
Abstract
Background
Bats are among the most widely distributed mammals worldwide and can represent hosts or reservoirs for a number of different pathogens. Bartonella spp. are opportunistic bacterial pathogens, which are transmitted by a large variety of arthropods. The aim of this study was to investigate the presence and host-associations of these Gram-negative bacteria in heart tissues of bats collected in four different countries from eastern and central Europe and to analyze their phylogenetic relationship with other bat-associated bartonellae.
Results
The results of this study show for the first time the presence of Bartonella spp. DNA in heart tissues of bats from central and eastern Europe. The overall prevalence of the infection was 1.38%. Phylogenetic analysis identified four new Bartonella spp. sequences, which were closely related with other Bartonella previously isolated from bats in Europe and North America.
Conclusions
The gltA sequences of Bartonella spp. showed considerable heterogeneity in the phylogenetic analysis resulting in six different clades. Our study demonstrated the presence of Bartonella spp. only in heart tissues of bats from Romania, with two new bat species recorded as hosts (Myotis cf. alcathoe and Pipistrellus pipistrellus).
Similar content being viewed by others


Background
Bats are among the most widespread mammalian species worldwide with high local diversity and abundance. They are divided in two suborders: Yinpterochiroptera with distribution especially in the tropical regions and Yangochiroptera more widely distributed and with higher species diversity [1]. They are unique among mammals, as they have the ability to fly, even for long distances during the migration periods [2, 3]. Moreover, they can live in dense colonies, sometimes consisting in several bat species. Bats can adapt to various environmental conditions, and act as potentially important reservoir hosts for multiple pathogens, including zoonotic ones [4]. Multiple studies demonstrated their role as natural reservoirs for different pathogens including viruses [5,6,7], bacteria [8, 9] and parasites [10,11,12].
The genus Bartonella is a relatively diverse group of Gram-negative, facultative intracellular, haemotropic, vector-borne, bacteria that infect a wide-range of mammals and have a global distribution. After infection, the bacteria eventually enter the erythrocytes and endothelial cells and can persist asymptomatically in a wide range of mammalian reservoir hosts such as rodents, insectivores, carnivores, and ungulates [13,14,15]. The infection is transmitted mainly by arthropod vectors including fleas [16], sand flies [17], lice [18], mites [19] and ticks [20, 21]. The transmission and evolution of Bartonella species in mammals is the result of a complex relationship between multiple hosts, vectors and pathogens. There are many species of Bartonella, some of them with a large host spectrum and zoonotic potential (i.e. B. henselae, B. grahamii, B. elizabethae, B. koehlerae and B. rochalimae) while some others are known only from single host species [22,23,24].
Bartonella spp. has been reported with different prevalence and a high genetic diversity in bats and bat flies [25,26,27,28,29]. However, the knowledge on the occurrence of Bartonella in tissues of bats is still scarce. In Europe there are two studies reporting the presence of Bartonella spp. in bat tissues, involving different species [30, 31]. Both are geographically located at the margins of the continent (UK vs Georgia). Bai et al. [9] found 35 % of 218 bats positive for Bartonella DNA and more than 25 genetic variants were identified. Urushadze et al. [30] investigated the presence of Bartonella in the blood of 212 live bats by culture followed by PCR and found a 49.5 % prevalence.
Considering all these, the aim of our study was to demonstrate the presence and diversity of Bartonella spp. in heart tissues of different species of bats from central and eastern Europe. We primarily targeted bat species which are rarely recorded in caves (and are less represented in epidemiological studies), with accent on building-dwelling bats, the group with the highest contact rate with humans and potentially posing a zoonotic risk.
Methods
A total of 435 carcasses were collected from different countries from central and eastern Europe (Austria, Czech Republic, Hungary and Romania) between 2001 and 2016 (Additional file 1: Table S1). The samples were collected from carcasses of bats accidentally killed (collision with man-made structures, road kills) or that had died of natural causes (e. g. hypothermia caused by early spring emergence) and stored in freezer at -20 °C (samples from Czech Republic, Hungary and Romania) or at -80 °C (samples from Austria) until their necropsy. From each bat the heart was collected, as this was the only tissue available from all animals. No live bat was harmed or killed for the purpose of this study. Bats were identified to species level using morphological keys [31]. Genomic DNA was extracted from 25 mg of heart tissue using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions using 200 μl of elution buffer and stored at -20 °C.
A PCR targeting the 370 bp of the gltA encoding gene was employed, using the following primers: CSH1f (5'-GCG AAT GAA GCG TGC CTA AA-3') and BhCS.1137 (5'-AAT GCA AAA AGA ACA GTA AAC A-3') [32]. The reactions were carried out in 25 μl reaction mixture which contained 12.5 μl 2× Green Master Mix (Rovalab GmBH, Teltow, Germany), 6.5 μl water, 1 μl of each primer (0.01 mM final concentration) and 4 μl aliquot of isolated DNA. The PCR was performed using the T1000™ Thermal Cycler (Bio-Rad, Hercules, CA, USA) with the following conditions: initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 52.5 °C for 30 s and extension at 72 °C for 30 s and a final extension at 72 °C for 10 min. For each set of reactions (45 samples), 2 negative controls (PCR water) and one positive control which was DNA obtained from a Bartonella henselae, strain (ID 54A) isolated from a cat from Israel [33]. Amplification products were visualized by electrophoresis on 1.5% agarose gel stained with RedSafe™ 20,000× Nucleic Acid Staining Solution (Chembio, St Albans, UK), and their molecular weight was assessed by comparison to a molecular marker (Hyperladder IV, Bioline, London, UK). PCR products were purified using a commercial kit (Isolate II PCR and Gel Kit, Bioline, London, UK) and sent for sequencing with the primers described above in both directions (Macrogen Europe, Amsterdam, Netherlands).
The sequences were compared with those available in GenBank using Basic Local Alignments Tool (BLAST) analysis. The evolutionary history was inferred by Maximum Likelihood method based on the Tamura-Nei model [34]. The gltA gene has been shown to be suitable for phylogenetic analysis among Bartonella species [35] and is currently the most widely used to detect Bartonella infection. Using the search query keywords ‘Bartonella bats gltA’, we retrieved from GenBank all the sequences available from bats and their ectoparasites. Furthermore, based on the available literature concerning bartonellae from bats, we produced a database, where, from each unique Bartonella gltA genotype found, we included data on the host species and the species of the ectoparasite, in the case they were present (Additional file 2: Table S2). For phylogenetic analyses, as the lengths of the downloaded gltA sequences were different, they were trimmed to a length of 232 base pairs. In total, the phylogenetic analysis included 210 unique Bartonella genotypes from bat flies as well as from bats belonging to 8 families from both suborders. Brucella melitensis was chosen as outgroup, as it is also an Alphaproteobacteria from the order Rhizobiales.
Statistical analysis was performed using EpiInfo™ 7 (CDC, USA) software. The overall prevalence of Bartonella spp., the prevalence at locality level and the prevalence for each bat species and their 95% confidence interval (95% CI) were calculated.
Results
Overall, 435 samples were tested for the presence of Bartonella spp. DNA. A total of 6 samples were positive (1.38%). The positive samples belonged to three bat species: Myotis cf. alcathoe (3/12; 25%), Nyctalus noctula (2/228; 0.88%) and Pipistrellus pipistrellus (1/68; 1.47%). The following species were negative (numbers of examined bats in parentheses): Barbastella barbastellus (n = 2); Eptesicus nilssonii (n = 1); E. serotinus (n = 6); Hypsugo savii (n = 9); Miniopterus schreibersii (n = 4); My.bechsteinii (n = 4); My. cf. brandtii (n = 3); My. daubentonii (n = 2); My. myotis (n = 6); My. cf. mystacinus (n = 4); My. nattereri (n = 1); Nyctalus leisleri (n = 5); Pipistrellus kuhlii (n = 8); Pi. nathusii (n = 28); Pi. pygmaeus (n = 5); Plecotus auritus (n = 7); Pl. austriacus (n = 1); Rhinolophus euryale (n = 9); R. ferrumequinum (n = 1); R. hipposideros (n = 1); and Vespertilio murinus (n = 20).
All positive samples (n = 6) originated from three locations in Romania: Muntele Puciosu (3/31; 9.68%), Cheile Bicazului (2/92; 2.17%) and Huda lui Papară (1/68; 1.47%) (Table 1).
The analysis of the sequences showed that two from Muntele Puciosu and two from Cheile Bicazului were identical to each other, resulting in 4 unique sequences. The four sequences differed from each other by 6–24 nucleotides (Table 2).
BLAST analysis of the gltA sequences showed 96– 98% similarity to different sequences, isolated from bats in Europe (Georgia, GenBank: KX300154.1 and KX300200.1; and UK, GenBank: AJ871614.1) (Table 3). All sequences were submitted to the GenBank database under the accession numbers MG914431-MG914434.
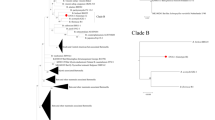
The global molecular phylogenetic analysis using the gltA sequences of Bartonella spp. isolated from bats in different parts of the world showed the presence of six major clades (Table 4, Fig. 1).

Phylogenetic tree of bat associated bartonellae. The evolutionary history was inferred by using the Maximum Likelihood method based on the Tamura-Nei model [34]. The bootstrap consensus tree inferred from 1000 replicates [56] is taken to represent the evolutionary history of the taxa analyzed [56]. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log-likelihood value. The analysis involved 210 nucleotide sequences. There were a total of 232 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 [57]
The first clade consisted in Bartonella spp. genotypes isolated from bats or bat flies in the Americas as well as sequences of the zoonotic pathogen B. mayotimonensis but also one of the sequences isolated from a bat in Romania. The other three sequences of Bartonella spp. in our study clustered in the second clade, together with various sequences isolated from Europe (Finland, France, Georgia, Spain and the UK) and four sequences isolated from North America. The third cluster consisted in different sequences isolated from the Old World (Asia, Europe and Africa). The fourth clade was the largest and most diverse and included sequences isolated from four different continents. The fifth clade comprised sequences from both Old World and New World, while the sixth clade consisted exclusively in sequences from South America, belonging to Yangochiroptera (Fig. 1).
Discussion
This study investigated the presence, prevalence and genetic diversity of Bartonella spp. in insectivorous bats from three different countries from central and eastern Europe and is the first evidence of the presence of these bacteria in heart tissues of bats from eastern and central Europe. This is the first study where My. cf. alcathoe and Pi. pipistrellus were found positive for Bartonella spp., while Ny. noctula was previously reported to harbour this group of pathogens [36, 37]. Multiple bat species may share the same Bartonella species without evident host specificity [38, 39] or they can harbour one or few Bartonella species-specific for a particular bat species [25, 36, 40, 41].
Reports of Bartonella infections are known from blood of bats from various countries across the world with different prevalence. High prevalence was reported in Georgia [30], Taiwan [42], Guatemala [38], Costa Rica [27], Kenya [40] and China [43], compared with a low prevalence in South Africa, Swaziland [29] and the USA [44]. Most of the studies were focused on the detection of Bartonella spp. in blood and bat associated ectoparasites [45, 46], but researchers from Argentina [47], France, Spain [37], Georgia [9] and the UK [36] tested also tissues for the presence of the bacteria. The positive bats from France, Spain and the UK, together with our positive samples belonged to the family Vespertilionidae, which contain high number of building-dwelling bats species. The positive bats from Argentina and Georgia belonged to three different bat families, the Molossidae, Rhinolophidae and Vespertilionidae, with all the analysed bats were cave-dwelling species. On the family level, the prevalence of Bartonella was estimated to be between 7.3% on species of the family Nycteridae and 54.4% on species of the Miniopteridae [37]. The report of low prevalence of Bartonella DNA in bats from Romania may be the result that we targeted only one molecular marker (the gltA gene) instead of multiple markers [48] and the majority of bat species analyzed are rarely parasitized by bat flies, which are suggested to be the main vectors for Bartonella sp. [49].
The global phylogenetic analysis of the sequences considered in this study showed that there is a high diversity among Bartonella isolated from bats and their ectoparasites. The distribution of Bartonella spp. in different bat families depends also on the geographical distribution of that particular family (Table 4). Three of the clades (I, II and VI) include only Bartonella spp. isolated from Yangochiroptera. The most diverse clade regarding the number of bat host families was clade IV: the Miniopteridae and Vespertilionidae (Yangochiroptera) and the Hipposideridae, Pteropodidae and Rhinolophidae (Yinpterochiroptera) (Table 4). Sequences of Bartonella spp. isolated from bats belonging to the family Vespertilionidae were present in five out of six clades (all except clade V), as this family is among the most diverse, widespread and well-studied. In Europe there are 44 bat species out of which 35 belong to Vespertilionidae [31] and all the studies conducted in this part of the Old World for detection of Bartonella spp. were focused mainly on this family [36, 37, 50, 51]. Our study was performed on various bat species, with the positive samples belonging to the family Vespertilionidae and the negative belonging to the families Miniopteridae and Rhinolophidae.
So far, the pathogenicity of bat-associated bartonellae to humans remains unknown, and further studies are needed to clarify their zoonotic potential. There are reports from Finland and the USA where different Vespertilionidae bats harboured the human pathogen B. mayotimonensis [44, 50], which was originally detected in the resected aortic valve of a 59-year-old patient from the USA [41]. Stuckey et al. [37] suggested that studies regarding the detection of Bartonella spp. in bats should be focused especially on those belonging to the Vespertilionidae (genera Nyctalus, Pipistrellus and Myotis), as the Bartonella isolated from these genera seem to be genetically related to B. mayotimonensis. Although all the positive samples from Romania were isolated from species of the family Vespertilionidae, our study did not reveal sequences related with any of the zoonotic Bartonella genotypes.
Diverse genetic variants of Bartonella were found in bats and their associated bat flies, suggesting that the latter may act as vectors. Bartonella spp. prevalence is higher in bat ectoparasites and have a much more genetic diversity compared with those isolated from the bats [26, 28, 38, 39, 42, 49, 50, 52,53,54,55].
Conclusions
This study showed that bats can harbour different strains of Bartonella spp., but with a low prevalence, reporting the presence of these bacteria in two new hosts (My. cf. alcathoe and Pi. pipistrellus). The molecular phylogenetic analysis conducted in this study revealed a high genetic diversity among Bartonella spp. isolated from bats in different parts of the world, with the presence of six major clades.
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
References
Teeling EC, Springer MS, Madsen O, Bates P, O’Brien SJ, Murphy WJ. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307:580–4.
Petit E, Mayer F. A population genetic analysis of migration: the case of the noctule bat (Nyctalus noctula). Mol Ecol. 2000;9:683–90.
Moussy C, Hosken DJ, Mathews F, Smith GC, Aegerter JN, Bearhop S. Migration and dispersal patterns of bats and their influence on genetic structure. Mamm Rev. 2013;43:183–95.
Moratelli R, Calisher CH. Bats and zoonotic viruses: can we confidently link bats with emerging deadly viruses? Mem Inst Oswaldo Cruz. 2015;110:1–22.
Dacheux L, Cervantes-Gonzalez M, Guigon G, Thiberge JM, Vandenbogaert M, Maufrais C, et al. A preliminary study of viral metagenomics of french bat species in contact with humans: identification of new mammalian viruses. PLoS One. 2014;9:e87194.
Kading RC, Schountz T. Flavivirus infections of bats: potential role in Zika virus ecology. Am J Trop Med Hyg. 2016;95:993–6.
Afelt A, Lacroix A, Zawadzka-Pawlewska U, Pokojski W, Buchy P, Frutos R. Distribution of bat-borne viruses and environment patterns. Infect Genet Evol. 2018;58:181–91.
Mühldorfer K, Speck S, Wibbelt G. Diseases in free-ranging bats from Germany. BMC Vet Res. 2011;7:61.
Bai Y, Urushadze L, Osikowicz L, McKee C, Kuzmin I, Kandaurov A, et al. Molecular survey of bacterial zoonotic agents in bats from the country of Georgia (Caucasus). PLoS One. 2017;12:e0171175.
Hornok S, Estók P, Kováts D, Flaisz B, Takács N, Szõke K, et al. Screening of bat faeces for arthropod-borne apicomplexan protozoa: Babesia canis and Besnoitia besnoiti-like sequences from Chiroptera. Parasit Vectors. 2015;8:441.
Hornok S, Szöke K, Kováts D, Estók P, Görföl T, Boldogh SA, et al. DNA of piroplasms of ruminants and dogs in ixodid bat ticks. PLoS One. 2016;11:e0167735.
Corduneanu A, Hrazdilová K, Sándor AD, Matei IA, Ionicǎ AM, Barti L, et al. Babesia vesperuginis, a neglected piroplasmid: new host and geographical records, and phylogenetic relations. Parasit Vectors. 2017;10:598.
Gerrikagoitia X, Gil H, García-Esteban C, Anda P, Juste RA, Barral M. Presence of Bartonella species in wild carnivores of northern Spain. Appl Environ Microbiol. 2012;78:885–8.
Gutiérrez R, Krasnov B, Morick D, Gottlieb Y, Khokhlova IS, Harrus S. Bartonella infection in rodents and their flea ectoparasites: an overview. Vector Borne Zoonotic Dis. 2015;15:27–39.
Chomel BB, Molia S, Kasten RW, Borgo GM, Stuckey MJ, Maruyama S, et al. Isolation of Bartonella henselae and two new Bartonella subspecies, Bartonella koehlerae subspecies boulouisii subsp. nov. and Bartonella koehlerae subspecies bothieri subsp. nov. from free-ranging californian mountain lions and bobcats. PLoS One. 2016;11:e0148299.
Zouari S, Khrouf F, M’ghirbi Y, Bouattour A. First molecular detection and characterization of zoonotic Bartonella species in fleas infesting domestic animals in Tunisia. Parasit Vectors. 2017;10:436.
Battisti JM, Lawyer PG, Minnick MF. Colonization of Lutzomyia verrucarum and Lutzomyia longipalpis sand flies (Diptera: Psychodidae) by Bartonella bacilliformis, the etiologic agent of Carrión’s disease. PLoS Negl Trop Dis. 2015;9:e0004128.
Gutiérrez R, Cohen L, Morick D, Mumcuoglu KY, Harrus S, Gottlieb Y. Identification of different Bartonella species in the cattle tail louse (Haematopinus quadripertusus) and in cattle blood. Appl Environ Microbiol. 2014;80:5477–83.
Loan HK, Van Cuong N, Takhampunya R, Klangthong K, Osikowicz L, Kiet BT, et al. Bartonella species and trombiculid mites of rats from the Mekong Delta of Vietnam. Vector Borne Zoonotic Dis. 2015;15:40–7.
Klangthong K, Promsthaporn S, Leepitakrat S, Schuster AL, McCardle PW, Kosoy M, et al. The distribution and diversity of Bartonella species in rodents and their ectoparasites across Thailand. PLoS One. 2015;10:e0140856.
Ereqat S, Nasereddin A, Vayssier-Taussat M, Abdelkader A, Al-Jawabreh A, Zaid T, et al. Molecular evidence of Bartonella species in ixodid ticks and domestic animals in Palestine. Front Microbiol. 2016;7:1217.
Bown KJ, Bennett M, Begon M. Flea-borne Bartonella grahamii and Bartonella taylorii in bank voles. Emerg Infect Dis. 2004;10:684–7.
Breitschwerdt EB, Maggi RG, Mozayeni BR, Hegarty BC, Bradley JM, Mascarelli PE. PCR amplification of Bartonella koehlerae from human blood and enrichment blood cultures. Parasit Vectors. 2010;3:76.
Berrich M, Kieda C, Grillon C, Monteil M, Lamerant N, Gavard J, et al. Differential effects of Bartonella henselae on human and feline macro- and micro-vascular endothelial cells. PLoS One. 2011;6:e20204.
Kamani J, Baneth G, Mitchell M, Mumcuoglu KY, Gutiérrez R, Harrus S. Bartonella species in bats (Chiroptera) and bat flies (Nycteribiidae) from Nigeria, West Africa. Vector Borne Zoonotic Dis. 2014;14:625–32.
Brook CE, Bai Y, Dobson AP, Osikowicz LM, Ranaivoson HC, Zhu Q, et al. Bartonella spp. in fruit bats and blood-feeding ectoparasites in Madagascar. PLoS Negl Trop Dis. 2015;9:e0003532.
Judson SD, Frank HK, Hadly EA. Bartonellae are prevalent and diverse in Costa Rican bats and bat flies. Zoonoses Public Health. 2015;62:609–17.
Olival KJ, Dittmar K, Bai Y, Rostal MK, Lei BR, Daszak P, et al. Bartonella spp. in a Puerto Rican bat community. J Wildl Dis. 2015;51:274–8.
Dietrich M, Tjale MA, Weyer J, Kearney T, Seamark ECJ, Nel LH, et al. Diversity of Bartonella and Rickettsia spp. in bats and their blood-feeding ectoparasites from South Africa and Swaziland. PLoS One. 2016;11:e0152077.
Urushadze L, Bai Y, Osikowicz L, McKee C, Sidamonidze K, Putkaradze D, et al. Prevalence, diversity, and host associations of Bartonella strains in bats from Georgia (Caucasus). PLoS Negl Trop Dis. 2017;11:e0005428.
Dietz C, Nill D, von Helversen O. Bats of Britain, Europe and Northwest Africa. London: A&C Black; 2009.
Birtles RJ, Raoult D. Comparison of partial citrate synthase gene (gltA) sequences for phylogenetic analysis of Bartonella species. Int J Syst Bacteriol. 1996;46:891–7.
Gutiérrez R, Morick D, Gross I, Winkler R, Abdeen Z, Harrus S. Bartonellae in domestic and stray cats from Israel: comparison of bacterial cultures and high-resolution melt real-time PCR as diagnostic methods. Vector Borne Zoonotic Dis. 2013;13:857–64.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–5.
Norman AF, Regnery R, Jameson P, Greene C, Krause DC. Differentiation of Bartonella-like isloates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J Clin Microbiol. 1995;33:1797–803.
Concannon R, Wynn-Owen K, Simpson VR, Birtles RJ. Molecular characterization of haemoparasites infecting bats (Microchiroptera) in Cornwall, UK. Parasitology. 2005;131:489–96.
Stuckey MJ, Boulouis HJ, Cliquet F, Picard-Meyer E, Servat A, Aréchiga-Ceballos N, et al. Potentially zoonotic Bartonella in bats from France and Spain. Emerg Infect Dis. 2017;23:539–41.
Bai Y, Kosoy M, Recuenco S, Alvarez D, Moran D, Turmelle A, et al. Bartonella spp. in bats, Guatemala. Emerg Infect Dis. 2011;17:1269–72.
Bai Y, Recuenco S, Gilbert AT, Osikowicz LM, Gómez J, Rupprecht C, et al. Prevalence and diversity of Bartonella spp. in bats in Peru. Am J Trop Med Hyg. 2012;87:518–23.
Kosoy M, Bai Y, Lynch T, Kuzmin IV, Niezgoda M, Franka R, et al. Bartonella spp. in bats, Kenya. Emerg Infect Dis. 2010;16:1875–81.
Lin EY, Tsigrelis C, Baddour LM, Lepidi H, Rolain JM, Patel R, et al. Candidatus Bartonella mayotimonensis and endocarditis. Emerg Infect Dis. 2010;16:500–3.
Lin JW, Hsu YM, Chomel BB, Lin LK, Pei JC, Wu SH, et al. Identification of novel Bartonella spp. in bats and evidence of Asian gray shrew as a new potential reservoir of Bartonella. Vet Microbiol. 2012;156:119–26.
Han HJ, Wen HL, Zhao L, Liu LM, Zhou CM, Qin XR, et al. Novel Bartonella species in insectivorous bats, Northern China. PLoS One. 2017;12:e0167915.
Lilley TM, Wilson CA, Bernard RF, Willcox EV, Vesterinen EJ, Webber QMR, et al. Molecular detection of Candidatus Bartonella mayotimonensis in North American bats. Vector Borne Zoonotic Dis. 2017;17:243–6.
Morse SF, Olival KJ, Kosoy M, Billeter S, Patterson BD, Dick CW, et al. Global distribution and genetic diversity of Bartonella in bat flies (Hippoboscoidea, Streblidae, Nycteribiidae). Infect Genet Evol. 2012;12:1717–23.
Davoust B, Marié JL, Dahmani M, Berenger JM, Bompar JM, Blanchet D, et al. Evidence of Bartonella spp. in blood and ticks (Ornithodoros hasei) of bats, in French Guiana. Vector Borne Zoonotic Dis. 2016;16:516–9.
Cicuttin GL, De Salvo MN, La Rosa I, Dohmen FEG. Neorickettsia risticii, Rickettsia sp. and Bartonella sp. in Tadarida brasiliensis bats from Buenos Aires, Argentina. Comp Immunol Microbiol Infect Dis. 2017;52:1–5.
McKee CD, Hayman DTS, Kosoy MY, Webb CT. Phylogenetic and geographic patterns of Bartonella host shifts among bat species. Infect Genet Evol. 2016;44:382–94.
Sándor AD, Földvári M, Krawczyk AI, Sprong H, Corduneanu A, Barti L, et al. Eco-epidemiology of novel Bartonella genotypes from parasitic flies of insectivorous bats. Microb Ecol. 2018; https://doi.org/10.1007/s00248-018-1195-z.
Veikkolainen V, Vesterinen EJ, Lilley TM, Pulliainen AT. Bats as reservoir hosts of human bacterial pathogen, Bartonella mayotimonensis. Emerg Infect Dis. 2014;20:960–7.
Lilley TM, Veikkolainen V, Pulliainen AT. Molecular detection of Candidatus Bartonella hemsundetiensis in bats. Vector Borne Zoonotic Dis. 2015;15:706–8.
Anh PH, Van Cuong N, Son NT, Tue NT, Kosoy M, Woolhouse MEJ, et al. Diversity of Bartonella spp. in bats, southern Vietnam. Emerg Infect Dis. 2015;21:1266–7.
Bai Y, Hayman DTS, McKee CD, Kosoy MY. Classification of Bartonella strains associated with straw-colored fruit bats (Eidolon helvum) across Africa using a multi-locus sequence typing platform. PLoS Negl Trop Dis. 2015;9:e0003478.
Reeves WK, Loftis AD, Gore JA, Dasch GA. Molecular evidence for novel Bartonella species in Trichobius major (Diptera : Streblidae) and Cimex adjunctus (Hemiptera : Cimicidae) from two southeastern bat caves, USA. J Vector Ecol. 2005;30:339–4.
Reeves WK, Beck J, Orlova MV, Daly JL, Pippin K, Revan F, et al. Ecology of bats, their ectoparasites, and associated pathogens on Saint Kitts Island. J Med Entomol. 2016;53(5):1218–25.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Acknowledgements
We are grateful for the help of our collaborators during field work and sample collection: Ioan Coroiu, Ioana Adriana Matei, Levente Barti, Lucie Dufková, Daniel Ruzek, Péter Estók, Sándor Boldogh, Irina Pocora, Viorel Pocora, Dragoş Ștefan Măntoiu, Marius-Alexandru Ciocănău and Teodor Dan Ursache. Special thanks to Ricardo Gutiérrez and Shimon Harrus for all the help regarding PCR protocols and for providing us the positive control.
Funding
This study was published under the frame of UEFISCDI project PCCDI 57/2018 and was supported by the János Bolyai Research Scholarship of Hungarian Academy of Science (to ADS).
Availability of data and materials
The data supporting the conclusion of this article are provided within the article and its additional files. The sequences were submitted to the GenBank database under the accession numbers MG914431-MG914434.
Author information
Authors and Affiliations
Contributions
AC, ADS and ADM wrote the manuscript, ADS, SH, ZB and KS collected the material for the study, ADS, SH, NL, ZB and KS helped in the identification of bat species, AC and AMI performed the necropsy, AC and AMI performed laboratory work and analysis of the data, ADS, AMI, SH, HPF and ADM participated in manuscript correction. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Samples distribution according to locality and species. (XLSX 14 kb)
Additional file 2:
Table S2. Detailed information regarding the sequences used in the phylogenetic analysis. (XLSX 29 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Corduneanu, A., Sándor, A.D., Ionică, A.M. et al. Bartonella DNA in heart tissues of bats in central and eastern Europe and a review of phylogenetic relations of bat-associated bartonellae. Parasites Vectors 11, 489 (2018). https://doi.org/10.1186/s13071-018-3070-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3070-7