
Abstract
Background
Humicola insolens is a filamentous fungus with high potential of producing neutral and heat- and alkali-resistant cellulase. However, the genetic engineering tools, particularly the genome-editing tool, are scarce, hindering the study of cellulase expression regulation in this organism.
Results
Herein, a CRISPR/Cas9 genome-editing system was established in H. insolens based on a hybrid 5S rRNA–tRNAGly promoter. This system is superior to the HDV (hepatitis delta virus) system in genome editing, allowing highly efficient single gene destruction in H. insolens with rates of deletion up to 84.1% (37/44). With this system, a putative pigment synthesis gene pks and the transcription factor xyr1 gene were disrupted with high efficiency. Moreover, the extracellular protein concentration and cellulase activity largely decreased when xyr1 was deleted, demonstrating for the first time that Xyr1 plays an important role in cellulase expression regulation.
Conclusions
The established CRISPR/Cas9 system is a powerful genetic operation tool for H. insolens, which will accelerate studies on the regulation mechanism of cellulase expression and engineering of H. insolens for higher cellulase production.
Similar content being viewed by others

Background
Lignocellulose is one of the most abundant renewable biomass resources on earth, which contains cellulose, hemicellulose, and lignin as its major components. Cellulase and hemicellulase degrade the two plant cell wall polysaccharides into simple sugars including mono sugars or oligosaccharides, which can be used by natural occurring or engineered brewer’s yeasts to produce ethanol and advanced biofuels. This leads to an eco-friendly solution to the current energy and environmental problems [1]. The thermophilic filamentous fungus Humicola insolens is thus regarded to be of high potential, because it has noticeable merits such as high growth temperature, fast growth rate, and excellent cellulase- and hemicellulase-producing ability [2]. Its cellulase system was similar to that of Trichoderma reesei. However, the straw degradation efficiency of the H. insolens cellulase was higher than that of T. reesei. In addition, the H. insolens cellulase has stable activity at high temperature [2, 3]. The high temperature-resistant β-glucosidase and xylanase expressed by H. insolens have been used in the wine industry for quality improvement, while its neutral cellulase has been used in the textile and washing industry [4]. By having excellent heat- and alkali-resistance, high cellulose degradation ability, and an optimal pH close to neutral, the cellulase expressed by H. insolens is a good complement to that from T. reesei [4,5,6,7]. However, the production level of H. insolens is low and cannot meet the need of biofuel industries. Previously, a T-DNA random insertional mutant library created by Agrobacterium tumefaciens-mediated transformation was established [8]. It was also discovered that mutation of the transcriptional regulator CreA did not greatly improve the ability of H. insolens to produce cellulase [9]. One main reason for the inefficiency of strain engineering is that there is no well-established genome-editing system, limiting the study of the regulating mechanisms of cellulase expression in H. insolens. There is an urgent need for a new technology to solve this problem.
With the advantages of high efficiency, versatility and ease of operation, the CRISPR/Cas9 technology is now widely used in functional genomics studies of filamentous fungi [10,11,12]. The essence of this technology is that, a small guide RNA (sgRNA) is designed to target and direct the Cas9 nuclease to bind and cleave a specific site in the chromosome [13]. sgRNA recognizes and complexes with the DNA in the targeting site. The complex is inserted into the gap between the nuclease recognition and cutting sites of Cas9, which will activate the cleavage activity of Cas9 and lead to cutting of the target site and forming a double-stranded break (DSB) [14, 15]. DSB is repaired by either the non-homologous end joining (NHEJ) or homology-directed repair (HDR) mechanisms [16].
Herein, the CRISPR/Cas9-based genome-editing technique was established for the first time in H. insolens. The technique contains a codon-optimized Cas9 nuclease-expressing cassette directed by a tef1 promoter, a sgRNA-expressing cassette with a prevailing tRNAGly element directed by a 5S rRNA promoter, and a donor DNA fragment with 600-bp homologous arms. Using this system, we greatly improved the genome editing efficiency in H. insolens and successfully disrupted the pks pigment synthesis gene and xyr1 transcription factor gene, with a maximum efficiency of 84.1% (37/44) and 78.3% (18/23), respectively. This method displayed great potential for genome editing in H. insolens and laid a foundation for functional genomics as well as construction of engineered strains with improved ability to produce cellulase.
Results and discussion
Construction of a CRISPR/Cas9 system for genome editing in H. insolens
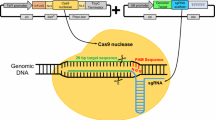
The CRISPR/Cas9 system used in H. insolens included a Cas9-expressing cassette, a sgRNA expression cassette, and a donor DNA fragment (Fig. 1). Abundant Cas9 protein and sgRNA are well-known to be critical to successful genome editing. Since expression of Cas9 depends heavily on the promoter, the strong and constitutive promoter Ptef1 has been successfully applied in Aspergilli (the Aspergillus nidulans Ptef1) and M. thermophile (the Myceliophthora thermophila Ptef1) [11, 17]. Therefore, the Cas9-expressing cassette containing the tef1 promoter, cas9 gene from Streptococcus pyogenes with two nuclear localization signals (NLS), and the trpC terminator was synthesized according to the sequence of Cas9-expressing cassette used in M. thermophila [17]. It was used herein to provide stable expression of Cas9 and proved to have a good effect on gene editing (Fig. 1; Table 1).

Schematic representation of the CRISPR/Cas9 genome editing systems in H. insolens. ① Cas9 expression cassette. Ptef1: tef1 promoter from M. thermophila. ② sgRNA expression cassette. P5S rRNA (light gray): the A. niger 5S rRNA gene with its 338-bp upstream promoter. HDV (light green): the hepatitis delta virus ribozyme; tRNAGly (purple): the transfer RNA for glycine from H. insolens. The dotted box frames up the structure of tRNAGly–sgRNA; the red arrows indicate excision sites by RNase P and RNase Z. ③ Donor DNA. armL (bright brown): the upstream homologous arm; armR (bright brown): downstream homologous arm; NGG (blue): the PAM sequence; DSB: double-strand break; Hyg (light brown): hygromycin B resistance gene expression cassette
For sgRNA expression in other fungi such as T. reesei, Neurospora crassa, Aspergillus fumigatus, Penicillium chrysogenum, and Pyricularia oryzae, typically an RNA polymerase III type U6 promoter was used to drive its transcription [12, 18,19,20,21]. However, it was sometimes difficult to identify a U6 promoter in many species and the use of a heterologous U6 promoter may otherwise reduce the efficiency of gene editing [22, 23]. In addition to the U6 promoter, there are also other types of promoters successfully used to initiate sgRNA transcription. In Aspergillus niger and Fusarium fujikuroi, a highly conserved 5S rRNA promoter was found to drive sgRNA transcription with higher efficiency than that using the U6 promoter [24, 25]. Furthermore, high gene editing efficiency can be obtained by using 5S rRNA along with its upstream 338-bp sequence as the promoter in A. niger [25]. Four tRNA promoters were more efficient than the U6 promoter in Ustilaginoidea virens [26]. A small RNA, i.e., tRNAGly, was regarded to be able to enhance transcription of the sgRNA as a potential enhancer of Pol III and also ensure precise release of sgRNA spacer-scaffold structure from the sgRNA expression cassette [27, 28]. In addition to change of the promoter, in recent years, ribozymes (an RNA-based nuclease) were included in the sgRNA expression cassette, expanding the use of other types of promoters. In Aspergilli, an HDV ribozyme was fused in the sgRNA expression cassette to liberate more sgRNA [11, 25]. Taken together, both the HDV ribozyme and the tRNAGly element were used in sgRNA expression cassette to improve the genome editing efficiency in different filamentous fungi. Since the U6 promoter of H. insolens was not identified at present, two strategies employing either the HDV ribozyme or tRNAGly element were used to improve sgRNA expression directed by the 5S rRNA (-338) promoter in this study (Fig. 1).
For the donor fragments, the length of homologous arms has a high impact on the efficiency of genome editing. It was reported that, when the homologous arms for lea1 were 600-bp (or more) in T. reesei, the genome editing efficiency reached 100% [10]. Therefore, we designed homologous arms with 600-bp and fused them with the marker gene-expressing cassette Pgpd-hyg-Tgpd (Fig. 1).
Selection of target genes for genome editing
The putative pks and xyr1 genes were predicted from the genome of H. insolens and selected as targets of genome editing in H. insolens using the CRISPR/Cas9 system. The 6694-bp pks gene is predicted to encode a putative polyketide synthase (Pks) containing 2167 amino acids, which is necessary for the black pigment melanin biosynthesis [29]. The Pks from H. insolens is homologous to Pestalotiopsis fici PfmaE [30], A. nidulans AnWA [31], A. niger AnAlbA [32], and A. fumigatus AfAlb1 [33] with amino acid sequence identity of 61.9%, 37.6%, 39.4%, and 34.8%, respectively (Additional file 1: Figure S1). The 3403-bp xyr1 of H. insolens encodes a transcription factor belonging to the MHR superfamily [34]. The predicted Xyr1 protein contains 969 amino acids typified by a Zn2Cys6 zinc finger motif [35]. The H. insolens Xyr1 has 78.7%, 74.2%, 55.6% and 48.4% amino acid sequence identity, respectively, to M. thermophila MtXyr1 [36], N. crassa NcXyr1 [37], T. reesei TrXyr1 [38], and A. nidulans AnXyr1 [39] (Additional file 1: Figure S2).
5StRNAGly–sgRNA is superior to 5SHDV–sgRNA in genome editing
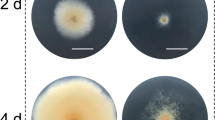
Similar to A. niger, the pks-destroyed mutants of H. insolens produced albino conidia (Fig. 2A). When the wild-type Y1 strain was used, the 5StRNAGly–sgRNA (where 5StRNAGly is an abbreviation for the 5SrRNA–tRNAGly construct) expression disrupted 57.8% (186/322, pks1) of the pks gene in transformants without addition of a donor DNA, while the 5SHDV–sgRNA (where 5SHDV is an abbreviation for the 5SrRNA-HDV construct) system had a disruption efficiency of only 23.5% (79/336, pks1) (Table 1). This trend was stable for other sgRNAs, pks2 and pks3, two other target sites of pks gene (Table 1). The 5StRNAGly–sgRNA system had an efficiency of 59.1% (194/328) for pks2 and 56.4% (208/369) for pks3, while the efficiency of 5SHDV–sgRNA system were only 22.4% (71/317, pks2) and 21.4% (60/281, pks3). Six pks gene mutations were verified by sequencing. These mutants displayed insertion mutations upstream of the PAM site with a single-nucleotide A (Fig. 2B). The efficiency of genome editing did not change much (56.2% (204/363) for 5StRNAGly–sgRNA and 26.6% (91/342) for 5SHDV–sgRNA) when the donor DNA was added.

Mutations into the pks gene in H. insolens as introduced by CRISPR/Cas9-mediated genome editing. A Disruption of pks was correlated to an albino conidia phenotype in H. insolens. The pks gene disrupted strains displayed pigment-less conidia and resultant white colonies as indicated by red arrows. B Sequence alignment of the pks gene in WT and the pks-edited mutants. The 20-nt protospacer sequence of pks in wild-type strain is represented by blue letters and PAM sequence is labeled by red letters with underline
In the wild-type Y1 strain, the numbers of transformation did not change significantly due to the addition of Cas9 expression cassette (617 transformants for donor DNA/Cas9 cotransformation versus 625 transformants for donor DNA only). Additionally, the controls without donor DNA using Cas9 expression cassette and 5SsgRNA–HDV (abbreviation for the 5SrRNA–sgRNA-HDV construct), 5SsgRNA (abbreviation for the 5SrRNA–sgRNA construct), tRNAGly–sgRNA systems all displayed low disruption efficiency for 7.0% (26/371), 4.1% (14/345) and 9.3% (35/375), respectively (Table 1). Apparently, the efficiency of genome editing using the 5StRNAGly–sgRNA system almost doubled that of 5SHDV–sgRNA (Table 1). This is worth noting since in A. niger, addition of the HDV ribozyme improves the genome editing efficiency [25]. Two reasons may account for the high efficiency of 5StRNAGly–sgRNA system in H. insolens. For one thing, tRNAGly can recruit the PolIII complex due to the existence of internal promoter elements box A and B and drive transcription of sgRNA by serving as a promoter [40]. For another, the structure of pre-tRNAGly can be recognized by RNase P and RNase Z and the site between tRNAGly and sgRNA spacer is efficiently cleaved [41]. These lead to releasing of active guide RNA molecules and explain the higher cleavage activity than observed for HDV.
In cells, the broken DNA double strands are repaired through either the nonhomologous end joining (NHEJ) pathway or homology-directed repair (HDR) pathway [42]. The NHEJ pathway is rapid and does not require a template [43]. In fungi, the Ku proteins play a key role in the NHEJ pathway for DNA repair. When the DNA strands are broken, the Ku70/Ku80 heterodimer recognizes and binds to the broken ends and recruits kinase, ligase, and other associated proteins for DNA repairing [16, 44]. In filamentous fungi, it has been reported that disrupting the gene encoding Ku70 can significantly improve the efficiency of homologous recombination [21, 45,46,47]. Therefore, we used the Y1/Δku70 strain to further test CRISPR/Cas9-mediated genome editing. In this strain, the gene-editing efficiency for pks using the 5StRNAGly–sgRNA system increased from 56.2% (204/363, in the WT strain) to 84.1% (37/44), tripling that of 5SHDV–sgRNA (25.6%, 10/39, Table 1). Disruption of xyr1 was all carried out by supplementing with the donor DNA (Fig. 3A) and this gene was disrupted at an efficiency of 78.1% (18/23) with 5StRNAGly–sgRNA system but at 26.1% (6/23) with the 5SHDV–sgRNA system (Table 1). This again demonstrated that the using tRNAGly was more efficient in genome editing than HDV. Disruption of xyr1 by homologous recombination was verified by diagnostic PCR: a 4824-bp specific fragment was amplified by PCR from the xyr1 disruption mutant, while a 1920-bp fragment was amplified from the wild-type Y1 and Y1/Δku70 strains (Fig. 3B).

Verification of xyr1 gene disruption in the Y1/Δku70 strain. A Schematic diagram of xyr1 disruption verification. hyg: hygromycin B resistance gene. B PCR verification of xyr1 disruption using the primer pair xyr1YS/xyr1YA. M: DNA molecular mass marker; lane 1: negative control; lane 2: H. insolens Y1; lane 3: Y1/Δku70; lanes 4–7: four representative xyr1 disrupted mutants
When the Y1/Δku70 strain was used as the host strain, the genome editing efficiency of the 5SHDV–sgRNA system was comparable for pks. In Y1/Δku70 the knockout efficiency was 25.6% (10/39), while in Y1 the efficiency was 26.6% (91/342) (Table 1). With this strain, the increase of genome editing efficiency of the 5StRNAGly–sgRNA system was obvious: 56.2% (204/363) in Y1 versus 84.1% (37/44) in Y1/Δku70. However, the efficiency still did not reach to nearly 100% as observed in Magnaporthe grisea and A. nidulans [48, 49]. Note that the genome editing efficiency of 11.0% (8/73) in Y1/Δku70 did not change significantly as compared with 6.3% (25/400) in Y1 when only the donor DNA for pks1 was used. There might be several reasons explaining the limited increase of genome editing efficiency in the ku70 disrupted strain. First, the expression of ku70 is so low that knockout of this gene does not have significant impact on the repair rate of NHEJ [50]. Second, the expression of HDR-related component genes is too low that the homology recombination efficiency cannot be improved even after the NHEJ pathway is repressed [51].
We also used the 5StRNAGly–sgRNA system in the Y1/Δku70 strain for multiplexing genome editing. In the trial to simultaneously destruct two genes, the successful rate of deleting pks and xyr1 dropped to 17.4% (Table 1). These results revealed that this system could be used for editing the cellulase expression-related genes in H. insolens.
Deletion of xyr1 decreased cellulase production in H. insolens
Xyr1 is the main transcription activator of cellulase expression in mainly lignocellulose-degrading filamentous fungi [52, 53]. However, the regulatory role of Xyr1 on fungal growth and cellulase expression is not consistent in all filamentous fungi due to its phosphorylation degree [54, 55]. In H. insolens, the role of xyr1 in cellulase expression regulation is not clear. Therefore, in this study, the xyr1 mutant of H. insolens obtained by CRISPR/Cas9-mediated gene disruption was further studied for growth phenotype and cellulase production.
On culturing at 42 °C in PDA, MMN, and YPD solid plates, the nascent hyphae were dense and clustered in the center of the colonies. At 24 h, the colony of Y1/Δku70Δxyr1 strain was significantly smaller than that of Y1 and Y1/Δku70 strains. This was also true for colonies grown at 72 h on these plates (Fig. 4A–C). When cultured in PDA, the sporulation of Y1/Δku70Δxyr1 was basically the same as those of wild-type and the Y1/Δku70 strains (Fig. 4A). Although the sporulation of Y1/Δku70Δxyr1 strain was slower, the length of its aerial hyphae appeared to increase (Fig. 4B) in the MMN medium. In YPD, Y1/Δku70Δxyr1 showed a layered state of mycelial extension in addition to aerial hyphae (Fig. 4C). Deletion of xyr1 and its homologous genes in other filamentous fungi such as T. reesei, M. thermophila and Aspergilli similarly altered the growth phenotypes depending on the carbon sources [55,56,57].

Characterization of the growth morphology of xyr1 disruption mutants. Colony growth and sporulation of the wild-type Y1, Y1/Δku70, and Y1/Δku70Δxyr1 strains cultured on PDA (A), MMN (B) and YPD (C) plates at 42 °C for 24 and 72 h, respectively
In flask fermentation, the wild-type Y1 and Y1/Δku70 strains produced almost the same amount of extracellular enzymes. However, both the cellulase and hemicellulase activities of the Y1/Δku70Δxyr1 were lower than those of the wild type (Fig. 5). The FPase activity of Y1/Δku70Δxyr1 reached the maximum on day 6 post-induction, 63.95% lower than that of WT (Fig. 5A). The enzymatic activities of endoglucanase, cellobiohydrolase, β-glucosidase, and xylanase of Y1/Δku70Δxyr1 were also significantly decreased to as low as 32.10%, 70.55%, 68.16%, and 59.45%, respectively, of the original strain (Fig. 5A–E). The biomass of Y1/Δku70Δxyr1 strain was slightly lower than that of Y1 throughout the culture. Y1/Δku70 was similar to Y1/Δku70Δxyr1 till day 4 post-induction, but then it grew to a level similar to that of the wild type (Fig. 5F). For Y1/Δku70Δxyr1, the highest FPase/biomass ratio was 0.34 U/mg, which was much lower than those of Y1 (0.80 U/mg) and Y1/Δku70 (0.74 U/mg) (Fig. 5G).

Assay of enzyme activities of H. insolens. The activities are FPase (A), CMCase (B), pNPCase (C), pNPGase (D), and xylanase (E). The biomass (F) and FPase/biomass (G) are also shown
An SDS-PAGE analysis indicated that, compared with Y1 and Y1/Δku70 strains, the major extracellular proteins with molecular masses lower than 70 kDa in Y1/Δku70Δxyr1 almost completely disappeared (Fig. 6). Taken together, the decrease of enzyme activity in H. insolens Y1/Δku70Δxyr1 was due to lowered ability to express the enzymes as well as the impaired biomass accumulation. These results were also consistent with the results of other filamentous fungi [56, 58], and for the first time, demonstrated that Xyr1 played a positive pleiotropic regulatory role in cellulase expression of H. insolens.

xyr1 deletion changed the extracellular protein profile in H. insolens, as analyzed by SDS-PAGE. M: protein molecular mass marker; lane 1: the H. insolens Y1 strain; lane 2: Y1/Δku70; lane 3: Y1/Δku70Δxyr1
The widespread Xyr1 and its homologs can activate transcription of cellulase and/or hemicellulase genes in many lignocellulose-degrading filamentous fungi [59]. It is well-known that, after xyr1 deletion, almost all cellulase and hemicellulase genes are unable to be induced in T. reesei [60]. However, deletion of xlnR (encoding a Xyr1 homolog) in Fusarium graminearum led to elevation of cellulase gene transcription [61]. The discrepancy in transcriptional regulation by Xyr1 is that the function of this transcription factor is mainly determined by its phosphorylation status, but also impacted by the interaction between Xyr1 and other transcription regulators [62,63,64].
Conclusion
In this study, an efficient CRISPR/Cas9 genome editing platform for H. insolens has been successfully developed for the first time, employing the 5S rRNA promoter and tRNAGly in sgRNA synthesis. This system proved to be highly effective when pks and xyr1 were used as two model target genes. The CRISPR–Cas9 system provides a technical platform for further study of the regulation mechanism of cellulase expression in H. insolens, which enables us to study the function of other transcriptional regulators and cellulase genes. It is expected to aid in promoting studies on regulation mechanisms of cellulase expression and engineering industrial strains with improved cellulase-producing ability.
Material and methods
Strains and culture media
The H. insolens Y1 (CGMCC 4573) and its engineered strains were cultured on potato dextrose agar (PDA) plates at 42 °C for 5 days for conidiation. The yeast extract–peptone–dextrose medium (YPD) was used for mycelia growth at 42 °C. For cellulase production in flask fermentation, H. insolens were cultured at 42 °C for 6 days in a modified Melin-Norkrans medium (MMN) (containing 1 g/L tryptone, 20 g/L yeast extract, 0.6 g/L MgSO4·7H2O and 20 g/L Avicel). For observation of colony phenotypes, 2 × 105 spores each of the H. insolens wild-type strain and its mutants were spotted and cultured on MMN, PDA, or YPD plates for 3 days at 42 °C. The Escherichia coli Top 10 (GenStar, Beijing, China) was grown at 37 °C for plasmid propagation in a Luria–Bertani (LB) broth supplemented with 100 μg/mL of ampicillin when necessary.
Plasmid construction
The expressing cassette Ptef1-NLS-cas9-NLS-TtrpC containing the tef1 promoter, a codon-optimized cas9 gene with two nuclear localization signals (NLS), and the trpC terminator was synthesized according to the sequence of Cas9-expressing cassette used in M. thermophila as described earlier [17] for expression in H. insolens.
Two different sgRNA-expressing cassettes were constructed in this study. First, the A. niger 5S rRNA along with its 338-bp upstream promoter sequence, an HDV ribozyme, and a sgRNA scaffold were all amplified from the plasmid psgRNA4.0 as described earlier [25]. The second construct was constructed by replacing the HDV ribozyme with the tRNAGly fragment (sequence listed in Additional file 1: Table S2) from H. insolens (Fig. 1), which was predicted out of the genome by using the online GtRNAdb server (http://gtrnadb.ucsc.edu/) and amplified from the genomic DNA of H. insolens using a pair of primers tRNAGlyS/tRNAGlyA (Additional file 1: Table S1). The sgRNA targeting sites for pks (GenBank accession number: MT875153) and xyr1 (MT720880) genes in H. insolens were analyzed using the sgRNACas9 tool [65]. For pks, three different sgRNAs were analyzed to verify the stability of gene editing efficiency. The 23-nt protospacer sequences were 5′-GGCCTCCTCCACATTTGAGCAGG-3′ (pks1, the underline letter represents the PAM sequence), 5′-TCCCCGAAGAGGAGAAATGCAGG-3′ (pks2), and 5′-GAGGAGAAATGCAGGCTGCTCGG-3′ (pks3), respectively. For xyr1, the 23-nt protospacer sequence was 5′-CCCTTATGGTCCTGCTGCCAGGG-3′. The sgRNA fragments were in vitro annealed using synthesized primers shown in Additional file 1: Table S1 and individually cloned into a pBlunt-simple cloning vector, which yielded the plasmids pBS-5SHDVsgRNA-pks1, pBS-5SHDVsgRNA-xyr1, pBS-5StRNAsgRNA-pks1, and pBS-5StRNAsgRNA-xyr1, respectively. The plasmids pBS-5SsgRNA-pks1-HDV, pBS-5SsgRNA-pks1, pBS-tRNAsgRNA-pks1, pBS-5SHDVsgRNA-pks2, pBS-5SHDVsgRNA-pks3, pBS-5StRNAsgRNA-pks2, pBS-5StRNAsgRNA-pks3 were constructed using the same method.
The 600-bp fragments flanking the targeting sites in donor DNAs (Fig. 1) were amplified by PCR from the H. insolens genomic DNA with primers shown in Table S1. The selection marker PTgpd-hyg containing the hygromycin resistance gene was amplified from the plasmid pAg1-hyg [8]. The 5′-flanking fragment, PTgpd-hyg, 3′-flanking fragment, and the NotI/XhoI-digested pBluescript KS were assembled using a Vazyme ClonExpress Ultra One Step Cloning Kit (Vazyme, Nanjing, China) to generate the plasmids pPTgpd-hyg-Δpks and pPTgpd-hyg-Δxyr1 containing the donor DNA fragments “donor-pks” (for pks deletion) and “donor-xyr1” (for xyr1 deletion), respectively.
Protoplast transformation of H. insolens
Both the H. insolens wild-type Y1 and the Y1/Δku70 strain were used as hosts in this study. Protoplast transformation of H. insolens was carried out as described earlier [66] with slight modifications. H. insolens strains were cultured on PDA medium at 42 °C for 3 days, and 107 spores were harvested and transferred to the YPD medium for a continued culture of 10 h. Lysing enzymes (5 mg/mL) from Trichoderma harzianum (Sigma, L-1412) was used for releasing protoplasts from mycelia. The PDA medium supplemented with 50 µg/ml of Hygromycin B and 0.44 M of sucrose was used to screen for successful transformants.
For pks disruption, a total of 20–30 μg DNA including the PCR products of Ptef1-NLS-cas9-NLS-TtrpC (10 μg, amplified with the primer pair Cas9S/Cas9A, Additional file 1: Table S1), 5SHDVsgRNA-pks1 or 5StRNAsgRNA-pks1 (10 μg, both amplified with primers 5SHPS/5SSA, Additional file 1: Table S1), with or without the donor for pks1 (10 μg, amplified with primers pksLS/pksRA, Additional file 1: Table S1) were mixed and added to the protoplasts of WT or Y1/Δku70. Transformants were screened on the PDA/hygromycin B plates and verified for DNA integration via PCR with primers shown in Additional file 1: Table S1. For xyr1 disruption, a total of 30 μg DNA fragments including the PCR products of Ptef1-NLS-cas9-NLS-TtrpC (10 μg, obtained with the primer pair Cas9S/Cas9A), 5SHDVsgRNA-xyr1 or 5StRNAsgRNA-xyr1 (10 μg each, both amplified with primers 5SHPS/5SSA), and donor-xyr1 (10 μg, amplified with the primer pair xyr1LS/xyr1RA) were similarly mixed and co-transformed into Y1/Δku70. The same procedure was used for transformations with other combinations of the DNA fragments (Table 1).
For simultaneous disruption of two genes, a total of 50 μg DNA including the PCR products of Ptef1-NLS-cas9-NLS-TtrpC, 5StRNAsgRNA-pks1, 5StRNAsgRNA-xyr1, donor (for pks1), and donor (for xyr1) (10 μg for each) were co-transformed into Y1/Δku70.
Assay of enzyme activity and SDS-PAGE
The mycelia of H. insolens Y1 and mutant strains were individually cultured in YPD and then transferred to MMN for cellulase induction [9]. The cellulase activities including the overall cellulase activity (FPase, using filter paper as the substrate), the endoglucanase (or carboxymethyl cellulose activity, using carboxylmethyl cellulose as the substrate), cellobiohydralase (using p-nitrophenyl-β-cellobioside as the substrate), β-glucosidase (using p-nitrophenyl-β-glucopyranoside as the substrate), and xylanase activity (using birchwood xylan as the substrate) in the culture supernatant were determined according to the methods described before [67]. The extracellular proteins were resolved by SDS-PAGE on 12% (w/v) polyacrylamide gels. Proteins were visualized by staining with Coomassie Brilliant Blue G-250.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files.
References
Soni SK, Sharma A, Soni R. Cellulases: role in lignocellulosic biomass utilization. In: Cellulases: methods and protocols. Methods Mol Biol. 2018;1796:3–23.
Karlsson J, Momcilovic D, Wittgren B, Schülein M, Tjerneld F, Brinkmalm G. Enzymatic degradation of carboxymethyl cellulose hydrolyzed by the endoglucanases Cel5A, Cel7B, and Cel45A from Humicola insolens and Cel7B, Cel12A and Cel45Acore from Trichoderma reesei. Biopolymers. 2002;63:32–40.
Schülein M. Enzymatic properties of cellulases from Humicola insolens. J Biotechnol. 1997;57:71–81.
Matsumoto H, Koganei K, Nishida N, Koyama Y, Saito S, Kataoka H, Ogihara J, Kasumi T. Cell dispersion culture for the effective growth of Humicola insolens and efficient enzyme production. J Biosci Bioeng. 2014;117:257–62.
Du Y, Shi P, Huang H, Zhang X, Luo H, Wang Y, Yao B. Characterization of three novel thermophilic xylanases from Humicola insolens Y1 with application potentials in the brewing industry. Bioresour Technol. 2013;130:161–7.
Xia W, Bai Y, Cui Y, Xu X, Qian L, Shi P, Zhang W, Luo H, Zhan X, Yao B. Functional diversity of family 3 β-glucosidases from thermophilic cellulolytic fungus Humicola insolens Y1. Sci Rep. 2016;6:27062.
Xu X, Li J, Zhang W, Huang H, Shi P, Luo H, Liu B, Zhang Y, Zhang Z, Fan Y. A neutral thermostable β-1,4-glucanase from Humicola insolens Y1 with potential for applications in various industries. PLoS ONE. 2015;10:e0124925.
Xu XX, Li JY, Shi PJ, Ji WL, Liu B, Zhang YH, Yao B, Fan YL, Zhang W. The use of T-DNA insertional mutagenesis to improve cellulase production by the thermophilic fungus Humicola insolens Y1. Sci Rep. 2016;6:9.
Xu X, Fan C, Song L, Li J, Chen Y, Zhang Y, Liu B, Zhang W. A novel CreA-mediated regulation mechanism of cellulase expression in the thermophilic fungus Humicola insolens. Int J Mol Sci. 2019;20:3693.
Liu R, Chen L, Jiang Y, Zhou Z, Zou G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 2015;1:15007–15007.
Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE. 2015;10:e0133085–e0133085.
Pohl C, Kiel JA, Driessen AJ, Bovenberg RA, Nygard Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synth Biol. 2016;5:754–64.
Makarova KS, Zhang F, Koonin EV. SnapShot: class 2 CRISPR–Cas systems. Cell. 2017;168:328-328.e321.
Jiang F, Doudna JA. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46:505–29.
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–7.
Shibata A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat Res. 2017;803–805:51–5.
Liu Q, Gao R, Li J, Lin L, Zhao J, Sun W, Tian C. Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering. Biotechnol Biofuels. 2017;10:1–1.
Arazoe T, Miyoshi K, Yamato T, Ogawa T, Ohsato S, Arie T, Kuwata S. Tailor-made CRISPR/Cas system for highly efficient targeted gene replacement in the rice blast fungus. Biotechnol Bioeng. 2015;112:2543–9.
Li Z, Yao G, Wu R, Gao L, Kan Q, Liu M, Yang P, Liu G, Qin Y, Song X, Zhong Y, Fang X, Qu Y. Synergistic and dose-controlled regulation of cellulase gene expression in Penicillium oxalicum. PLoS Genet. 2015;11(9):e1005509.
Matsu-ura T, Baek M, Kwon J, Hong C. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Genet Biol. 2015;2:4.
Zhang C, Meng X, Wei X, Lu L. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet Biol. 2016;86:47–57.
Gao Y, Zhao Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol. 2014;56:343–9.
Katayama T, Tanaka Y, Okabe T, Nakamura H, Fujii W, Kitamoto K, Maruyama JI. Development of a genome editing technique using the CRISPR/Cas9 system in the industrial filamentous fungus Aspergillus oryzae. Biotechnol Lett. 2016;38:637–42.
Shi TQ, Gao J, Wang WJ, Wang KF, Xu GQ, Huang H, Ji XJ. CRISPR/Cas9-based genome editing in the filamentous fungus Fusarium fujikuroi and its application in strain engineering for gibberellic acid production. ACS Synth Biol. 2019;8:445–54.
Zheng X, Zheng P, Zhang K, Cairns T, Meyer V, Sun J, Ma Y. 5S rRNA promoter for guide RNA expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger. ACS Synth Biol. 2018;8(7):1568–74.
Liang Y, Han Y, Wang C, Jiang C, Xu JR. Targeted deletion of the USTA and UvSLT2 genes efficiently in Ustilaginoidea virens with the CRISPR-Cas9 system. Front Plant Sci. 2018;9:699.
Chen C, Liu J, Duan C, Pan Y, Liu G. Improvement of the CRISPR-Cas9 mediated gene disruption and large DNA fragment deletion based on a chimeric promoter in Acremonium chrysogenum. Fungal Genet Biol. 2020;134:103279.
Xie K, Minkenberg B, Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA. 2015;112:3570–5.
Woo PCY, Tam EWT, Chong KTK, Cai JJ, Tung ETK, Ngan AHY, Lau SKP, Yuen KY. High diversity of polyketide synthase genes and the melanin biosynthesis gene cluster in Penicillium marneffei. FEBS J. 2010;277:3750–8.
Zhang P, Zhou S, Wang G, An Z, Liu X, Li K, Yin WB. Two transcription factors cooperatively regulate DHN melanin biosynthesis and development in Pestalotiopsis fici. Mol Microbiol. 2019;112:649–66.
Fujii I, Watanabe A, Sankawa U, Ebizuka Y. Identification of Claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. Chem Biol. 2001;8:189–97.
Obermaier S, Müller M. Biaryl-forming enzymes from Aspergilli exhibit substrate-dependent stereoselectivity. Biochemistry. 2019;58:2589–93.
Jackson JC, Higgins LA, Lin X. Conidiation color mutants of Aspergillus fumigatus are highly pathogenic to the heterologous insect host Galleria mellonella. PLoS ONE. 2009;4:e4224.
Ámon J, Fernández-Martín R, Bokor E, Cultrone A, Kelly JM, Flipphi M, Scazzocchio C, Hamari Z. A eukaryotic nicotinate-inducible gene cluster: convergent evolution in fungi and bacteria. Open Biol. 2017;7:170199.
Mello-de-Sousa TM, Rassinger A, Pucher ME, dos Santos CL, Persinoti GF, Silva-Rocha R, Poças-Fonseca MJ, Mach RL, Nascimento Silva R, Mach-Aigner AR. The impact of chromatin remodelling on cellulase expression in Trichoderma reesei. BMC Genom. 2015;16:588.
Berka RM, Grigoriev IV, Otillar R, Salamov A, Grimwood J, Reid I, Ishmael N, John T, Darmond C, Moisan MC, Henrissat B, Coutinho PM, Lombard V, Natvig DO, Lindquist E, Schmutz J, Lucas S, Harris P, Powlowski J, Bellemare A, Taylor D, Butler G, de Vries RP, Allijn IE, van den Brink J, Ushinsky S, Storms R, Powell AJ, Paulsen IT, Elbourne LD, Baker SE, Magnuson J, Laboissiere S, Clutterbuck AJ, Martinez D, Wogulis M, de Leon AL, Rey MW, Tsang A. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat Biotechnol. 2011;29:922.
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, Rehman B, Elkins T, Engels R, Wang S, Nielsen CB, Butler J, Endrizzi M, Qui D, Ianakiev P, Bell-Pedersen D, Nelson MA, Werner-Washburne M, Selitrennikoff CP, Kinsey JA, Braun EL, Zelter A, Schulte U, Kothe GO, Jedd G, Mewes W, Staben C, Marcotte E, Greenberg D, Roy A, Foley K, Naylor J, Stange-Thomann N, Barrett R, Gnerre S, Kamal M, Kamvysselis M, Mauceli E, Bielke C, Rudd S, Frishman D, Krystofova S, Rasmussen C, Metzenberg RL, Perkins DD, Kroken S, Cogoni C, Macino G, Catcheside D, Li W, Pratt RJ, Osmani SA, DeSouza CP, Glass L, Orbach MJ, Berglund JA, Voelker R, Yarden O, Plamann M, Seiler S, Dunlap J, Radford A, Aramayo R, Natvig DO, Alex LA, Mannhaupt G, Ebbole DJ, Freitag M, Paulsen I, Sachs MS, Lander ES, Nusbaum C, Birren B. The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003;422:859–68.
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EG, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, Schoch CL, Yao J, Barabote R, Nelson MA, Detter C, Bruce D, Kuske CR, Xie G, Richardson P, Rokhsar DS, Lucas SM, Rubin EM, Dunn-Coleman N, Ward M, Brettin TS. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol. 2008;26:553–60.
Tamayo EN, Villanueva A, Hasper AA, de Graaff LH, Ramón D, Orejas M. CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genet Biol. 2008;45:984–93.
White RJ. Transcription by RNA polymerase III: more complex than we thought. Nat Rev Genet. 2011;12:459–63.
Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–60.
Krappmann S. Gene targeting in filamentous fungi: the benefits of impaired repair. Fungal Genet Biol. 2007;21:25–9.
Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344:189–93.
Lieber MR, Wilson TE. SnapShot: nonhomologous DNA end joining (NHEJ). Cell. 2010;142:496–496.
Liu H, Wang G, Li W, Liu X, Li E, Yin WB. A highly efficient genetic system for the identification of a harzianum B biosynthetic gene cluster in Trichoderma hypoxylon. Microbiol. 2018;164:769–78.
Liu Q, Zhang Y, Li F, Li J, Sun W, Tian C. Upgrading of efficient and scalable CRISPR–Cas-mediated technology for genetic engineering in thermophilic fungus Myceliophthora thermophila. Biotechnol Biofuels. 2019;12:293.
Ninomiya Y, Suzuki K, Ishii C, Inoue H. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA. 2004;101:12248–53.
Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA, Oakley BR. A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics. 2006;172:1557–66.
Villalba F, Collemare J, Landraud P, Lambou K, Brozek V, Cirer B, Morin D, Bruel C, Beffa R, Lebrun MH. Improved gene targeting in Magnaporthe grisea by inactivation of MgKU80 required for non-homologous end joining. Fungal Genet Biol. 2008;45:68–75.
Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010;38:5075–87.
Cao M, Gao M, Ploessl D, Song C, Shao Z. CRISPR-mediated genome editing and gene repression in Scheffersomyces stipitis. Biochem J. 2018;13:1700598.
Furukawa T, Shida Y, Kitagami N, Mori K, Kato M, Kobayashi T, Okada H, Ogasawara W, Morikawa Y. Identification of specific binding sites for XYR1, a transcriptional activator of cellulolytic and xylanolytic genes in Trichoderma reesei. Fungal Genet Biol. 2009;46:564–74.
Sun J, Tian C, Diamond S, Glass NL. Deciphering transcriptional regulatory mechanisms associated with hemicellulose degradation in Neurospora crassa. Eukaryot Cell. 2012;11:482–93.
Battaglia E, Klaubauf S, Vallet J, Ribot C, Lebrun MH, de Vries RP. Xlr1 is involved in the transcriptional control of the pentose catabolic pathway, but not hemi-cellulolytic enzymes in Magnaporthe oryzae. Fungal Genet Biol. 2013;57:76–84.
Klaubauf S, Narang HM, Post H, Zhou M, Brunner K, Mach-Aigner AR, Mach RL, Heck AJR, Altelaar AFM, de Vries RP. Similar is not the same: Differences in the function of the (hemi-) cellulolytic regulator XlnR (Xlr1/Xyr1) in filamentous fungi. Fungal Genet Biol. 2014;72:73–81.
dos Santos Gomes AC, Falkoski D, Battaglia E, Peng M, Nicolau de Almeida M, Coconi Linares N, Meijnen JP, Visser J, de Vries RP. Myceliophthora thermophila Xyr1 is predominantly involved in xylan degradation and xylose catabolism. Biotechnol Biofuels. 2019;12:220.
Raulo R, Kokolski M, Archer DB. The roles of the zinc finger transcription factors XlnR, ClrA and ClrB in the breakdown of lignocellulose by Aspergillus niger. AMB Express. 2016;6:5.
Cao Y, Zheng F, Wang L, Zhao G, Chen G, Zhang W, Liu W. Rce1, a novel transcriptional repressor, regulates cellulase gene expression by antagonizing the transactivator Xyr1 in Trichoderma reesei. Mol Microbiol. 2017;105:65–83.
Rauscher R, Würleitner E, Wacenovsky C, Aro N, Stricker AR, Zeilinger S, Kubicek CP, Penttilä M, Mach RL. Transcriptional regulation of xyn1, encoding Xylanase I, in Hypocrea jecorina. Eukaryot Cell. 2006;5:447–56.
Stricker AR, Grosstessnerhain K, Würleitner E, Mach RL. Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and d-xylose metabolism in Hypocrea jecorina. Eukaryot Cell. 2006;5:2128–37.
Brunner K, Lichtenauer AM, Kratochwill K, Delic M, Mach RL. Xyr1 regulates xylanase but not cellulase formation in the head blight fungus Fusarium graminearum. Curr Genet. 2007;52:213–20.
Aro N, Pakula T, Penttilä M. Transcriptional regulation of plant cell wall degradation by filamentous fungi. FEMS Microbiol Ecol. 2005;29:719–39.
Coradetti ST, Craig JP, Xiong Y, Shock T, Tian C, Glass NL. Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. Proc Natl Acad Sci USA. 2012;109(19):7397–402.
Lichius A, Seidl-Seiboth V, Seiboth B, Kubicek CP. Nucleo-cytoplasmic shuttling dynamics of the transcriptional regulators XYR1 and CRE1 under conditions of cellulase and xylanase gene expression in Trichoderma reesei. Mol Microbiol. 2014;94:1162–78.
Xie S, Shen B, Zhang C, Huang X, Zhang Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE. 2014;9(6):e100448–e100448.
Yang F, Gong Y, Liu G, Zhao S, Wang J. Enhancing cellulase production in thermophilic fungus Myceliophthora thermophila ATCC42464 by RNA interference of cre1 gene expression. J Microbiol Biotechnol. 2015;25:1101–7.
Fan C, Xu X, Song L, Guan W, Li J, Liu B, Shi P, Zhang W. The use of Agrobacterium-mediated insertional mutagenesis sequencing to identify novel genes of Humicola insolens involved in cellulase production. 3 Biotech. 2018;8(3):153.
Acknowledgements
The authors thank Dr. Xiaomei Zheng (Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences) for providing the plasmid psgRNA4.0.
Funding
This study was funded by the National Key Research and Development Program of China (2021YFC2100204), the Agricultural Science and Technology Innovation Program (ASTIP) and Modern Agriculture Biotechnology System (CARS-41).
Author information
Authors and Affiliations
Contributions
XX and HH conceived and designed the experiments; CF, WZ and WJ performed the experiments; CF, YZ and BL analyzed the data; WZ and HL contributed reagents/materials/analysis tools; CF and XS wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Table S1: Primers used in this study; Table S2: The DNA sequence of the tRNAGly from H. insolens; Figure S1: Multiple amino acid sequence alignment of Pks from H. insolens with four Pks homologs; Figure S2: Multiple amino acid sequence alignment of Xyr1 from H. insolens with four Xyr1 homologs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, C., Zhang, W., Su, X. et al. CRISPR/Cas9-mediated genome editing directed by a 5S rRNA–tRNAGly hybrid promoter in the thermophilic filamentous fungus Humicola insolens. Biotechnol Biofuels 14, 206 (2021). https://doi.org/10.1186/s13068-021-02057-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-021-02057-y