
Abstract
Lignocellulose is the most abundant organic carbon polymer on the earth. Its decomposition and conversion greatly impact the global carbon cycle. Furthermore, it provides feedstock for sustainable fuel and other value-added products. However, it continues to be underutilized, due to its highly recalcitrant and heterogeneric structure. Microorganisms, which have evolved versatile pathways to convert lignocellulose, undoubtedly are at the heart of lignocellulose conversion. Numerous studies that have reported successful metabolic engineering of individual strains to improve biological lignin valorization. Meanwhile, the bottleneck of single strain modification is becoming increasingly urgent in the conversion of complex substrates. Alternatively, increased attention has been paid to microbial consortia, as they show advantages over pure cultures, e.g., high efficiency and robustness. Here, we first review recent developments in microbial communities for lignocellulose bioconversion. Furthermore, the emerging area of synthetic ecology, which is an integration of synthetic biology, ecology, and computational biology, provides an opportunity for the bottom-up construction of microbial consortia. Then, we review different modes of microbial interaction and their molecular mechanisms, and discuss considerations of how to employ these interactions to construct synthetic consortia via synthetic ecology, as well as highlight emerging trends in engineering microbial communities for lignocellulose bioconversion.
Similar content being viewed by others

Background
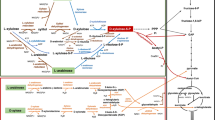
Lignocellulose, which is chiefly composed of cellulose, hemicellulose and lignin, is the most abundant organic carbon polymer in the biosphere. Take lignin for instance, with an estimated 300 billion tons present globally and an annual increase of ~ 20 billion tons [1]. Its decomposition and conversion have a significant impact on the global carbon cycle. Moreover, this abundance enables its use as a feedstock for sustainable fuel and chemical production (e.g., ethanol, nylon, lipid and polyhydroxyalkanoate (PHA)), addressing energy and environmental concerns (Fig. 1) [1,2,3]. However, lignocellulose has remained the underutilized renewable biomass due to its complex chemical structure. This is especially true for lignin. Unlike cellulose and hemicellulose, which consists of carbohydrate monomers, lignin is composed of three phenylpropanoid units, which are linked by various, strong carbon–carbon and ether bonds, to form natural barriers against decay and give plants their rigidity and structure (Fig. 1) [1]. So far, lignin is removed during the pretreatment process at lignocellulosic biorefineries and used as fuel to supply heat and power through burning. Thus, lignin valorization not only provides opportunity to produce value-added chemicals and fuels, but also improves the efficiency of the current lignocellulosic biorefineries.

The structure of lignin and microbial mediated lignin bioconversion. Lignin is the most recalcitrant component of lignocellulose. It’s composed of G, S, and H phenylpropanoid constitutes, which are linked by five linkage types. These common linkages are cleaved by lignolytic enzymes (e.g., laccases, peroxidases, and redox accessory enzymes) for lignin depolymerization. Subsequently, aromatic compounds from G-, H-, and S-type lignin are degraded via microbial funnel pathways to generate the key intermediate aromatic compounds protocatechuate, catechol, and gallate. These intermediates are further catabolized to synthesize value-added bioproducts e.g., polyamide and nylon. Alternatively, they enter TCA cycle and fatty acid metabolism for PHA or lipid synthesis
Microorganisms, which are present in nearly all habitats on the Earth, have evolved different, yet complementary, mechanisms to degrade these polymers. Therefore, they provide green and energy-efficient routes for lignocellulose bioconversion [4, 5].
-
i)
Various enzymes have been identified as lignocellulose biocatalysts. Fungi and bacteria release cohorts of carbohydrate-active enzymes (CAZymes, e.g., glycoside hydrolases (GHs)) to synergistically hydrolyze (hemi) cellulose. For instance, aerobic fungi secrete ample and distinct cellulases, as noncomplexed cellulase mixtures, to hydrolyze cellulose, while some anaerobic bacteria, e.g., Clostridium strains, produce cell-bound cellulosomes for efficient cellulose hydrolysis [6]. Moreover, microorganisms have diverse sets of oxidoreductase enzymes for lignin oxidization [4, 7,8,9]. Recently, bacterial enzymes have been reported to play a key role in lignin oxidization [9, 10]. Our lab indicated that the dye-decolorizing peroxidases (DypBs) in Pseudomonas putida A514 showed a unique Mn2+ independent lignin depolymerization activity and exhibited synergistic lignin degradation with P. putida cells [11]. Furthermore, we bio-designed two secretory apparatuses for enhanced extracellular laccase expression to promote lignin degradation [12].
-
ii)
Highly versatile metabolic pathways contribute to achieve lignocellulose conversion. With the development of systems biology and synthetic biology, we and others have developed various genetic engineering strategies to engineer metabolic pathways for enhancement of lignocellulose bioconversion [4, 13,14,15]. Shaw et al. deleted genes involved in organic acid formation in Thermoanaerobacterium saccharolyticum to significantly improve cellulosic ethanol production [16]. Jin et al. introduced a rationally designed hydroxylase system in Rhodococcus opacus to accumulate gallate, used o-demethylation systems to convert multiple lignin-derived methoxy aromatics to gallate, and engage an aryl side-chain oxidase to broaden the substrate spectrum [17]. As a result, they greatly improved lignin to gallate conversion. We also applied systems biology to guide design of lignin-to-PHA bioconversion in P. putida [18] and then, developed a strategy to simultaneously improve cell growth and PHA production in P. putida from a lignin derivative [19]. Moreover, for enhancement of ferulic acid (a lignin derivative)-to-PHA bioconversion, we developed a CRISPR/Cas9n-based genome editing tool to metabolically engineer P. putida [20]. These studies laid an essential foundation in understanding the molecular mechanisms of lignocellulose bioconversion and further engineering microorganisms for enhancement of lignocellulose bioprocess.
With numerous studies reporting successful metabolic engineering of individual strains, the bottleneck of single strain modification is becoming more urgent in the conversion of complex substrates, because lignocellulose conversion involves multiple enzymes and pathways that generally do not exist within, nor can be feasibly introduced into, a single strain. In natural environments, microorganisms form communities, where each member executes specialized sub-functions to synergistically perform the complete lignocellulose bioconversion [21]. Moreover, in contrast to pure cultures, microbial communities exhibit the property of resilience, which can increase resistance to environmental perturbations [22, 23]. Undoubtedly, natural microbial communities can provide clues to expand microbial engineering to mixed consortia. This review discusses current developments in microbial consortia for lignocellulose degradation, microbial interaction modes and molecular mechanisms, considerations of how to construct synthetic consortia, as well as the trends in designing and engineering synthetic microbial communities for lignocellulose bioconversion.
Development of microbial consortia for lignocellulose degradation
As stated previously, consortium-based approaches are a promising avenue for lignocellulose bioconversion [24, 25]. To date, “top-down” enrichment and “bottom-up” synthetic communities are the two major routes in developing microbial consortia for lignocellulose degradation (Fig. 2).

The current strategies to develop microbial consortia for lignocellulose bioconversion. A “Top-down” selection and enrichment strategy. After manipulation via primary screening, re-screening, and identification, a group of microbial consortia are developed. B–D “Bottom-up” reconstruction of co-cultures. B “bottom-up” co-cultures are constructed based on potential interactions predicted by the genomic sequencing of “top-down” enriched consortia [25]. C A combination strategy is used to develop fungal-bacterial microbial consortia, based on as many strains as possible with as few experiments as possible [40]. D A reductive-screening approach with ecological strategies is developed to acquire an effective lignocellulose-degrading minimally active microbial consortium [37]. FMG: functional metabolic groups
“Top-down” selection and enrichment
“Top-down” selection and enrichment (e.g., dilution-to-stimulation method) is the most common strategy to obtain microbial consortia with lignocellulose degradation properties from natural environments [26, 27]. In this route, microbial samples from different environmental sources are collected, screened, and enriched after generations of subculture. Through primary screening and rescreening, the composition of the microbial communities gradually changes to adapt to the current conditions. Finally, stabilized microbial communities are generated, where members synergistically interact to degrade lignocellulose (Fig. 2A). Fang et al. employed this method to develop the microbial consortium DM-1 from tree trimmings [28]. DM-1 included Mesorhizobium, Cellulosimicrobium, Pandoraea, Achromobacter, and Stenotrophomones as the predominant genera and efficiently degrades lignin (28.7%) and cellulose (10.2%). Cortes-Tolalpa cultured a salt marsh soil microbiome in six sequential cycles on fresh wheat straw and four cycles on pre-digested wheat straw. Consequently, a consortium, which was selected on the latter, highly-recalcitrant substrate, improved cellulose (64.2%) and lignin (61.4%) degradation [29]. The dominant microorganisms in this consortium were bacteria Joostella marina, Flavobacterium beibuense, Algoriphagus ratkowskyi, Pseudomonas putida, and Halomonas meridiana, whereas Sarocladium strictum was the single fungal strain, indicating that bacteria have a major role in lignocellulose degradation under saline conditions. Gilmore et al. selected a stable native consortium for efficient lignocellulose-to-methane bioconversion via antibiotic treatment and serial cultivation, dominating by an anaerobic fungus (Piromyces), a bacterium (Sphaerochaeta) and two methanogenic archaea (Methanosphaera and Methanocoprpusculum) (Fig. 2A) [25]. Via the “top-down” strategy, our lab also obtained several lignocellulose degradation microbial communities from coastal waters of the East China Sea and revealed several previously unrecognized marine bacterial lignin-degraders, e.g., Yangia, Pelagibaca, Salipiger, Celeribacter, and Vibrio [30]. To date, microbial consortia with the ability to degrade lignocellulose have been acquired from forest soil, canal sediment, decaying wood, sea sediment, and coastal sea water [28, 31,32,33,34]. Moreover, these consortia commonly exhibited higher lignocellulose degradation and production of value-added products than single strain. The DM-1 consortium and the community, enriched from salt marsh soil, showed 28–64% lignin consumption [28, 29], whereas pure culture (e.g., R. opacus PD630 and Pseudomonas putida A514) showed 15–18% lignin consumption [12, 35]. In addition, the consortium, acquired by Gilmore et al., produced 0.75–1.9-fold more methane-rich biogas than a monoculture of fungi from the community [25].
Metagenomic sequencing analysis has further revealed key lignocellulose degrading genes and pathways in lignocellulose-degrading consortia. This greatly boosted the study of lignin bio-degradation, which has not been fully characterized, in contrast to the firm understanding of cellulose degradation. For instance, metagenomic sequencing analysis demonstrated that the lignin-degrading consortium (LigMet), enriched from a sugarcane farm, employed peroxidases, dye-peroxidases, laccases, carbohydrate esterases, and lignocellulosic auxiliary activities to oxidize lignin, and utilized benzoate-to-catechol degradation pathway, catechol ortho-/meta-cleavage pathway, and phthalate degradation pathway to catabolize lignin-derived aromatic compounds [36]. In addition, metagenomic and quantitative stable isotope probing experiments suggested aryl alcohol oxidase genes were the most significantly correlated with the enriched bacterial lignin degradation consortium from forest soils [21]. Interestingly, metagenomic sequencing detected that bacteria in Eastern Mediterranean Sea potentially utilized the phenylacetyl-CoA pathway for lignin degradation [34].
These “top-down” enriched microbial consortia, which contributed to lignocellulose bioconversion, have improved our understanding of microbial strains and key genes/pathways for lignocellulose degradation. However, it’s worth noting that consortia developed via this strategy are complex, and may contain thousands of fungal, bacterial, and archaeal members. Such complexity makes it difficult to untangle the interactive network that is responsible for the conversion process. In addition, not all effective strains from the native enrichments can be cultured in the laboratory, and further, may cause instability of community composition and function in these natural communities during sequential enrichment cultivation.
“Bottom-up” co-cultures
To address this issue of complexity and instability, various studies about “bottom-up” route have been reported to construct consortia with a limited number of culturable strains for more efficient lignocellulose conversion. First, “bottom-up” reconstruction can be guided by “top-down” enrichment. As stated earlier, Gilmore et al. acquired a native consortium for lignocellulose-to-methane bioconversion, via the “top-down” enrichment method (Fig. 2A) [25]. The microbial composition and potential syntrophic mechanisms, which were identified by high-throughput sequencing, guided the design of a minimal consortium (Fig. 2B) [25]. This resulted in a minimal, but effective community, including fungi Neocallimastix californiae and Anaeromyces robustus with the methanogen Methanobacterium bryantii. In addition, Díaz-García et al. developed a combined “top-down” enrichment strategy, coupling dilution-to-stimulation and dilution-to-extinction, to build a minimal and effective lignocellulolytic microbial consortium (MELMC), where two bacterial species (Pseudomonas sp. and Paenibacillus sp.) are highly abundant (> 99%) [27]. Second, a method for efficient combination of diverse microbial strains have been reported to construct communities for lignocellulose degradation. Co-cultivation of microorganisms with complementary activities is a simple, yet effective strategy to construct efficient consortia [37, 38]. For instance, co-cultures of cellulolytic Clostridium thermocellum with ethanol-producing Thermoanaerobacter strain significantly improved ethanol production in the defined medium with 1% of cellulose, ~ 65 mM by co-cultures vs < 13 mM by C. thermocellum mono-culture [39]. However, microbial interactions are varied and complicated. Thus, a large number of experiments are required to examine the various effects of different microbial combinations. To overcome this limitation of intensive labor, Hu et al. reported a method for screening possible microbial interactions based on as many strains as possible, but with as few experiments as possible (Fig. 2C) [40]. In this strategy, multiple cellulolytic fungal strains were, firstly, combined in various ways to develop a synergistic fungal community, which exhibited high lignocellulosic enzyme activity. Next, a screened microbial community, which included thousands of strains, was introduced as an additional “member” of this community. Direct microbial interactions and the key strains in such interactions were identified by measuring lignocellulolytic enzyme activity and high-throughput sequencing technology [40]. As a result, two fungal (Trichoderma and Aspergillus) and 16 bacterial strains (Bacillus, Enterococcus, Lactococcus, Acinetobacter, and Pseudomonas) were designated as the final members of a synergistic microbial consortium with improved capacity for lignocellulolytic enzyme production (Fig. 2C). Among them, the β-glucosidase activity of the microbial community was 197% higher than that of the signal fungal strain, Trichoderma reesei [40]. Compared to the simple combinations of a limited number of strains, this approach could identify beneficial interactions among microorganisms and further improve those interactions by optimizing the composition and structure of the microbial community. Third, a reductive-screening approach, combined with ecological strategies, was utilized to acquire a minimal active microbial consortium (Fig. 2D). Enrichment of microorganisms is performed first, followed by isolation, identification, and metabolic characterization. Subsequently, a set of strains representing these groups are used to construct minimal active microbial consortia (MAMC). Each consortium contains different species, which vary in the number of functional groups, metabolic potential, and degradation capacity (Fig. 2D). Via this reductive-screening method, Puentes-Téllez and Falcao Salles identified 45 soil bacterial strains and classified them to four functional metabolic groups [37]. Finally, they successfully developed an effective MAMC with a 96.5% degradation rate. The MAMC contained all 18 species, where Stenotrophomonas maltophilia, Paenibacillus sp., Microbacterium sp., Chryseobacterium taiwanense, and Brevundimonas sp. are the major lignocellulose degraders [37].
These developed microbial consortia, via either “top–bottom” enrichment or “bottom-up” combination methods, employ microbial interactions to efficiently perform the task of lignocellulose conversion. With the advances in metagenomic and transcriptomic sequencing, it’s convenient for us to understand the microbial composition, function genes, and dynamics of these communities. However, there is not yet a clear, basic mechanistic understanding of the individuals that drive the overall function and ecological principles in these communities. To understand these complex communities and further manipulate them with defined functions, we should explore microbial interactions (e.g., modes and mechanisms) and how these properties can be applied to the rational design of bottom-up synthetic microbial communities, instead of current strategies of co-culturing microorganisms via random combinations or intuition about simple metabolite exchange.
Microbial interaction modes and molecular mechanisms
Synergistic microbial interaction modes
Microbial interactions are believed to be central to the survival, stability, and productivity of communities. There are six different categories of pairwise interaction modes, including neutralism (0/0), commensalism (+/0), amensalism (−/0), mutualism (+/+), competition (−/−), and parasitism or predation (±) [41]. In this review, we focus on the symbiotic interactions (commensalism and mutualism) in microbial consortia, which drive members to cooperate and execute different, yet complementary tasks.
The essence of microbial synergistic interactions is the sharing and exchange of public goods. Public goods (e.g., enzymes, amino acids, vitamins and detoxification agents) are products that are costly to produce, but provide a benefit to members of a community, especially to neighbors of the producer [42,43,44]. For lignocellulosic degradation consortia, carbohydrate monomers and aromatic monomers, which can be directedly utilized by the members, are representative public goods. The sharing of public goods creates an opportunity for cooperative interactions, one of which is mutualism (+/+). It is the win–win relationship of symbiotic associations, in which each member derives benefits from one another (Fig. 3A) [41, 45]. Metabolic division of labor (DOL) is a typical mutualistic interaction, where distinct populations perform different, but complementary, metabolic tasks to diminish the metabolic burden on each population [46]. This was demonstrated in the synthetic lignocellulose-degrading microbial community, including Pseudomonas putida, Cellulomonas fimi, and Methylorubrum extorquens [47]. P. putida is a lignin degrader, while C. fimi is a cellulose degrader and these organisms work together to degrade lignocellulose (Fig. 3A). Moreover, metabolic cross-feeding is also a well-known DOL, where different groups exchange costly metabolites to the benefit of both interacting partners. In this consortium, P. putida produced formaldehyde and C. fimi generated organic acids to support the growth of M. extorquens. In turn, M. extorquens supplied methionine and iron to P. putida and C. fimi (Fig. 3A) [47].

Synergistic microbial interaction modes and mechanisms. A Synergistic microbial interaction modes. Mutualism and commensalism are presented. CBHI, cellobiohydrolase I; CBHII, cellobiohydrolase II; EGI, endoglucanase I. Three cellulases which are produced by T. reesei, hydrolyze cellulose to soluble oligosaccharides. B Microbial interaction mechanisms. These include contact-independent and contact-based interactions. Contact-independent interactions indicate that molecules (e.g. QS signal molecules) are released by diffusion or transported by efflux pumps. Contact-based interactions suggest that molecules are exchanged by physical cell–cell contact, e.g., pili, nanotubes, vesicles. AHLs: QS signal molecules. AHL signals, which are produced by LasI family, bind LasR, thus activating it. The activated complex modulated transcription of biofilm synthesis genes. Direct electron transfer is based on membrane bound c-type cytochromes or pili, indirect electron transfer is mediated by electron shuttles
With the succession of microbial communities, public goods could be exploited by selfish cheaters, which generates another synergistic interaction, commensalism (+/0). In this relationship one partner acquires benefits from the other, while the other one is in unaffected (neither harmful nor beneficial) [45]. In lignocellulose degrading microbial community, these opportunistic organisms are termed “sugar cheaters” [26]. Minty et al. constructed a fungal-bacterial consortia (TrEc), where Trichoderma reesei secreted cellulase enzymes to hydrolyze lignocellulose into soluble saccharides, as the public goods, and Escherichia coli metabolized soluble saccharides into isobutanol [48]. In this consortia, T. reesei acted as a cooperator (+) by secreting metabolically expensive cellulase, while E. coli was a “sugar cheater” (0), which directly utilized the saccharides without bearing the burden of cellulase production (Fig. 3A). Together, the two types of synergistic interactions enable communities to better accomplish tasks that are more metabolically intensive than pure cultures could.
Interaction mechanisms in microbial consortia
To achieve the above stated interactions, microorganisms mostly employ two major types of interaction mechanisms, contact-independent and contact-based interactions. Contact-independent interactions rely on the release and sensing of chemical molecules (Fig. 3B) [49]. Quorum-Sensing (QS) is one of the major means to program cell–cell contact-independent communication [50]. Bacteria produce and release small hormone-like signal molecules, termed autoinducers (AI), into the environment, while neighboring microbial cells detect and respond to these molecules to alter gene expression and synchronize community-wide behavior [51]. QS mediate intra- and inter-species communication, control many microbial physiological behaviors (e.g., competence, symbiotic interactions, motility, and biofilm formation) as well as coordinate population-level behavior, thus playing a key role in lignocellulose conversion (Fig. 3B) [23, 50]. For instance, QS regulates biofilm synthesis, which can be utilized to improve lignocellulose conversion [52, 53]. On one hand, the concentration of cell-associated hydrolytic enzymes at the biofilm-substrate interface can be increased to improve saccharification [53, 54]. On the other hand, symbiotic biofilms, which consists of aerobic fungi (e.g., T. reesei) and anaerobic bacteria (e.g., L. pentosus), is an ideal strategy for simultaneous (hemi) cellulose hydrolysis and products (e.g., lactic acid) generation [55, 56]. Beyond QS signal molecules, a large repertoire of metabolites are actively or passively diffused among microorganisms to establish cell–cell interactions, including small (e.g., H2, CH4, CO2, and lactate) and large molecules (peptides and proteins) (Fig. 3B) [49]. In addition, electron exchanges also occur between bacteria via contact-independent interactions (Fig. 3B) [57]. Such extracellular electron transfer, via soluble electron shuttles, fuels cellulose hydrolysis and lignin oxidation [58, 59].
Meanwhile, microorganisms have evolved contact-dependent delivery systems (e.g., pili, nanotubes, outer membrane vesicles and channels) to transfer metabolites, including amino acids, proteins, DNA, and other small molecules (Fig. 3B). In addition to these macromolecules, intraspecies and interspecies electrons can be transferred via direct contact-based interactions, mediated by outer membrane cytochromes and conductive pili [45, 60]. For instance, Staphylococcus aureus can oxidize cellulose and generate the electricity in MFC (microbial fuel cells), via directed electron transfer [61, 62].
Contact-independent communication allows molecules to reach many neighboring cells, as opposed to only one cell at a time (e.g., cell-to-cell contact). Therefore, it enables them to serve as nutrients or cues to nearby cells, and, in effect, lead to behavior similar to that of a multicellular entity [49]. However, unintended third parties may intercept the signal or catabolize the metabolite, causing the exchanged molecules to be degraded or lost during the diffusion process [63]. In contrast, contact-dependent communication systems can overcome this issue, although they cannot simultaneously connect to multiple neighboring cells. Such synergistic microbial interaction modes and mechanisms provide insights to the following “bottom-up” design of synthetic consortia for lignocellulosic bioconversion.
Synthetic ecology provides an opportunity to “bottom-up” design synthetic microbial consortia for efficient lignocellulose bioconversion
With the rapid development of high-throughput sequencing, the top-down study of microbial communities efficiently reveals their composition and function, further guiding us to obtain minimal, yet efficient, synthetic consortia. However, the contributions of individual members and the highly inter-connected networks of metabolic and ecological interactions are still unclear, which greatly restricts development of the “bottom-up” study of microbial consortia to enhance lignocellulose bioconversion. Synthetic ecology, which is the integration of synthetic biology, ecology, and computational biology, presents a feasibility to rationally engineer microorganisms at the population-scale. There are three important factors that should be considered for “bottom-up” synthetic ecology study of microbial consortia. These factors and corresponding methods/tools are discussed below.
Current cooperation strategies and computational approaches to design cooperation
Cooperation in consortia is the first key factor to achieve efficient and stable bioconversion (Fig. 4A). So far, synthetic microbial consortia mainly employ three strategies to engineer intercellular cooperation. The first is an aggregation strategy (Fig. 4A), where every member in the consortium can individually perform the task, but each one underperforms the community. Cortes-Tolalpa et al. employed this strategy to develop an efficient microbial community for lignocellulosic biomass degradation. They utilized Citrobacter freundii and Sphingobacterium multivorum, both of which are lignocellulosic degraders. S. multivorum w15 secreted extracellular enzymes (e.g., cellobiohydrolases and β-xylosidases) to generate public goods, while C. freundii so4 provided redox power and metabolic intermediates for S. multivorum w15 [64]. Hence, compared to each pure culture, this coculture showed significantly increased enzymatic activities (e.g., cellobiohydrolases, mannosidases and xylosidases) and biomass (18.2-fold). The second strategy is a metabolic division of labor (DOL) (Fig. 4A). He et al. co-cultured cellulolytic C. thermocellum LQR1 and ethanol-producing Thermoanaerobacter sp. X514 to enhance cellulosic bioethanol fermentation [39]. LQR1 secreted the cellulosome to hydrolyze cellulose and produce soluble monosaccharides/oligosaccharides for the growth of LQR1 and X514, while X514 produced ethanol. They worked together to achieve cellulosic ethanol production. The third strategy is a boosting method (Fig. 4A). Via a cross-feeding interaction, members without the defined functions (e.g., lignocellulose degradation and product biosynthesis) contribute to the positive performances of other members with those functions. As stated previously, in the three-species lignocellulose-degrading consortia, P. putida and C. fimi were lignocellulose degraders, while M. extorquens removed a toxic burden and fed back iron and methionine to the degraders for improvement of lignocellulose degradation (Fig. 3A) [47].

Key factors of design and engineering microbial communities for lignocellulosic conversion. A Microbial cooperation can be designed to perform the task of lignocellulose bioconversion. Computational and mathematical models are effective tools for genome-wide explore task assignment. B Spatial assortment contributes to microbial cooperation to degrade lignocellulose. Biofilms, formed by degraders or producers, provide an interface in which multiple species synergize for lignocellulose conversion. In addition, modified bioreactors can achieve spatial separation for microbial members which have different growth conditions. C Robustness and stability of communities can be maintained by spatial self-organization and regulation of relative cooperator/cheater benefits. D Overview of pipeline for the automated design of synthetic microbial communities [95]. It provides a promising avenue for the effective and easy design of microbial communities for lignocellulosic conversion
The essence of design cooperation is understanding how metabolic tasks can be rationally allotted to individual members to achieve a desirable productive manner at a community-level. However, microorganisms have complex cross-feeding strategies for multiple metabolites, either simultaneously, or in an environment-dependent manner [65, 66], undoubtedly leading to the manual design of cooperation to be very challenging. Mathematical models and computational approaches in synthetic ecology have been developed to characterize microbial interactions and complex metabolic networks at the genome level. Minty et al. developed a comprehensive ordinary differential equation (ODE) modeling framework for the TrEc consortium (consisting of cellulolytic specialist T. reesei and isobutanol producer E. coli), including growth kinetic model, cellulase secretion model and saccharide uptake model for T. reesei, as well as growth model, saccharide uptake model and isobutanol production for E. coli [48]. Fifty parameters and variables for the concentration of microbial biomass, enzymes, isobutanol, soluble oligosaccharides, and each possible cellulose polysaccharide were introduced into the models to capture salient features of the TrEc consortium. It suggested that competition between T. reesei and E. coli for soluble saccharides was a key ecological interaction to drive community behavior, indicating an inherent tradeoff between cellulose hydrolysis rate and isobutanol yield [48]. Through this experimentally validated mathematical modeling framework, key parameters (e.g., growth kinetics, substrate uptake kinetics, and population ratio), which controlled consortium behavior, were identified and optimized. As a result, isobutanol yields of this consortium increased to 62% of theoretical maximum, reaching titers of 1.88 g/L [48]. In addition, in silico genome-scale metabolic models (GSMM) have been constructed to combine experimental and omics data sets in a systematic manner to understand complex microbial metabolism [67,68,69]. GSMM are constructed in a bottom-up fashion based on genomic and bibliomic data, thus representing the biochemical, genetic, and genomic (BiGG) knowledge for the target organism (Fig. 4A) [70]. Based on GSMM, a variety of flux balance analyses (FBA) were performed, including parsimonious flux balance, dynamic flux balance, and flux variability analysis [65, 68, 71, 72]. Such flux analysis can determine how a cell should optimally allocate nutrients so that the growth rate is maximized [73, 74]. Moreover, it can be extended to microbial communities to study metabolic interactions and predict community-wide behaviors [75]. Hanly et al. employed dynamic flux balance modeling to improve the consumption of glucose/xylose mixtures (the main enzymatic products of cellulose degradation) in Saccharomyces cerevisiae and E. coli co-cultures [76]. In this consortium, S. cerevisiae consumes glucose, while E. coli strain ZSC113 catabolizes xylose. Inhibitory interactions were observed: S. cerevisiae metabolizes some compounds, which are toxic to E.coli, and produces ethanol that inhibits the growth of E. coli. Thus, the efficiency of glucose/xylose utilization was not high. The dynamic flux balance model suggested that optimization of co-culture inoculum can address such problem, as optimized inoculum can accelerate xylose uptake rates at the expense of decrease glucose uptake. As a result, the co-culture system can exhaust 16 g/L glucose and 8 g/L xylose at 9.8 h, compared to original 11.5 h batch time [76]. Additional in silico genome-scale models have been developed to simulate better programs about microbial interactions [77]. For instance, division of labor in metabolic networks (DOLMNs) were reported to explore how metabolic interdependencies between metabolically different E. coli strains [65]. A multiphase multi-objective dynamic genome-scale model of was constructed to provide insights into how different redox balancing was achieved among Saccharomyces strains [78]. These models offer a systematic approach to expand on the engineering intercellular cooperation for lignocellulosic bioconversion.
Spatiotemporal assortment and related biotechnology
Spatial and temporal assortment should be considered to promote cooperation, although microbial communities are generally cultured in well-mixed systems. Various microbial spatial and temporal segregations exist in nature. For example, aerobic and anaerobic microbial populations are separated by oxic/anoxic niches [22], whereas biofilms provide structured microenvironments [53, 56, 79, 80]. Such spatial organization of microbes generates locally heterogeneous subpopulations, which can acquire different resources, and further enhance local interactions. Hence, considerations of spatial disposition should improve system robustness and productivity (Fig. 4B). Biofilm-based separation shed light on enhancement of lignocellulosic bioconversion. For a cellulose degrader, biofilms can not only strengthen physical contact between microbial cells and cellulose substrate, but also enhance concentration of cellulases at the biofilm-substrate interface to promote hydrolysis rates, e.g., 70% higher cellulase activity by Aspergillus niger biofilm, compared to suspended A. niger culture [53, 81]. For a product producer, biofilm also increased biochemical production, due to the increment of surface area for mass transfer and cell resistance against adverse environment [53, 56]. Higher ethanol concentration (5.16 g/L and 5.33 g/L) was produced from rice husk hydrolysate (RHH) via Scheffersomyces stipitis in a biofilm reactor (BR) and continues plastic composite support (PCS)-BR, respectively [82, 83]. For microbial consortia, biofilm further provides a layered microbial structure, in which multispecies synergize for simultaneous delignification and saccharification or co-fermentation of pentose and hexose (Fig. 4B) [52]. The biofilm of T. reesei, which was co-cultured with S. stipitis (cellulose degrader) and S. cerevisiae (ethanol producer), can contribute to ethanol production (9.8 g/L) from the undetoxified dilute acid pretreated wheat straw [84]. Moreover, biofilm mediated spatial separation provides a strategy to resolve the physiological incompatibility of microorganisms. In the cellulose to lactate conversion system, aerobic T. reesei, which secretes cellulase to hydrolyze cellulose, formed biofilm on the surface of tubular membrane. As a result, biofilm consumed oxygen and generated anaerobic microenvironment for the growth of anaerobic Lactobacillus pentosus for lactate production (34.7 g/L) [53].
Additional positive effect of spatial separation was achieved via modified bioreactors (Fig. 4B). Fu et al. used beads in a modified fermenter to achieve spatial separation between the bacterium Zymomonas mobilis and the yeast Pichia stipitis for efficient lignocellulosic ethanol production (Fig. 4B) [85]. Z. mobilis can ferment glucose, while P. stipitis can ferment xylose. However, viable Z. mobilis inhibited xylose fermentation by P. stipitis, due to the oxygen deprivation by Z. mobilis. Thus, Fu et al. developed a novel co-culture process, where immobilized Z. mobilis beads were used, instead of free Z. mobilis in the co-culture, to decrease the interaction between these two strains (Fig. 4B). As a result, the efficiency of the xylose fermentation was significantly enhanced. Moreover, ethanol yield achieved 0.477 g/g, which is more than 96% of the theoretical value. In addition, in a lignocellulosic-to-biodiesel production system, C. thermocellum anaerobically hydrolyzes lignocellulose into hexose and pentose [86]. Next, Z. mobilis and P. stipitis anaerobically ferment hexose and pentose to ethanol, whereas downstream A. baylyi aerobically converts ethanol to biodiesel. The four strains were cultured in different bioreactors to maintain the most suitable growth conditions (e.g., temperature and oxygen) for each strain and avoid cross-inhibition, while hollow fiber bridges connected the bioreactors to exchange intermediate products (Fig. 4B). It is worth mentioning that microfluidic and microwell platform also can efficiently control spatial structure and metabolite communication at micron scale [22, 87]. Such high spatial resolution techniques should offer an attractive alternative for microbial spatial assortment to improve lignocellulosic bioconversion processes, especially those that are upstream.
In addition to spatial segregation, temporal separation also could be used to resolve physiological or metabolic incompatibilities. As mentioned previously, Z. mobilis and P. stipitis have cross-inhibition. Fu et al. also developed the sequential culture strategy to boost glucose/xylose co-fermentation. Free Z. mobilis was first inoculated to consume glucose and subsequently free P. stipitis was inoculated to ferment xylose when Z. mobilis was inactivated [85]. Moreover, sequential activities (e.g., gene expression) in distinct growth phases can achieve temporal separation, although synthetic consortia have yet to be specifically designed with this concept [22]. The gene circuits, which have feedback loop architecture, could have temporally coordinated oscillations across the population, through intercellular communication mediated by chemical molecules. Well-known quorum sensing and quenching, which produce chemicals to sequential turn on/off gene expression, is an ideal circuit to design temporal separation of lignocellulosic conversion microbial communities in the future [88, 89].
Sub-population ratio control for community robustness and stability
Stability of microbial consortia is essential, yet challenging. For microbial consortia in nature, the environment causes community change, where consortia may experience environmental fluctuations and exposure to competitive species. Moreover, succession of microbial consortia changes the community due to genome evolution and horizontal gene transfer [90]. For synthetic microbial communities, sub-population ratio, especially ratio of cooperator-cheater, is the important factor that interferes with the community stability. Cooperators produce enzymes, which catalyze substrates, and have a lower growth rate than that of cheaters, as they consume more energy to synthesize enzymes. In case that enzymatic products are equally shared by cooperators and cheaters. Cheaters would dominate the system and cause collapse of the microbial community. In contrast, “Snowdrift game” suggests that cheaters and cooperators can stably coexist when cooperators can acquire enough net benefits in systems [91]. Two strategies have been utilized to protect the interests of cooperator in lignocellulose bioconversion microbial consortia. (i) Spatial self-organization contributes to cooperation (Fig. 4C) [92]. Positive assortment can enable cooperation without being taken over by cheaters. Take cellulosomes for instance, previous studies demonstrated that the cell-bound cellulosome had 2.7–4.7-folds higher hydrolysis rate than their free counterparts, containing the same cellulases [93]. This should be the result of substantially higher substrate retention at cell surface and enhancement of the local hydrolysate concentration. Thus, in the co-culture system of C. thermocellum (a cooperator) and Thermoanaerobacter (a cheater), the cooperation of cellulose-to-ethanol conversion was stable, since higher rewards (hydrolysates) are received by C. thermocellum and favor its growth. Such spatial self-organization of cellulases in the cooperator contributes to co-exist of the two strains. (ii) Regulation of relative cooperator/cheater benefits is another strategy to resist cheaters and maintain cooperation (Fig. 4C). Minty et al. constructed a dynamic model to perform a stability analysis on a simplified version of the TrEc consortium model for cellulosic isobutanol production. Their analysis suggested that ecological parameters, including growth kinetics, substrate uptake kinetics and carbon flow partition, determined the tradeoff between cellulose hydrolysis rate and product yields. Thus, manipulation of these parameters can control the relative cooperation/cheating benefits to maintain the stability of the TrEc consortium (T. reesei/E. coli) [48].
Moreover, precise population control is also a desirable method to maintain the stability of communities. Genetic parts, e.g., QS systems and bacteriocins, are useful elements to manipulate the growth rate or subpopulation fitness [45, 94, 95]. These elements should provide genetic tools to further construct synthetic microbial community for lignocellulose bioconversion.
Automated and computational design of synthetic microbial consortia
With the increasing number of potential types of engineered interactions and community members, manual judgement of these considerations is cumbersome and inefficient, representing a major hurdle in synthetic microbial community. Automated design of synthetic microbial communities has been recently developed to counter this hurdle [95]. This methodology enables researchers to automatically build up computational circuit design, including engineer metabolic interdependencies mutualism and employ QS/bacteriocins to control community stability. Hence, model selection can be computationally performed to identify the most promising designs [95] (Fig. 4D). This framework of design-build-test cycle can generate impactful rules and heuristics for building a robust and stable synthetic community with a desired behavior, laying foundation to further design lignocellulose bioconversion consortia.
Conclusion and perspectives
Bottom-up synthetic ecology study of microbial communities, which precisely consider the properties of individual functions, interactions, communities, and applications, yield a better understanding for diversity, interactions, and dynamics at the systems level. Hence, it presents an exciting opportunity to rationally engineer microbial consortia for lignocellulose bioconversion. However, we still lack the critical knowledge and technology to effectively characterize the contributions of individual members, reveal the highly inter-connected networks of metabolic and ecological interactions, as well as perform genetic manipulation at the systems-level. There are two major barriers, which need to be addressed in the next step. First, it’s difficult to gain a fine-scale understanding of functions of individual microbial species and a subsequent large-scale understanding of ecosystem function. So far, this has mainly relied on genome-scale and computational modeling approaches. The qualities of these in silico models greatly depend on the accuracy and completeness of information about metabolic networks and biochemical roles. For most microbial species in natural environments, such information is missing and requires significant time and effort to accumulate. Thus, it is restricted to constructed synthetic communities which are comprised a few (e.g., 2–3 strains) well-characterized species. Development of automated approaches for metabolic reconstructions of unmodelled strains would alleviate this bottleneck. Alternatively, it would be desirable for the extension of computational approaches to design communities which contain novel species without genome-scale metabolic reconstructions. Second, it’s challenging to genetically manipulate microbial communities with a wide range of specificities and magnitudes. So far, genetic engineering is focused at the level individual strains. In contrast to breakthroughs in high-throughput sequencing to obtain molecular information of microbiomes, microbiome genetic modulation has not seen widespread success. With the recent revolution in genome engineering toolboxes [96], genetic modification of a community’s metagenomic content provides an avenue to achieve the desired manipulations [97]. Moreover, directly manipulating in situ bacterial communities in an open and changing environment could offer an exciting route for lignocellulose bioconversion.
Availability of data and materials
Not applicable.
Abbreviations
- PHA:
-
Polyhydroxyalkanoate
- GHs:
-
Glycoside hydrolases
- DypBs:
-
B type dye-decolorizing peroxidases
- LigMet:
-
Lignin-degrading consortium
- MELMC:
-
Minimal and effective lignocellulolytic microbial consortium
- MAMC:
-
Minimal active microbial consortia
- QS:
-
Quorum-Sensing
- AI:
-
Autoinducers
- MFC:
-
Microbial fuel cells
- ODE:
-
Ordinary differential equation
- GSMM:
-
Genome-scale metabolic models
- BiGG:
-
Biochemical, genetic, and genomic knowledge
- FBA:
-
Flux balance analyses
- DOLMNs:
-
Division of labor in metabolic networks
References
Becker J, Wittmann C. A field of dreams: lignin valorization into chemicals, materials, fuels, and health-care products. Biotechnol Adv. 2019;37(6):107360.
Van Aelst K, Van Sinay E, Vangeel T, Zhang Y, Renders T, Van den Bosch S, Van Aelst J, Sels BF. Low molecular weight and highly functional RCF lignin products as a full bisphenol a replacer in bio-based epoxy resins. Chem Commun (Camb). 2021;57(46):5642–5.
Linger JG, Vardon DR, Guarnieri MT, Karp EM, Hunsinger GB, Franden MA, Johnson CW, Chupka G, Strathmann TJ, Pienkos PT, et al. Lignin valorization through integrated biological funneling and chemical catalysis. Proc Natl Acad Sci USA. 2014;111(33):12013–8.
Liu Z-H, Le RK, Kosa M, Yang B, Yuan J, Ragauskas AJ. Identifying and creating pathways to improve biological lignin valorization. Renew Sust Energ Rev. 2019;105:349–62.
Bugg TD, Ahmad M, Hardiman EM, Singh R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol. 2011;22(3):394–400.
You C, Zhang XZ, Sathitsuksanoh N, Lynd LR, Zhang YH. Enhanced microbial utilization of recalcitrant cellulose by an ex vivo cellulosome-microbe complex. Appl Environ Microbiol. 2012;78(5):1437–44.
Lubbers RJM, Dilokpimol A, Visser J, Mäkelä MR, Hildén KS, de Vries RP. A comparison between the homocyclic aromatic metabolic pathways from plant-derived compounds by bacteria and fungi. Biotechnol Adv. 2019;37(7):107396.
Bugg TD, Ahmad M, Hardiman EM, Rahmanpour R. Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep. 2011;28(12):1883–96.
de Gonzalo G, Colpa DI, Habib MH, Fraaije MW. Bacterial enzymes involved in lignin degradation. J Biotechnol. 2016;236:110–9.
Rashid GM, Taylor CR, Liu Y, Zhang X, Rea D, Fulop V, Bugg TD. Identification of manganese superoxide dismutase from Sphingobacterium sp. T2 as a novel bacterial enzyme for lignin oxidation. ACS Chem Biol. 2015;10(10):2286–94.
Lin L, Wang X, Cao L, Xu M. Lignin catabolic pathways reveal unique characteristics of dye-decolorizing peroxidases in Pseudomonas putida. Environ Microbiol. 2019;21(5):1847–63.
Cao L, Lin L, Sui H, Wang H, Zhang Z, Jiao N, Zhou J. Efficient extracellular laccase secretion via bio-designed secretory apparatuses to enhance bacterial utilization of recalcitrant lignin. Green Chem. 2021;23(5):2079–94.
Kohlstedt M, Starck S, Barton N, Stolzenberger J, Selzer M, Mehlmann K, Schneider R, Pleissner D, Rinkel J, Dickschat JS, et al. From lignin to nylon: cascaded chemical and biochemical conversion using metabolically engineered Pseudomonas putida. Metab Eng. 2018;47:279–93.
Shen L, Kohlhaas M, Enoki J, Meier R, Schönenberger B, Wohlgemuth R, Kourist R, Niemeyer F, van Niekerk D, Bräsen C, et al. A combined experimental and modelling approach for the Weimberg pathway optimisation. Nat Commun. 2020;11(1):1098.
Lin L, Xu J. Dissecting and engineering metabolic and regulatory networks of thermophilic bacteria for biofuel production. Biotechnol Adv. 2013;31(6):827–37.
Shaw AJ, Podkaminer KK, Desai SG, Bardsley JS, Rogers SR, Thorne PG, Hogsett DA, Lynd LR. Metabolic engineering of a thermophilic bacterium to produce ethanol at high yield. Proc Natl Acad Sci U S A. 2008;105(37):13769–74.
Cai C, Xu Z, Zhou H, Chen S, Jin M. Valorization of lignin components into gallate by integrated biological hydroxylation, O-demethylation, and aryl side-chain oxidation. Sci Adv. 2021;7(36):eabg4585.
Lin L, Cheng Y, Pu Y, Sun S, Li X, Jin M, Pierson EA, Gross DC, Dale BE, Dai SY, et al. Systems biology-guided biodesign of consolidated lignin conversion. Green Chem. 2016;18(20):5536–47.
Wang X, Lin L, Dong J, Ling J, Wang W, Wang H, Zhang Z, Yu X. Simultaneous improvements of Pseudomonas cell growth and polyhydroxyalkanoate production from a lignin derivative for lignin-consolidated bioprocessing. Appl Environ Microbiol. 2018;84(18):01469–1418.
Zhou Y, Lin L, Wang H, Zhang Z, Zhou J, Jiao N. Development of a CRISPR/Cas9n-based tool for metabolic engineering of Pseudomonas putida for ferulic acid-to-polyhydroxyalkanoate bioconversion. Commun Biol. 2020;3(1):020–0824.
Wilhelm RC, Singh R, Eltis LD, Mohn WW. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 2019;13(2):413–29.
Lindemann SR, Bernstein HC, Song HS, Fredrickson JK, Fields MW, Shou W, Johnson DR, Beliaev AS. Engineering microbial consortia for controllable outputs. ISME J. 2016;10(9):2077–84.
Abisado RG, Benomar S, Klaus JR, Dandekar AA, Chandler JR. Bacterial quorum sensing and microbial community interactions. mBio. 2018;9(3):e02331-02317.
Ali SS, Abomohra AE, Sun J. Effective bio-pretreatment of sawdust waste with a novel microbial consortium for enhanced biomethanation. Bioresour Technol. 2017;238:425–32.
Gilmore SP, Lankiewicz TS, Wilken SE, Brown JL, Sexton JA, Henske JK, Theodorou MK, Valentine DL, O’Malley MA. Top-down enrichment guides in formation of synthetic microbial consortia for biomass degradation. ACS Synth Biol. 2019;8(9):2174–85.
Jiménez DJ, Dini-Andreote F, DeAngelis KM, Singer SW, Salles JF, van Elsas JD. Ecological insights into the dynamics of plant biomass-degrading microbial consortia. Trends Microbiol. 2017;25(10):788–96.
Díaz-García L, Huang S, Spröer C, Sierra-Ramírez R, Bunk B, Overmann J, Jiménez DJ. Dilution-to-Stimulation/Extinction method: a combination enrichment strategy to develop a minimal and versatile lignocellulolytic bacterial consortium. Appl Environ Microbiol. 2021. https://doi.org/10.1128/AEM.02427-20.
Fang X, Li Q, Lin Y, Lin X, Dai Y, Guo Z, Pan D. Screening of a microbial consortium for selective degradation of lignin from tree trimmings. Bioresour Technol. 2018;254:247–55.
Cortes-Tolalpa L, Norder J, van Elsas JD, Falcao Salles J. Halotolerant microbial consortia able to degrade highly recalcitrant plant biomass substrate. Appl Microbiol Biotechnol. 2018;102(6):2913–27.
Wang X, Lin L, Zhou J. Links among extracellular enzymes, lignin degradation and cell growth establish the models to identify marine lignin-utilizing bacteria. Environ Microbiol. 2021;23(1):160–73.
Wang C, Dong D, Wang H, Müller K, Qin Y, Wang H, Wu W. Metagenomic analysis of microbial consortia enriched from compost: new insights into the role of Actinobacteria in lignocellulose decomposition. Biotechnol Biofuels. 2016;9:22.
Díaz-García L, Bugg TDH, Jiménez DJ. Exploring the lignin catabolism potential of soil-derived lignocellulolytic microbial consortia by a gene-centric metagenomic approach. Microb Ecol. 2020;80(4):885–96.
Cortes-Tolalpa L, Jiménez DJ, de Lima Brossi MJ, Salles JF, van Elsas JD. Different inocula produce distinctive microbial consortia with similar lignocellulose degradation capacity. Appl Microbiol Biotechnol. 2016;100(17):7713–25.
Woo HL, Hazen TC. Enrichment of bacteria from eastern mediterranean sea involved in lignin degradation via the phenylacetyl-coa pathway. Front Microbiol. 2018;9:922.
Xie S, Sun S, Lin F, Li M, Pu Y, Cheng Y, Xu B, Liu Z, da Costa SL, Dale BE, et al. Mechanism-guided design of highly efficient protein secretion and lipid conversion for biomanufacturing and biorefining. Adv Sci. 2019;6(13):1801980–1801980.
Moraes EC, Alvarez TM, Persinoti GF, Tomazetto G, Brenelli LB, Paixao DAA, Ematsu GC, Aricetti JA, Caldana C, Dixon N, et al. Lignolytic-consortium omics analyses reveal novel genomes and pathways involved in lignin modification and valorization. Biotechnol Biofuels. 2018;11(75):018–1073.
Puentes-Téllez PE, Falcao Salles J. Construction of effective minimal active microbial consortia for lignocellulose degradation. Microb Ecol. 2018;76(2):419–29.
Jiménez DJ. Chaib De Mares M, Salles JF: Temporal expression dynamics of plant biomass-degrading enzymes by a synthetic bacterial consortium growing on sugarcane bagasse. Front Microbiol. 2018;9:299.
He Q, Hemme CL, Jiang H, He Z, Zhou J. Mechanisms of enhanced cellulosic bioethanol fermentation by co-cultivation of Clostridium and Thermoanaerobacter spp. Bioresour Technol. 2011;102(20):9586–92.
Hu J, Xue Y, Guo H, Gao MT, Li J, Zhang S, Tsang YF. Design and composition of synthetic fungal-bacterial microbial consortia that improve lignocellulolytic enzyme activity. Bioresour Technol. 2017;227:247–55.
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–50.
Wang M, Liu X, Nie Y, Wu XL. Selfishness driving reductive evolution shapes interdependent patterns in spatially structured microbial communities. ISME J. 2020;15(5):1387–401.
West SA, Griffin AS, Gardner A, Diggle SP. Social evolution theory for microorganisms. Nat Rev Microbiol. 2006;4(8):597–607.
Andersen SB, Marvig RL, Molin S, Krogh Johansen H, Griffin AS. Long-term social dynamics drive loss of function in pathogenic bacteria. Proc Natl Acad Sci U S A. 2015;112(34):10756–61.
Song H, Ding MZ, Jia XQ, Ma Q, Yuan YJ. Synthetic microbial consortia: from systematic analysis to construction and applications. Chem Soc Rev. 2014;43(20):6954–81.
Tsoi R, Wu F, Zhang C, Bewick S, Karig D, You L. Metabolic division of labor in microbial systems. Proc Natl Acad Sci U S A. 2018;115(10):2526–31.
Lee JA, Baugh AC, Shevalier NJ, Strand B, Stolyar S, Marx CJ. Cross-feeding of a toxic metabolite in a synthetic lignocellulose-degrading microbial community. Microorganisms. 2021;9(2):321.
Minty JJ, Singer ME, Scholz SA, Bae CH, Ahn JH, Foster CE, Liao JC, Lin XN. Design and characterization of synthetic fungal-bacterial consortia for direct production of isobutanol from cellulosic biomass. Proc Natl Acad Sci U S A. 2013;110(36):14592–7.
Phelan VV, Liu W-T, Pogliano K, Dorrestein PC. Microbial metabolic exchange—the chemotype-to-phenotype link. Nat Chem Biol. 2012;8(1):26–35.
Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–46.
Bruger EL, Snyder DJ, Cooper VS, Waters CM. Quorum sensing provides a molecular mechanism for evolution to tune and maintain investment in cooperation. ISME J. 2021;15(4):1236–47.
Wang ZW, Chen S. Potential of biofilm-based biofuel production. Appl Microbiol Biotechnol. 2009;83(1):1–18.
Jiang Y, Liu Y, Zhang X, Gao H, Mou L, Wu M, Zhang W, Xin F, Jiang M. Biofilm application in the microbial biochemicals production process. Biotechnol Adv. 2021;48:107724.
Mangwani N, Dash HR, Chauhan A, Das S. Bacterial quorum sensing: functional features and potential applications in biotechnology. J Mol Microbiol Biotechnol. 2012;22(4):215–27.
Shahab RL, Luterbacher JS, Brethauer S, Studer MH. Consolidated bioprocessing of lignocellulosic biomass to lactic acid by a synthetic fungal-bacterial consortium. Biotechnol Bioeng. 2018;115(5):1207–15.
Germec M, Demirci A, Turhan I. Biofilm reactors for value-added products production: an in-depth review. Biocatal Agr Biotech. 2020;27:101662.
Stolyar S, Van Dien S, Hillesland KL, Pinel N, Lie TJ, Leigh JA, Stahl DA. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol. 2007;3:92.
Kracher D, Scheiblbrandner S, Felice AKG, Breslmayr E, Preims M, Ludwicka K, Haltrich D, Eijsink VGH, Ludwig R. Extracellular electron transfer systems fuel cellulose oxidative degradation. Science. 2016;352(6289):1098–101.
Pham LTM, Kim SJ, Kim YH. Improvement of catalytic performance of lignin peroxidase for the enhanced degradation of lignocellulose biomass based on the imbedded electron-relay in long-range electron transfer route. Biotechnol Biofuels. 2016;9(1):247.
Zhao J, Li F, Cao Y, Zhang X, Chen T, Song H, Wang Z. Microbial extracellular electron transfer and strategies for engineering electroactive microorganisms. Biotechnol Adv. 2020;53:107682.
Kumar R, Singh L, Zularisam AW. Exoelectrogens: recent advances in molecular drivers involved in extracellular electron transfer and strategies used to improve it for microbial fuel cell applications. Renew Sust Energ Rev. 2016;56:1322–36.
Bhuvaneswari A, Krishnaraj RN, Berchmans S. Metamorphosis of pathogen to electrigen at the electrode/electrolyte interface: direct electron transfer of Staphylococcus aureus leading to superior electrocatalytic activity. Electrochem Commun. 2013;34:25–8.
Pande S, Shitut S, Freund L, Westermann M, Bertels F, Colesie C, Bischofs IB, Kost C. Metabolic cross-feeding via intercellular nanotubes among bacteria. Nat Commun. 2015;6:6238.
Cortes-Tolalpa L, Salles JF, van Elsas JD. Bacterial synergism in lignocellulose biomass degradation—complementary roles of degraders as influenced by complexity of the carbon source. Front Microbiol. 2017;8:1628.
Thommes M, Wang T, Zhao Q, Paschalidis IC, Segrè D. Designing metabolic division of labor in microbial communities. mSystems. 2019;4(2):e00263-00218.
Pacheco AR, Moel M, Segrè D. Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems. Nat Commun. 2019;10(1):103.
Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z, Takeuchi R, Nomura W, Zhang Z, Mori H, et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat Biotechnol. 2017;35(10):904–8.
Thiele I, Palsson B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5(1):93–121.
Nogales J, Mueller J, Gudmundsson S, Canalejo FJ, Duque E, Monk J, Feist AM, Ramos JL, Niu W, Palsson BO. High-quality genome-scale metabolic modelling of Pseudomonas putida highlights its broad metabolic capabilities. Environ Microbiol. 2020;22(1):255–69.
Feist AM, Palsson B. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol. 2008;26(6):659–67.
Price ND, Papin JA, Schilling CH, Palsson BO. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21(4):162–9.
Garcia S, Thompson RA, Giannone RJ, Dash S, Maranas CD, Trinh CT. Development of a genome-scale metabolic model of Clostridium thermocellum and its applications for integration of multi-omics datasets and computational strain design. Front Bioeng Biotechnol. 2020;8:772.
Schilling CH, Edwards JS, Letscher D, Palsson B. Combining pathway analysis with flux balance analysis for the comprehensive study of metabolic systems. Biotechnol Bioeng. 2000;71(4):286–306.
Henson MA, Hanly TJ. Dynamic flux balance analysis for synthetic microbial communities. IET Syst Biol. 2014;8(5):214–29.
Klitgord N, Segrè D. Ecosystems biology of microbial metabolism. Curr Opin Biotechnol. 2011;22(4):541–6.
Hanly TJ, Urello M, Henson MA. Dynamic flux balance modeling of S. cerevisiae and E. coli co-cultures for efficient consumption of glucose/xylose mixtures. Appl Microbiol Biot. 2012;93(6):2529–41.
Enyeart PJ, Simpson ZB, Ellington AD. A microbial model of economic trading and comparative advantage. J Theor Biol. 2015;364:326–43.
Henriques D, Minebois R, Mendoza SN, Macías LG, Pérez-Torrado R, Barrio E, Teusink B, Querol A, Balsa-Canto E. A multiphase multiobjective dynamic genome-scale model shows different redox balancing among yeast species of the Saccharomyces genus in fermentation. mSystems. 2021;6(4):e0026021.
Lee KW, Periasamy S, Mukherjee M, Xie C, Kjelleberg S, Rice SA. Biofilm development and enhanced stress resistance of a model, mixed-species community biofilm. ISME J. 2014;8(4):894–907.
Mukhi M, Vishwanathan AS. Beneficial biofilms: a mini-review of strategies to enhance biofilm formation for biotechnological applications. Appl Environ Microbiol. 2021. https://doi.org/10.1128/AEM.01994-21.
Villena GK, Gutiérrez-Correa M. Production of cellulase by Aspergillus niger biofilms developed on polyester cloth. Lett Appl Microbiol. 2006;43(3):262–8.
Bader NB, Germec M, Turhan I. Ethanol production from different medium compositions of rice husk hydrolysate by using Scheffersomyces stipitis in a repeated-batch biofilm reactor and its modeling. Process Biochem. 2021;100:26–38.
Bader NB, Germec M, Turhan I. Scheffersomyces stipitis biofilm reactor for ethanol production from acid-pretreated/detoxified and glucose- or xylose-enriched rice husk hydrolysate under a continuous process. Biomass Convers Bior. 2021;11(6):2909–21.
Brethauer S, Studer MH. Consolidated bioprocessing of lignocellulose by a microbial consortium. Energ Environ Sci. 2014;7(4):1446–53.
Fu N, Peiris P, Markham J, Bavor J. A novel co-culture process with Zymomonas mobilis and Pichia stipitis for efficient ethanol production on glucose/xylose mixtures. Enzyme Microb Tech. 2009;45(3):210–7.
Ben Said S, Or D. Synthetic microbial ecology: engineering habitats for modular consortia. Front Microbiol. 2017;8:1125.
Kim HJ, Boedicker JQ, Choi JW, Ismagilov RF. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci U S A. 2008;105(47):18188–93.
Garcia-Ojalvo J, Elowitz MB, Strogatz SH. Modeling a synthetic multicellular clock: repressilators coupled by quorum sensing. Proc Natl Acad Sci U S A. 2004;101(30):10955–60.
Whiteley M, Diggle SP, Greenberg EP. Progress in and promise of bacterial quorum sensing research. Nature. 2017;551(7680):313–20.
Niehus R, Mitri S, Fletcher AG, Foster KR. Migration and horizontal gene transfer divide microbial genomes into multiple niches. Nat Commun. 2015;6:8924.
Gore J, Youk H, van Oudenaarden A. Snowdrift game dynamics and facultative cheating in yeast. Nature. 2009;459(7244):253–6.
Momeni B, Waite AJ, Shou W. Spatial self-organization favors heterotypic cooperation over cheating. eLife. 2013;2:e00960.
Lu Y, Zhang YH, Lynd LR. Enzyme-microbe synergy during cellulose hydrolysis by Clostridium thermocellum. Proc Natl Acad Sci U S A. 2006;103(44):16165–9.
Brenner K, Karig DK, Weiss R, Arnold FH. Engineered bidirectional communication mediates a consensus in a microbial biofilm consortium. Proc Natl Acad Sci U S A. 2007;104(44):17300–4.
Karkaria BD, Fedorec AJH, Barnes CP. Automated design of synthetic microbial communities. Nat Commun. 2021;12(1):672.
McCarty NS, Ledesma-Amaro R. Synthetic biology tools to engineer microbial communities for biotechnology. Trends Biotechnol. 2019;37(2):181–97.
Sheth RU, Cabral V, Chen SP, Wang HH. Manipulating bacterial communities by in situ microbiome engineering. Trends Genet. 2016;32(4):189–200.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (91951116), National Key Research and Development Project (2019YFA0606704) and Fundamental Research Funds of Shandong University (2019GN101).
Author information
Authors and Affiliations
Contributions
LL conceptualized, wrote and edited the manuscript. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The author read and approved the final manuscript.
Competing interests
The author declares that she has no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, L. Bottom-up synthetic ecology study of microbial consortia to enhance lignocellulose bioconversion. Biotechnol Biofuels 15, 14 (2022). https://doi.org/10.1186/s13068-022-02113-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-022-02113-1