
Abstract
Background
The cloacal tapeworm Cloacotaenia megalops (Hymenolepididae) is one of the most common cestode parasites of domestic and wild ducks worldwide. However, limited information is available regarding its epidemiology, biology, genetics and systematics. This study provides characterisation of the complete mitochondrial (mt) genome of C. megalops.
Methods
The complete mt genome of C. megalops was obtained by long PCR, sequenced and annotated.
Results
The length of the entire mt genome of C. megalops is 13,887 bp; it contains 12 protein-coding, 2 ribosomal RNA and 22 transfer RNA genes, but lacks an atp8 gene. The mt gene arrangement of C. megalops is identical to that observed in Anoplocephala magna and A. perfoliata (Anoplocephalidae), Dipylidium caninum (Dipylidiidae) and Hymenolepis diminuta (Hymenolepididae), but differs from that reported in taeniids owing to the position shift between the tRNA (L1) and tRNA (S2) genes. The phylogenetic position of C. megalops was inferred using Maximum likelihood and Bayesian inference methods based on the concatenated amino acid data for 12 protein-coding genes. Phylogenetic trees showed that C. megalops is sister to Anoplocephala spp. (Anoplocephalidae) + Pseudanoplocephala crawfordi + Hymenolepis spp. (Hymenolepididae) indicating that the family Hymenolepididae is paraphyletic.
Conclusions
The complete mt genome of C. megalops is sequenced. Phylogenetic analyses provided an insight into the phylogenetic relationships among the families Anoplocephalidae, Hymenolepididae, Dipylidiidae and Taeniidae. This novel genomic information also provides the opportunity to develop useful genetic markers for studying the molecular epidemiology, biology, genetics and systematics of C. megalops.
Similar content being viewed by others


Background
The cloacal tapeworm, Cloacotaenia megalops Nitzsch in Creplin, 1829 (Cestoda: Hymenolepididae), is one of the most common hymenolepidid tapeworms parasitising waterfowl, with a global distribution. The life-cycle of this tapeworm is complex. Seed shrimpos (Ostracoda) act as intermediate hosts and many waterfowl species (including ducks, geese and swans) serve as definitive hosts [1]. In China, C. megalops is considered as a predominant cestode species in ducks and geese [2, 3].
Comparison of entire mitochondrial (mt) genomes has been used for reconstructing phylogenetic relationships among parasitic Platyhelminthes [4, 5], including cestodes [6–11]. Cestode mt genomes usually encode 36 genes, including 12 protein-coding genes, 2 ribosomal RNA (rRNA) genes and 22 transfer RNA (tRNA) genes [6–11]. Cestoda is a large class of parasitic flatworms with many species representing a health danger for animals and humans worldwide. Despite the availability of advanced DNA technologies and bioinformatic methods, there is still a paucity of knowledge of mt genomes for many tapeworms of socioeconomic importance, such as the members of the family Hymenolepididae. Although complete mt genomes are available for Hymenolepis diminuta [8], H. nana (=Rodentolepis nana or Vampirolepis nana) [9], and Pseudanoplocephala crawfordi [10], no mt genomes are available from the genus Cloacotaenia. Furthermore, little is known about the epidemiology, genetics and biology of the type-and only species of this genus, C. megalops.
The taxonomic status of C. megalops has been controversial for many years, and is still debated. Czaplinski & Vaucher [12] considered Cloacotaenia a synonym of Hymenolepis but Makarikov et al. [13] have recently restored the independent status of the genus Cloacotaenia based on remarkable morphological differences between C. megalops and Hymenolepis (sensu stricto). To tackle these issues, in the present study, the complete mt genome of C. megalops was determined and its phylogenetic relationships with selected cestode species were inferred based on analysis of the concatenated mt amino acid sequences.
Methods
Parasites and DNA extraction
Cloacotaenia megalops were collected from the cloaca of ducks from a small abattoir in Xinjiang Uygur Autonomous Region, China. The adult tapeworm was isolated from cloaca of a duck. Cestode identification was conducted by morphological criteria including the features of the scolex and mature and gravid proglottids [14]; the scolex was observed in stereoscan photographs and mature and gravid proglottids were examined after hematoxylin staining. The remaining fragment was fixed in 70 % alcohol and stored at -20 °C until use. Total genomic DNA was extracted from one of these specimens using Tissue DNA Kit (OMEGA, Doraville, USA) according to the manufacturer’s instructions.
PCR amplification and sequencing
Three pairs of PCR primers (Additional file 1: Table S1) were designed based on well-conserved regions within the mt genomes of tapeworms [6, 7]. These primers were used to amplify three overlapping segments of the complete mt genome of C. megalops by long PCR technology. Long PCR reactions (50 μl) were conducted in 5.0 μl 10× LA Mixture (Takara), 10 pmol of each primer (1 μl), 1.5 μl of DNA sample and 41.5 μl of H2O in a thermocycler (Eppendorf, Hamburg, Germany) under the following conditions: 94 °C for 5 min (initial denaturation), followed by 35 cycles of 98 °C for 10 s (denaturation), 50 °C for 20 s (annealing), and 68 °C for 8 min (extension), and with a final extension step at 68 °C for 10 min. Amplicons were examined on 0.8 % agarose gels stained with ethidium bromide. PCR products were subsequently sent to Sangon Biotech Co. Ltd. (Shanghai, China) for sequencing using a primer-walking strategy.
Sequence analyses
Sequences were assembled using CAP3 Server online. The complete mt genome of C. megalops was aligned against the complete mt genome sequences of H. diminuta and A. perfoliata using the computer program MAFFT 7.122 [15] to identify gene boundaries. Each gene was translated into its amino acid sequence using the flatworm mt genetic code (Translation table 9) in MEGA 5 [16]. The translation start and stop codons were identified based on the similarity of the gene lengths and usual codons between H. diminuta and A. perfoliata mt genomes. Twenty-two tRNA genes were predicted using the program tRNAscan-SE [17] and then confirmed by recognizing anticodon sequences and potential secondary structures by visual inspection, and two rRNA genes were identified by comparison with that of H. diminuta and A. perfoliata [7, 8].
Phylogenetic analyses
A total of 20 tapeworm species were selected for phylogenetic analyses using one trematode Schistosoma japonicum (GenBank accession number NC_002544) as the outgroup [18]. The 12 amino acid sequences of protein-coding genes were aligned independently using MAFFT 7.122. Ambiguously aligned sites and regions were excluded using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server) [19]. Phylogenetic analyses were performed using Bayesian inference (BI) and Maximum likelihood (ML) methods. The Akaike information criterion as implemented in ProtTest 2.4 [20] was used to choose the most suitable model of evolution. BI was performed in MrBayes using the MtArt + I + G + F model of evolution. BI was set up to perform two runs, each of four simultaneous chains for the Monte Carlo Markov Chain. In each run, the number of generations was set to 1,000,000 and a tree was sampled every 100 generations in MrBayes 3.1.1 [21]; the average standard deviation of split frequencies of less than 0.01 and the potential scale reduction factor approaching 1 were used to ensure the convergence of the two runs. The first 25 % of the trees were discarded as ‘burn-in’. A 50 % majority rule consensus tree was used to calculate Bayesian posterior probabilities (Bpp). ML analysis was conducted using PhyML 3.0 [22]. A BioNJ tree was used as a starting tree to search for the ML tree with the MtArt + I + G model of evolution. The subtree pruning and regrafting method was chosen. The middle of each discretized substitution rate class was determined using the median. ML analyses were checked on the basis of 100 bootstrap replicates (Bf). Phylograms were drawn using the program FigTree v.1.4 (http://tree.bio.ed.ac.uk/software/figtree).
Results and discussion
Features of the C. megalops mt genome
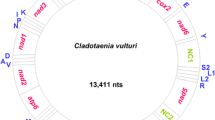
The complete mt genome of C. megalops is a 13,887 bp (KU641017) long circular DNA molecule (Fig. 1). All 36 genes expected for tapeworm mt genomes have been identified. This AT rich (71.6 %) mt genome includes 12 protein-coding genes (atp6, cox1–3, cytb, nad1–6 and nad4L), 22 tRNA genes and two rRNA genes, but lacks an atp8 gene (Table 1). Thirty-six mt genes are transcribed from the same direction (Fig. 1). The size of C. megalops mt genome is similar to other tapeworm mt genomes, such as H. diminuta (13,900 bp) [8], H. nana (13,764 bp) [9] and P. crawfordi (14,192 bp) [10]. The mt gene arrangement of C. megalops is identical to that in Anoplocephala magna and A. perfoliata (Anoplocephalidae), Dipylidium caninum (Dipylidiidae) and Hymenolepis diminuta (Hymenolepididae), but differs from that in taeniids owing to the position shift between the tRNA (L1) and tRNA (S2) genes. The nucleotide composition of the entire mt genome of C. megalops is A = 26.4 %, T = 45.2 %, G = 18.7 % and C = 9.7 %.

The organization of mitochondrial genome of Cloacotaenia megalops. All genes are transcribed in the same direction and the 22 tRNA genes are shown by a single-letter abbreviation of their corresponding amino acid. The two leucine tRNA genes are designated by L1 (CUN) and L2 (UUR), respectively, and two serine tRNA genes by S1 (AGN) and S2 (UCN), respectively. Gene scaling is only approximate
Annotation
A total of 3352 amino acids are encoded in the C. megalops mt genome. The aggregate length of all of the 12 protein-coding genes is 10,092 bp. In terms of the length of individual protein-coding genes, cox1 gene is the largest (1593 bp) and nad4L gene is the shortest (261 bp). In this mt genome, all protein-coding genes use ATG as start codon (Table 1). All protein-coding genes have complete termination codons (TAA and TAG) (Table 1). However, some studies have indicated that the incomplete termination codons T or TA are present in the protein-coding genes of some tapeworm mt genomes [7, 23]. A total of 22 tRNA (ranging from 56 to 72 nucleotides in length) genes were identified. Their predicted secondary structures (not shown) are similar to those in H. diminuta and A. perfoliata [8]. The tRNA-Cys gene separates rrnL from rrnS. The size of the rrnL gene is 959 bp and the size of the rrnS gene is 722 bp (Table 1). One larger non-coding region (NC2; 446 bp) is located between the tRNA-Gly and cox3 genes, and one shorter non-coding region (NC1; 221 bp) is located between the tRNA-Tyr and tRNA-Ser genes (Table 1; Fig. 1). In the NC1 region, there were two sets of short inverted repeats and one set of long inverted repeats (33 bp), each of them could be folded into a stem-loop hairpin structure (Additional file 2: Figure S1A). The NC2 region consists of six identical tandem repeats with 31 bp sequences (Additional file 2: Figure S1B). Similar stable hairpin structures and tandem repeats in C. megalops may play the same role as those in vertebrates, which have been shown to initiate replication and transcription [24].
Sequence comparisons
Pairwise comparisons of C. megalops mt protein-coding genes with those of three other hymenolepidid tapeworms revealed 12.9–35.3 % differences in the nucleotide sequences, and 10.6–43.6 % differences in amino acid sequences (Table 2). Among twelve protein-coding genes, cox1 and cytb genes were relatively conserved whilst nad5 and nad6 genes were the most different in all four species (Table 2). These results are useful to design primers to capture high sequence variability within and between mt genes of these species as genetic markers for population genetics and diagnostics.
Phylogenetic analyses
Phylogenetic analyses showed three distinct groups with high statistical support (Bpp = 1.0; Bf = 98 %) with C. megalops as a sister taxon to Anoplocephala spp. (Anoplocephalidae) + P. crawfordi + Hymenolepis spp. (Hymenolepididae) (Fig. 2), indicating that the family Hymenolepididae is paraphyletic. The result is consistent with the maximum likelihood analysis in the study by von Nickisch-Rosenegk et al. [25] in which 12S rDNA data of a wider set of taxa representing a larger number of families. The families Taeniidae, Anoplocephalidae and Diphyllobothriidae were monophyletic with maximum support in all analyses (Bpp = 1.0; Bf = 100 %) (Fig. 2), in agreement with previous studies [7, 9, 26]. In addition, our results show that H. diminuta is more closely related to P. crawfordi than to H. nana (Fig. 2).

Phylogenetic relationships among 19 species of tapeworms. Phylogenetic tree was inferred by Bayesian inference and Maximum likelihood analysis from deduced amino acids of 12 protein-coding genes using Schistosoma japonicum as the outgroup. Bayesian posterior probability (Bpp) and bootstrapping frequency (Bf) values are shown at nodes
In this study, the phylogenetic tree revealed that C. megalops is distantly related to the genus Hymenolepis, supporting the view of Makarikov et al. [13] who preferred to retain the independent status of Cloacotaenia based on the morphological characteristics. The present results also suggest that Pseudanoplocephala crawfordi is a member of the genus Hymenolepis as shown in previous studies [10, 27]. Hymenolepis nana is one of the most common tapeworms infecting humans and rodents. However, there is confusion regarding the nomenclature of this species [28]. A taxonomic revision of hymenolepidids with armed rostellae was suggested by Vaucher [29] in which Hymenolepis nana was regarded as a member of the genus Rodentolepis. This classification is currently accepted by some cestode taxonomists [12]. Despite the nomenclature being revised, the name H. nana persists in textbooks and medical papers [30]. Additionally, whether the Anoplocephalidae should be nested among species of the Hymenolepididae should also be rigorously evaluated in further studies based on more extensive taxon sampling of hymenolepidids. This will also help a better understanding of the evolution of hymenolepidid cestodes and a re-evaluation of the morphological traits employed in their systematics.
Conclusions
The complete mt genome of C. megalops is characterised. Phylogenetic analyses of the concatenated amino acid sequence dataset for 12 protein-coding mt genes of C. megalops and selected cestode representatives indicated that the family Hymenolepididae is paraphyletic. This mt genome provides a unique genetic marker for studying the molecular biology, genetics and systematics of C. megalops.
Abbreviations
- BI:
-
Bayesian inference
- Bpp:
-
Bayesian posterior probabilities
- ML:
-
Maximum likelihood
- Mt:
-
Mitochondrial
References
Green AJ, Georgiev BB, Brochet AL, Gauthier-Clerc M, Fritz H, Guillemain M. Determinants of the prevalence of the cloacal cestode Cloacotaenia megalops in teal wintering in the French Camargue. Eur J Wildlife Res. 2011;57:275–81.
Liao DJ, Li L, Dai ZJ, Lin Y, Yang GY, Mao GQ. Livestock tapeworm and acanthocephalan list of Southwest District in China. Chinese J Vet Parasitol. 2002;10:14–7.
Long WB, Yang GS, Yang GL. Investigation of parasitic species of duck in Qian dongnan in China. Chinese J Vet Parasitol. 2002;10:38–40.
Brabec J, Kostadinova A, Scholz T, Littlewood DT. Complete mitochondrial genomes and nuclear ribosomal RNA operons of two species of Diplostomum (Platyhelminthes: Trematoda): a molecular resource for taxonomy and molecular epidemiology of important fish pathogens. Parasit Vectors. 2015;8:336.
Bachmann L, Fromm B, Patella de Azambuja L, Boeger WA. The mitochondrial genome of the egg-laying flatworm Aglaiogyrodactylus forficulatus (Platyhelminthes: Monogenoidea). Parasit Vectors. 2016;9(1):285.
Jia WZ, Yan HB, Guo AJ, Zhu XQ, Wang YC, Shi WG, et al. Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics. 2010;11:447.
Guo A. The complete mitochondrial genome of Anoplocephala perfoliata, the first representative for the family Anoplocephalidae. Parasit Vectors. 2015;8:549.
von Nickisch-Rosenegk M, Brown WM, Boore JL. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that Platyhelminths are Eutrochozoans. Mol Biol Evol. 2001;18:721–30.
Cheng T, Liu GH, Song HQ, Lin RQ, Zhu XQ. The complete mitochondrial genome of the dwarf tapeworm Hymenolepis nana - a neglected zoonotic helminth. Parasitol Res. 2016;115:1253–62.
Zhao GH, Wang HB, Jia YQ, Zhao W, Hu XF, Yu SK et al. The complete mitochondrial genome of Pseudanoplocephala crawfordi and a comparison with closely related cestode species. J Helminthol. 2016;90:588-95
Guo A. Complete mitochondrial genome of Anoplocephala magna solidifying the species. Korean J Parasitol. 2016;54(3):369–73.
Czaplinski B, Vaucher C. Family Hymenolepididae Ariola, 1899. In: Khalil LF, Jones A, Bray RA, editors. Keys to the cestode parasites of vertebrates. Wallingford: CAB International; 1994. p. 595–663.
Makarikov AA, Nims TN, Galbreath KE, Hoberg EP. Hymenolepis folkertsi n. sp. (Eucestoda: Hymenolepididae) in the oldfield mouse Peromyscus polionotus (Wagner) (Rodentia: Cricetidae: Neotominae) from the southeastern Nearctic with comments on tapeworm faunal diversity among deer mice. Parasitol Res. 2015;114:2107–17.
Nowak MR, Krolaczyk K, Kavetska KM, Pilarczyk B. Morphological features of Cloacotaenia megalops (Nitzsch in Creplin, 1829) (Cestoda, Hymenolepididae) from different hosts. Wiad Parazytol. 2011;57:31–6.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Le TH, Blair D, McManus DP. Mitochondrial DNA sequences of human schistosomes: the current status. Int J Parasitol. 2000;30:283–90.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704.
Nakao M, Abmed D, Yamasaki H, Ito A. Mitochondrial genomes of the human broad tapeworms Diphyllobothrium latum and Diphyllobothrium nihonkaiense (Cestoda: Diphyllobothriidae). Parasitol Res. 2007;101:233–6.
Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992;141:173–216.
von Nickisch-Rosenegk M, Lucius R, Loos-Frank B. Contributions to the phylogeny of the Cyclophyllidea (Cestoda) inferred from mitochondrial 12S rDNA. J Mol Evol. 1999;48:586–96.
Taleb-Hossenkhan N, Bhagwant S. Molecular characterization of the parasitic tapeworm Bertiella studeri from the island of Mauritius. Parasitol Res. 2012;110:759–68.
Jia YQ, Yan WC, Du SZ, Song JK, Zhao W, Zhao YX, et al. Pseudanoplocephala crawfordi is a member of genus Hymenolepis based on phylogenetic analysis using ribosomal and mitochondrial DNA sequences. Mitochondrial DNA A DNA MappSeq Anal. 2016;27:1688–92.
Macnish MG, Morgan UM, Behnke JM, Thompson RC. Failure to infect laboratory rodent hosts with human isolates of Rodentolepis (= Hymenolepis) nana. J Helminthol. 2002;76(1):37–43.
Vaucher C. Revision of the genus Vampirolepis Spasskij, 1954 (Cestoda: Hymenolepididae). Mem Inst Osw Cruz. 1992;87:299–304.
Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am. 1996;25:637–53.
Acknowledgements
The author wishes to thank editor and reviewers for providing language help and constructive suggestions in this manuscript.
Funding
Project support was provided by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840).
Availability of data and material
The datasets supporting the conclusions of this article are included within the article and its additional files. The complete mt genome sequence is submitted to the GenBank database under accession no. KU641017.
Competing interests
The author declares that she has no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Table S1. Primers used to amplify PCR fragments for Cloacotaenia megalops. (DOC 37 kb)
Additional file 2:
Figure S1. Putative secondary structures for the two non-coding regions in Cloacotaenia megalops mtDNA. The NC1 (A) consists of two identical repeats of 34 nt shown in the box. The NC2 region (B) consists of six identical tandem repeats of a 31 nt sequence and part of the seventh repeat (10 nt). Arrows represent inverted repeats. (DOC 330 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, A. Characterization of the complete mitochondrial genome of the cloacal tapeworm Cloacotaenia megalops (Cestoda: Hymenolepididae). Parasites Vectors 9, 490 (2016). https://doi.org/10.1186/s13071-016-1782-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-016-1782-0