distinct microbial communities. Further investigation on the influence of variations in laboratory environment on changes in microbiota could be important in understanding both the effect of laboratory conditions in shaping the midgut bacteria of colonized mosquitoes, and how this contributes to potential differences in experimental results between laboratories. With 10 generations of breeding field-caught mosquitoes in field-water under laboratory conditions, approximately 50% of the ‘natural’ microbiota was conserved compared to 38% in mosquitoes reared using dechlorinated tap water. The study did not show a significant difference in these percentages, likely due to small sample size.
Continuous breeding in field water may help replace some bacteria and introduce new ones that have not already been observed in the founding population, as observed in the unexpected increase in the number of bacterial families in the fifth generation of field-bred mosquitoes. Other bacteria may also be consistently lost once under laboratory conditions (Additional file 1: Table S2). The field-water returned to the laboratory at each collection point was not tested for bacteria in this study, but the dynamics of this environment to both natural biotic and abiotic factors could explain the variations observed from one field group to another.
The use of dechlorinated tap water, which is a standard practice in mosquito insectaries, poses an initial bottleneck for mosquito colonization as was observed in our study. Chlorine is an effective bacteria-inactivating and killing agent [16, 17]. This effect resulted in a small number of replicates in our tap-water reared as compared to field-water samples. That notwithstanding, these samples were able to demonstrate the reduction of bacterial families and relative abundance in samples brought into the laboratory after ten generations, consistent with reports of loss of bacterial populations in laboratory colonized mosquitoes [7, 8].
The use of balances on our dataset enabled the identification of major taxa whose relative abundances were most significant in explaining compositional variations. Most notable among these are the four taxonomically identified genera that formed part of the largest balance (y0): Thorsellia, Mesorhizobium, Microbacterium and Sphingomonas. These co-occurring bacteria became most pronounced in tap-water reared samples despite rarely occurring at baseline. This is indicative of these bacterial taxa having a proliferation advantage when many others have been lost due to some selective pressure [18]. The great extent of bacteria loss during laboratory rearing results in distinct profiles dominated by few species compared to field-caught mosquitoes [19], as demonstrated in our study. Again, the small number of tap-water reared samples limited the extent to which this could be observed. Despite this Microbacterium and Sphingomonas showed distinctly in our tap-water reared mosquitoes and demonstrated significant difference in abundance. Both bacterial taxa have been identified in field-caught and lab-reared Anopheles species [20, 21].
The persistence and increase in the incidence of Thorsellia in laboratory colonized mosquitoes cannot be ignored. This bacterial genus has been isolated from the midguts of some Anopheles malaria vectors [8, 22, 23] and Culex mosquitoes [24]. They are known to increase growth in blood medium [23], hence could potentially be involved in blood digestion in mosquitoes. Besides the midgut, Thorsellia spp. are also found to inhabit the reproductive tracts of both sexes of Anopheles gambiae and An. coluzzii [25], necessitating further investigations on the functions of these bacteria in malaria mosquitoes.
Conclusions
We have demonstrated the conservation of field-derived bacterial community in mosquitoes maintained under laboratory conditions for ten generations by field-water replacement. This study also confirms the loss of microbial profile when mosquitoes are bred in tap water, which is a standard laboratory practice. The ability to breed large populations of mosquitoes for controlled experiments could help provide answers to the contribution of microbiota to vector competencies to disease transmission.
Methods
Mosquito samples and experimental set-up
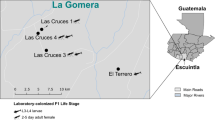
Late (3rd and 4th) instars and pupae of An. gambiae (s.l.) were sampled together with water from a breeding site in peri-urban Accra and transported to the laboratory in plastic containers. Three to four batches of mosquito samples were collected from the field within 3–4 days of each other. The pupae were separated into cups and placed in cages with no source of sugar meal for the emerging adults. The larvae were transferred into larval trays and maintained without adding larval food for a maximum of 3 days under standard insectary conditions. Remaining larvae were discarded. Emerging pupae were collected from the larval trays each day and transferred into cages wiped with 70% ethanol. For each batch of field collection, 30 one-day-old non-sugar-fed females were stored at -20 °C until needed for midgut dissection. The remaining adult mosquitoes in the cage were offered 10% glucose through cotton balls for 4–5 days and blood-fed.
F1 eggs collected from each batch of field collection were divided into two groups and placed in larval trays for the experiments. One group was placed in a tray containing field water (collected the previous day and sieved through a cloth to ensure mosquito eggs and early stage larvae were removed before use) and the other in dechlorinated tap water (chlorinated tap water left standing for at least 24 h). To standardize the set-up and prevent overcrowding, the egg to water ratio of 1 egg to 20 ml was maintained. Yeast and larval food were added to all trays daily. The water level was replenished with one-third of water (as appropriate for each tray) every other day to prevent drying. The larvae were observed daily, and the dead were removed. Emerging adult mosquitoes were maintained through generations as described above on sugar and blood. At 5 and 10 generations, emerged 1-day-old unfed female mosquitoes were sub-sampled (~30 female adults) for midgut dissections.
Midgut preparation and sequencing
Midguts were dissected from stored female adult mosquitoes sampled from F0 (baseline), F5 and F10 generations. A sample contained 5 dissected mosquito midguts, and for each treatment replicate a maximum of 4 samples (i.e. 20 midguts) was obtained. The total number of samples obtained for each experimental group (F0, Field_F5, Field_F10, Lab_F5, Lab_F10) ranged between 4 and 20 (Additional file 3: Table S5). Three sham samples were prepared during dissections as previously described [15]. Genomic DNA was extracted from each replicate and the 3 shams using QiaAmp Micro DNA kit (Qiagen, Hilden, Germany) according to manufacturer’s recommendations. The bacterial 16S V1-3 region was sequenced for 55 DNA samples (Additional file 3: Table S5) on an Illumina MiSeq platform (Pretoria, South Africa) using the primers 27F (5'-AGA GTT TGA TCM TGG CTC AG-3') and 534R (5'-ATT ACC GCG GCT GCT GG-3').
Quality filtering, which included trimming the sequences to retain bases with > 20 Phred score and demultiplexing, was performed, and a total of 527,005 sequences were retrieved. The length of the retained sequences ranged between 35–295 bp. We performed further sequence filtering to remove very low sequence lengths, which could be problematic in downstream data analyses, such as taxonomic assignments [26]. A total of 513,720 sequences with lengths between 150–295 bp were extracted. The minimum and maximum sequence count per sample was 1276 and 36,171, respectively, with a mean of 9340.
Screening sequences for potential ‘contamination’
Analyses of microbial data were performed using QIIME 2 (https://qiime2.org) following the “Moving Pictures” tutorials [27]. The paired end reads for all samples were imported into qiime2 and the sequence summaries were visualized by using the qiime demux summarize plugin. The resulting Interactive Quality Plot was examined to truncate the sequences in both read directions at base positions where the read quality fell below the threshold of 20. Using this cut-off criterion resulted in pruning the forward and reverse sequences at 295 and 246 base positions, respectively, and produced a set of output representative sequences (32,504 in total). These were aligned and masked to remove highly variable positions and used to build a mid-point rooted tree for phylogenetic diversity analyses. The Naive Bayes classifier was trained on SILVA 128 [28, 29] 97% OTUs, with taxonomic reference set to extract and include the target sequence between the forward and reverse primer. Taxonomic classification was performed for representative sequences with classify-sklearn [30] in the qiime2 feature-classifier plugin [31].
The resulting dataset was screened at the bacterial family level for possible ‘contamination’. To do this, the average relative abundance of bacterial families was calculated for the shams and those > 0.01 (Additional file 4: Tables S6, S7) were analysed for correlation with initial DNA concentration as previously described [15, 32]. No bacterial taxon was identified as a contaminant in our dataset.
Analyses of experimental samples
The inclusion of the sham sequences could potentially influence downstream analyses, therefore a second dataset of 470,772 for the 52 test samples was created, which excluded the shams from the initial 513,720 sequences. These were taken through the processing and analyses pipeline as described above. The cut-off criterion for pruning the forward and reverse sequences changed to positions 291 and 247, respectively, and resulted in 52,312 sequences. Sequences designated as ‘Unidentified bacteria’ and ‘Unassigned’ were excluded from dataset used for the diversity and differential composition analyses.
Rarefaction for diversity analyses was performed at a sampling depth of 250 sequences per sample, which resulted in the loss of 8 samples with low sequence counts. The remaining 44 samples were analysed for taxon richness, evenness and diversity using the Faith’s phylogenetic diversity (PD), Pielou’s and Shannon-Weaver indices, and explored for observed operational taxonomic units (OTUs). The significance of alpha diversity was determined using Kruskal-Wallis test, accepting only adjusted P-values < 0.05 as significant.
Analysis of composition of microbiomes (ANCOM) was used to identify differential relative abundance of bacterial genera [33], and balance trees to evaluate changes (growth or decline) in microbial sub-communities between experimental groups [34]. As both methods of analysis are sensitive to less informative features (sequence grouping), the taxa frequency table was filtered to remove bacterial classifications with less than 10 reads and those observed in less than 3 samples in the study. To tolerate the zero frequencies of bacterial counts, a pseudo-count composition table was created by adding a count of 1 to every value. For ANCOM, the composition table was log-transformed and significance determined from f-scores and ‘W’ statistics. The f-score measures the strength of the difference of a feature between groups. A high score indicates the more likelihood that the null hypothesis (the average of the feature in all groups are the same) can be rejected. The ‘W’ statistics indicate the number of times a feature is detected to be significantly different across groups. Principal balances were built with unsupervised hierarchical clustering and isometric log ratio (ILR) transformation [35] to group features based on how frequently they co-occur. An ordinary least square regression model was fitted to the balances using the different generation of samples as the only predictor variable. Coefficient P-values were accepted at a stringent significance level of 0.01.
Abbreviations
- ANCOM:
-
Analysis of Composition of Microbiomes
- OTU:
-
Operational Taxonomic Unit
References
WHO. Global vector control response 2017–2030. Geneva: World Health Organization; 2017. p. 52.
Dennison NJ, Jupatanakul N, Dimopoulos G. The mosquito microbiota influences vector competence for human pathogens. Curr Opin Insect Sci. 2014;3:6–13.
Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, Mulenga M, et al. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science. 2011;332:855–8.
Bahia AC, Dong Y, Blumberg BJ, Mlambo G, Tripathi A, Benmarzouk-Hidalgo OJ, et al. Exploring Anopheles gut bacteria for Plasmodium blocking activity. Environ Microbiol. 2014;16:2980–94.
Ramirez JL, Short SM, Bahia AC, Saraiva RG, Dong Y, Kang S, et al. Chromobacterium Csp_P reduces ralaria and dengue infection in vector mosquitoes and has entomopathogenic and in vitro anti-pathogen activities. PLoS Pathog. 2014;10:e1004398.
Guégan M, Zouache K, Démichel C, Minard G, Tran Van V, Potier P, et al. The mosquito holobiont: fresh insight into mosquito-microbiota interactions. Microbiome. 2018;6:49.
Boissière A, Tchioffo MT, Bachar D, Abate L, Marie A, Nsango SE, et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012;8:e1002742.
Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK. Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi - an Asian malarial vector. BMC Microbiol. 2009;9:96.
Romoli O, Gendrin M. The tripartite interactions between the mosquito, its microbiota and Plasmodium. Parasit Vectors. 2018;11:200.
Lindh JM, Borg-Karlson A-K, Faye I. Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop. 2008;107:242–50.
Coon KL, Vogel KJ, Brown MR, Strand MR. Mosquitoes rely on their gut microbiota for development. Mol Ecol. 2014;23:2727–39.
Wang Y, Gilbreath TM, Kukutla P, Yan G, Xu J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One. 2011;6:e24767.
Hörtnagl P, Pérez MT, Zeder M, Sommaruga R. The bacterial community composition of the surface microlayer in a high mountain lake. FEMS Microbiol Ecol. 2010;73:458–67.
Pennington MJ, Prager SM, Walton WE, Trumble JT. Culex quinquefasciatus larval microbiomes vary with instar and exposure to common wastewater contaminants. 2016;6:21969.
Akorli J, Gendrin M, Pels NAP, Yeboah-Manu D, Christophides GK, Wilson MD. Seasonality and locality affect the diversity of Anopheles gambiae and Anopheles coluzzii midgut microbiota from Ghana. PLoS One. 2016;11:e0157529.
Huang J, Wang L, Ren N, Ma F. Juli. Disinfection effect of chlorine dioxide on bacteria in water. Water Res. 1997;31:607–13.
Virto R, Mañas P, Alvarez I, Condon S, Raso J. Membrane damage and microbial inactivation by chlorine in the absence and presence of a chlorine-demanding substrate. Appl Environ Microbiol. 2005;71:5022–8.
Stubbendieck RM, Vargas-Bautista C, Straight PD. Bacterial communities: interactions to scale. Front Microbiol. 2016;7:1234.
Hegde S, Khanipov K, Albayrak L, Golovko G, Pimenova M, Saldaña MA, et al. Microbiome interaction networks and community structure from laboratory-reared and field-collected Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus mosquito vectors. Front Microbiol. 2018;9:2160.
Dong Y, Manfredini F, Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 2009;5:e1000423.
Ngo CT, Aujoulat F, Veas F, Jumas-Bilak E, Manguin S. Bacterial diversity associated with wild caught Anopheles mosquitoes from Dak Nong Province, Vietnam using culture and DNA fingerprint. PLoS One. 2015;10:e0118634.
Lindh JM, Terenius O, Faye I. 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl Environ Microbiol. 2005;71:7217–23.
Briones AM, Shililu J, Githure J, Novak R, Raskin L. Thorsellia anophelis is the dominant bacterium in a Kenyan population of adult Anopheles gambiae mosquitoes. ISME J. 2008;2:74–82.
Duguma D, Rugman-Jones P, Kaufman MG, Hall MW, Neufeld JD, Stouthamer R, et al. Bacterial communities associated with Culex mosquito larvae and two emergent aquatic plants of bioremediation importance. PLoS One. 2013;8:e72522.
Segata N, Baldini F, Pompon J, Garrett WS, Truong DT, Dabiré RK, et al. The reproductive tracts of two malaria vectors are populated by a core microbiome and by gender- and swarm-enriched microbial biomarkers. Sci Rep. 2016;6:24207.
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Commun. 2013;10:57–60.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Yilmaz P, Parfey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42:D643–8.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in python. J Mach Learn Res. 2011;12:2825–30.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with qiime 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90.
Jervis-Bardy J, Leong LE, Marri S, Smith RJ, Choo JM, Smith-Vaughan HC, et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome. 2015;3:19.
Mandal S, Van TW, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 2015;26:27663.
Morton JT, Sanders J, Quinn RA, McDonald D, Gonzalez A, Vázquez-Baeza Y, et al. Balance trees reveal microbial niche differentiation. mSystems. 2017;2:e00162–16.
Egozcue JJ, Pawlowsky-Glahn V, Mateu-Figueras G, Barceló-Vidal C. Isometric logratio transformations for compositional data analysis. Math Geol. 2003;35:279–300.
Acknowledgements
We thank the farmers who gave permission for the collection of mosquitoes and water from breeding sites in the field. We are also grateful to staff of Vestergaard NMIMR Vector Labs for their support in mosquito housing and breeding.
Funding
This work was supported by WACCBIP Postdoctoral Fellowship funds to JA from a DELTAS Africa grant (DEL-15-007: Awandare). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS) Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (107755/Z/15/Z: Awandare) and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. Raw sequence reads used in this study are available at Dryad Digital Repository under the accession doi:10.5061/dryad.98jj7gk
Author information
Authors and Affiliations
Contributions
JA designed, conducted and coordinated experiments. PAN, GWA, RKE and NAPP conducted experiments. JA analysed data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. ANCOM results. Table S2 Calculation of bacterial genera conservation in laboratory-colonized samples. (XLSX 17 kb)
Additional file 2:
Table S3. Simplicial linear regression summary. Table S4. Regression coefficients and P-values. (XLSX 15 kb)
Additional file 3:
Table S5. Details of 55 samples submitted for metagenomic sequencing. (XLSX 9 kb)
Additional file 4:
Table S6. Relative abundance of bacterial families in shams. Table S7. Relative abundance of bacterial families in experimental samples. (XLSX 36 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Akorli, J., Namaali, P.A., Ametsi, G.W. et al. Generational conservation of composition and diversity of field-acquired midgut microbiota in Anopheles gambiae (sensu lato) during colonization in the laboratory. Parasites Vectors 12, 27 (2019). https://doi.org/10.1186/s13071-019-3287-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3287-0