
Abstract
Background
Ralstonia eutropha is an important bacterium for the study of polyhydroxyalkanoates (PHAs) synthesis and CO2 fixation, which makes it a potential strain for industrial PHA production and attractive host for CO2 conversion. Although the bacterium is not recalcitrant to genetic manipulation, current methods for genome editing based on group II introns or single crossover integration of a suicide plasmid are inefficient and time-consuming, which limits the genetic engineering of this organism. Thus, developing an efficient and convenient method for R. eutropha genome editing is imperative.
Results
An efficient genome editing method for R. eutropha was developed using an electroporation-based CRISPR-Cas9 technique. In our study, the electroporation efficiency of R. eutropha was found to be limited by its restriction-modification (RM) systems. By searching the putative RM systems in R. eutropha H16 using REBASE database and comparing with that in E. coli MG1655, five putative restriction endonuclease genes which are related to the RM systems in R. eutropha were predicated and disrupted. It was found that deletion of H16_A0006 and H16_A0008-9 increased the electroporation efficiency 1658 and 4 times, respectively. Fructose was found to reduce the leaky expression of the arabinose-inducible pBAD promoter, which was used to optimize the expression of cas9, enabling genome editing via homologous recombination based on CRISPR-Cas9 in R. eutropha. A total of five genes were edited with efficiencies ranging from 78.3 to 100%. The CRISPR-Cpf1 system and the non-homologous end joining mechanism were also investigated, but failed to yield edited strains.
Conclusions
We present the first genome editing method for R. eutropha using an electroporation-based CRISPR-Cas9 approach, which significantly increased the efficiency and decreased time to manipulate this facultative chemolithoautotrophic microbe. The novel technique will facilitate more advanced researches and applications of R. eutropha for PHA production and CO2 conversion.
Similar content being viewed by others


Background
Ralstonia eutropha H16, also known as Cupriavidus necator H16, is a Gram-negative β-proteobacterium that is ubiquitously present in soil and freshwater environments [1]. It has attracted considerable research interest due to its significant economic potential [2] and CO2 fixation ability [3, 4]. This facultative chemolithoautotrophic bacterium is a metabolically versatile organism that can grow well under both lithoautotrophic and heterotrophic conditions [1]. Under lithoautotrophic conditions, it fixes CO2 via the Calvin–Benson–Bassham (CBB) cycle, which comprises enzymes encoded by the two CBB operons [1]. The energy used to implement CO2 fixation and maintain cell growth is generated by energy-conserving hydrogenases, which oxidize molecular H2 and thereby reduce NAD+ to form NADH [1]. The lithoautotrophic R. eutropha has great potential as a chassis for the study of CO2 fixation and development of microbial cell factories for syngas utilization. Under heterotrophic conditions, several organic substrates, such as fructose, gluconate, N-acetylglucosamine and some organic acids, are utilized as carbon sources [1]. R. eutropha grows to very high cell densities (281 g/L) under nutritionally rich conditions, and accumulates large amounts of PHAs (232 g/L) when the nitrogen or phosphate source is limited [2]. This characteristic makes it an attractive host for the synthesis of industrially relevant PHA materials.
In recent years, R. eutropha was engineered to produce biofuels, such as branched-chain alcohols [3], methyl ketones [5], hydrocarbons [6] and isopropanol [7]. In addition, R. eutropha can also be cultured under lithoautotrophic conditions to produce PHAs [8, 9] and biofuels [3, 5]. Although R. eutropha can already be engineered, convenient and efficient synthetic-biology tools are still underdeveloped compared to those available for model organisms such as Escherichia coli and Saccharomyces cerevisiae, and current manipulation techniques are still inefficient and time-consuming [10]. One of the major reasons for this is the low transformation efficiency of R. eutropha [11]. While common heat-shock transformation is not feasible in R. eutropha, electroporation has also been used rarely in previous publications, due to its extraordinarily low efficiency, which is several orders of magnitude lower than that of E. coli [11]. These properties make cell-to-cell transconjugation almost the only way to transfer plasmids into R. eutropha, which is not ideal for genome manipulation.
There are currently two techniques for genome editing of R. eutropha, one of which is based on group II introns, which is complicated and is consequently rarely used [10]. Another method was designed to integrate a suicide plasmid, via a single crossover recombination event [12, 13]. The integrating plasmid is transferred to R. eutropha via conjugation from a special host—E. coli S17-1 [14]. Transconjugants carrying the integrated plasmid are selected using proper antibiotics, and strains that have lost the integration vector via a second single crossover are selected in rich medium containing sucrose using sacB as the negative selection marker. This method which would take an average of 2–3 weeks to delete a single gene is not only time-consuming, but is also not very efficient [13].
Microbial genome editing techniques have progressed significantly due to the extensive research conducted on the CRISPR system (clustered regularly interspaced short palindromic repeats), derived from the RNA-guided immune systems found in many bacteria and archaea [15,16,17]. CRISPR-Cas9 and CRISPR-Cpf1 are Class 2 CRISPR-Cas systems, and are further classified as types II and V, respectively [18]. The systems recognize unique sequences and generate double strand breaks (DSBs) at the target locus, after which the DSBs is repaired either through NHEJ or HR [17]. CRISPR-Cas assisted genome editing tools have already been developed for a number of bacteria, including but not limited to Streptococcus pneumoniae [16], E. coli [19], Streptomyces [20, 21], Lactobacillus reuteri [22], Clostridium [23], Bacillus subtilis [24] and Corynebacterium glutamicum [25, 26]. However, this technique has not been developed in R. eutropha to date. Thus, we aimed to develop a convenient and efficient CRISPR-Cas assisted genome editing method for R. eutropha, preferentially using fast transformation methods, omitting the need for conjugation.
Methods
Strains and culture conditions
Escherichia coli S17-1 [14] was used for plasmid maintenance and conjugation with R. eutropha, and was cultured at 37 °C in Luria–Bertani medium (LB, 10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) with 100 µg/mL streptomycin or 50 µg/mL kanamycin if necessary. R. eutropha H16 [1] was the parent strain for genetic modifications and was cultured aerobically at 30 °C in LB with 10 µg/mL gentamicin or 200 µg/mL kanamycin for plasmid maintenance. All strains used in this study are listed in Table 1.
Plasmid construction
Primers (Additional file 1: Table S4) were designed using the j5 DeviceEditor [27] and synthesized by Genewiz (Beijing, China). DNA polymerase, BsaI restriction endonuclease and T4 ligase were purchased from Takara (Dalian, China), New England Biolabs (USA) and Thermo-Fisher Scientific (USA), respectively. The plasmids used in this study were constructed via Golden Gate [28] or Gibson [29] assembly, and cloned directly into E. coli S17-1. Plasmids used in this study are in Additional file 1: Table S3.
Plasmid extraction
Plasmids in E. coli or R. eutropha were extracted using AxyPrep Plasmid Miniprep Kit (AXYGEN, China) according to the manuscript with some minor modifications. For R. eutropha, after extraction reagents were added to the collected sample from 1 mL LB medium and the mixture was centrifuged at 13,000×g for 20 min. While for E. coli, centrifugation was performed at 12,000×g for 10 min. To facilitate the following step of electroporation, the eluent solution of the kit was replaced by sterile deionized water for plasmid elution. The plasmid solution obtained from the adsorbing column was used for eluting another column to improve the plasmid concentration.
Preparation of competent cells
For E. coli, the procedure was performed using a previously described protocol [19]. To prepare R. eutropha competent cells, the method described by Hae-Chul Park [11] was employed with some modifications. The procedure used in this study was performed as follows. An aliquot of a glycerol cryopreservation stock was streaked onto an LB plate with 10 µg/mL gentamicin and incubated for 48 h at 30 °C, after which a single colony was picked up and incubated aerobically in LB medium at 30 °C and 200 rpm. The resulting seed culture was transferred into 100 mL LB and cultured to an optical density at 600 nm (OD600) of 0.6–0.8, after which it was chilled on ice for 5–10 min. Cells were harvested by centrifugation at 3000×g and 4 °C for 5 min and washed three times with ice-cold sterile 10% glycerol. The cell pellet collected from 100 mL bacteria solution was resuspended in 0.6 mL 10% glycerol and aliquoted into sterile 1.5 mL tubes. Then the competent cells were used immediately or frozen in liquid nitrogen and preserved at − 80 °C.
Conjugation and electroporation
Conjugation was performed as follows. The E. coli S17-1 donor harboring the transferable plasmid was cultured in 10 mL LB at 37 °C for 12 h, and the R. eutropha recipient was cultured in 10 mL LB at 30 °C for 24 h, before they were mixed and centrifuged at 3000×g for 5 min. The supernatant was removed and the cell pellet was washed with 30 mL LB, after which the cells were resuspended in 100 µL LB, dropped onto LB plates without antibiotics and incubated at 30 °C for 24 h. Subsequently, a portion of the mixed bacterial lawn was resuspended in LB, plated on LB agar plates with 200 µg/mL kanamycin and 10 µg/mL gentamicin, and incubated at 30 °C for 48 h.
For the electroporation of R. eutropha, 8 µL of high-quality plasmid DNA (~ 400 ng) was added to 100 µL of competent cells, and transferred into a pre-chilled 2-mm electroporation cuvette (Bio-Rad, USA), and incubated on ice for 5 min, after which electroporation was performed at a voltage of 2.3 kV. Immediately afterward, 1 mL LB with 10 mg/mL fructose was added to the cells, and the resulting suspension was transferred to a sterile 1.5 mL centrifuge tube. After incubation at 30 °C for 2 h, the cells were spread on LB agar plates with 200 µg/mL kanamycin and 10 mg/mL fructose, and incubated at 30 °C for 48 h.
Gene knockout (or integration) via pK18mobsacB
Two homologous templates which were ~ 500 bp, respectively, were cloned into the pK18mobsacB plasmid backbone via Golden Gate [28] or Gibson [29]. For integration, the target gene was cloned between the two homologous templates. The knockout plasmid was transformed into E. coli S17-1, then identified and transferred to R. eutropha via conjugation. Single colonies were cultured in LB with 10 µg/mL gentamicin and 200 µg/mL kanamycin at 30 °C. A pair of primers with one bound to the genome and another to the plasmid was used for colony PCR to identify strains with the knockout plasmid integrated. Then corrected strains were incubated in LB without NaCl and kanamycin, but with 100 mg/mL sucrose at 30 °C for 72 h. Strains were streaked on LB plate (without NaCl and kanamycin, but with 50 mg/mL sucrose), then resistance against kanamycin was investigated, and single colonies with no resistance against kanamycin were identified by PCR. H16_A0006, H16_A0008-9, H16_A0014, PHG170 deletion and rfp integration were performed by this method.
Plasmid for gene editing via CRISPR-Cas system
All plasmids used for R. eutropha gene editing via CRISPR-Cas system were derived from the broad-range-host plasmid pBBR1MCS2 [30] and constructed via Golden Gate. The csa9 gene was amplified from the plasmid pCas9 (Addgene number 42876) [16, 19] and driven by the arabinose-inducible pBAD promoter, while the corresponding sgRNA was transcribed from a constitutive promoter (ctaggtttatacataggcgagtactctgttatggagtcagatcttagc). 20 bp sequence (take rfp for example, catgcgtttcaaagttcgta) was selected as the guide sequence. The cpf1 gene was amplified from the plasmid pFnCpf1_min (Addgene number 69975) [31] and driven by the arabinose-inducible pBAD promoter, while the corresponding sgRNA was transcribed from the constitutive promoter BBa_J23109. A 24 bp sequence (take rfp for example, caaagttcgtatggaaggttccgt) was selected as the guide sequence. Genes ligD and ku70 were amplified from pCas9(Ts)-NHEJ [32] provided by Prof. Qingsheng Qi (Shandong University, Ji’nan).
Genome editing via CRISPR-Cas9
Plasmid for gene editing was amplified in E. coli S17-1 and electroporated into R. eutropha. A single colony from the transformation plate was cultured in LB with 2 mg/mL arabinose and 200 µg/mL kanamycin for 120–168 h to induce the editing process, after which the cells were spread on LB plates with the same concentrations of arabinose and kanamycin to identify edited strains by colony PCR. Afterward, plasmid curing was performed by growing the cells in LB without kanamycin at 30 °C for 24 h and confirmed by testing for loss of resistance against kanamycin.
Results and discussions
Enhancing electroporation efficiency of R. eutropha by identification and deletion of key restriction endonuclease genes
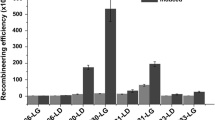
One of the major problems hindering the genome editing of R. eutropha is its low electroporation efficiency. To identify the main reasons for this problem, the plasmid pBBR1-rfp was transferred into either E. coli S17-1 or R. eutropha H16, extracted and electroporated into R. eutropha H16 again. This plasmid was derived from the broad-host-range plasmid pBBR1MCS2 [30], by introducing a red fluorescent protein (rfp) gene driven by the constitutive promoter BBa_J23100, which made its carriers exhibit visible red color. Intriguingly, it was found that the electroporation efficiency of pBBR1-rfp extracted from R. eutropha was 1677 times higher than that of its counterpart from E. coli S17-1 (Fig. 1a). The two plasmids had the same DNA sequence, but were likely modified by different RM systems in E. coli and R. eutropha, which indicated that the R. eutropha RM systems may be the major cause of the low electroporation efficiency.

Electroporation efficiencies of different plasmid DNA in various R. eutropha strains. a Electroporation efficiencies of pBBR1-rfp extracted from E. coli S17-1 or R. eutropha H16 in R. eutropha H16. pBBR1-rfp(S17), pBBR1-rfp extracted from E. coli S17-1. pBBR1-rfp(H16), pBBR1-rfp extracted from R. eutropha H16. b Electroporation efficiencies of pBBR1-rfp in H16 and engineered R. eutropha strains. pBBR1-rfp was extracted from E. coli S17-1. c Electroporation efficiencies of pBBR1-rfp(NM) in R. eutropha H16 and C5. pBBR1-rfp(NM), non-methylated pBBR1-rfp. d Electroporation efficiencies of pBBR1-rfp from E. coli MG1655 and MG18 in R. eutropha H16. pBBR1-rfp(MG1655), pBBR1-rfp extracted from E. coli MG1655. pBBR1-rfp(MG18), pBBR1-rfp extracted from E. coli MG18. All experiments were repeated four times and the error bars represent standard deviations. The significance of differences was calculated by one-way ANOVA using SPSS18.0 software. Asterisks indicate a significant difference compared with the control (**p < 0.01, highly significant difference, *p < 0.05, significant difference)
To investigate this hypothesis, we searched the genome of R. eutropha using the prediction tool based on the REBASE database [33] and identified four bona fide RM systems. Within these five putative restriction endonuclease genes, H16_A0006, H16_A 0008, H16_A0009, H16_A0014 and PHG170 (H16_A0008 and H16_A0009 may encode two subunits of a single endonuclease) were predicted by comparing with the RM systems of E. coli MG1655 (Additional file 1: Table S1). To determine their effects on the transformation efficiency, the putative restriction endonuclease genes were knocked out individually, yielding four R. eutropha RM knockout strains C1, C2, C3 and C4. The electroporation efficiencies of the wild-type strain H16 and the four knockout strains were tested using the plasmid pBBR1-rfp extracted from E. coli S17-1. The results showed that while C3 and C4 did not show an obvious enhancement of electroporation efficiency, the efficiencies of the strains C2 and C1 were, respectively, improved 4 and an astonishing 1658 times over H16 (Fig. 1b). This result indicated that endonucleases encoded by the genes H16_A0006 and H16_A0008-9 were indeed the major reason for the low transformation efficiency of R. eutropha. Subsequently, strain C5 was constructed by disrupting the H16_A0008-9 gene in strain C1, which led to an electroporation efficiency that was 1697 times higher than that of H16 strain, even if no significant improvement was evident compared to C1 (Fig. 1b). mcrBC in E. coli, which is homologous to the H16_A0008-9 in R. eutropha H16, was disrupted to enhance transformation efficiency in some laboratory strains [34], such as DH10B, DH12S, DM1, and HB101. Therefore, the C5 strain with a similar double knockout was selected for future research.
To investigate the electroporation efficiency of an in vitro constructed plasmid with no methylation, linear pBBR1-rfp was generated by PCR and ligated using the Golden Gate assembly method to obtain non-methylated plasmid DNA of pBBR1-rfp(NM). The resulting non-methylated material was individually electroporated into C5 and H16, which revealed that the electroporation efficiency of C5 with in vitro constructed DNA was 343 times higher than that of H16 (Fig. 1c). Thus, the deletion of H16_A0006 or H16_A0006 along with H16_A0008-9 enabled the efficient electrotransformation of R. eutropha, regardless of the methylation status of the plasmid DNA.
On the other hand, the genes H16_A0004-5, which are adjacent to H16_A0006, were predicted to encode the putative methyltransferase and specificity subunits of the RM system. Hence, the MG18 strain was constructed by integrating H16_A0004-5 with the constitutive promoter BBa_J23100 and an RBS into the poxB loci of the E. coli MG1655 genome, which should have enabled it to methylate plasmids according to the methylation-protection pattern of R. eutropha. However, when pBBR1-rfp extracted from MG1655 and MG18 were electroporated into R. eutropha H16, there was no significant difference in electroporation efficiency (Fig. 1d). These results indicated that H16_A0004-5 expressed in E. coli MG1655 alone is not sufficient to properly methylate the plasmid DNA in this study.
Optimal expression strategy with fructose as the suppression factor
Due to the relatively low transformation efficiency of R. eutropha, even in case of the C5 strain, linear DNA could not be used as the donor template for genome editing. To simplify the transformation process, a single plasmid was employed to carry all the functional parts of the CRISPR-Cas-based editing tool. The precise expression control of cas9 and sgRNA obtained with this strategy was the key for successful genome editing [19]. In this study, the arabinose-inducible pBAD promoter was selected to express the cas genes (cas9 and cpf1 [31]). pBBR1-pBAD-rfp [6] carrying pBAD-controlled rfp was used to transform the C5 strain to investigate the optimal expression level, which increased with the increase of l-arabinose concentration in LB, and the maximal expression was achieved at 2 mg/mL l-arabinose (Fig. 2a). Under these conditions, the rfp expression level was three orders of magnitude higher than in the control (Fig. 2b), which should be suitable for the expression of the cas genes, which are large (~ 4 kb) and have an extremely low GC content (~ 30%). In contrast to E. coli, glucose did not suppress the basal expression of pBAD, since it is not transported into R. eutropha H16 [35]. Interestingly, we found that the expression of pBAD was significantly suppressed by fructose (Fig. 2b). Hence, an expression strategy was developed that used pBAD as the inducible promoter for the expression of the cas genes, and fructose was added to decrease its basal expression.

Expression levels of pBAD-controlled rfp under different induction conditions. a Expression levels of pBAD-rfp induced by different concentrations of l-arabinose. b Expression levels of pBAD-rfp suppressed by fructose. All experiments were performed in LB and repeated three times to obtain mean values and standard deviations
Development of the CRISPR-Cas9 genome editing method for R. eutropha
To develop an efficient genome editing method for R. eutropha, both CRISPR-Cas9 and CRISPR-Cpf1 systems were evaluated. The systems were individually cloned into the pBBR1MCS2 vector backbone using Golden Gate or Gibson assembly. The expression of cas9 or cpf1 on the plasmids was driven by the pBAD promoter, while the corresponding sgRNA was transcribed from a constitutive promoter. By searching the genome sequence, H16_B2352 and H16_B2355 were found to encode gene products with 40 and 28% similarity to LigD and Ku70 from Mycobacterium tuberculosis H37Rv (Additional file 1: Table S2), which may encode a putative ATP-dependent DNA ligase and non-homologous end-binding protein, respectively [36]. This indicated that R. eutropha may possess a native NHEJ mechanism. To investigate whether NHEJ-based editing mediated by the CRISPR-Cas system is functional in the strain, rfp was integrated into the downstream of phaP1 to obtain the strain C5rfp, which exhibits red fluorescence and can be conveniently detected using a microplate reader, whereby the red fluorescence would disappear if the rfp gene was disrupted. The two plasmids pBBR1-Cas9 and pBBR1-Cpf1, respectively, harboring the CRISPR-Cas9 and CRISPR-Cpf1 systems, were constructed, confirmed and transferred into R. eutropha via electroporation or conjugation. It should be noted that the electroporation and conjugation efficiencies of pBBR1-Cas9 and pBBR1-Cpf1 were significantly lower than those of pBBR1-rfp, which were about two to three orders of magnitude lower than the latter. Moreover, few transformants were acquired by electroporation when fructose was not added, which may be due to the large size of the plasmids (> 9 kb) and leaky expression of the cas genes. However, when 10 mg/mL fructose was added to the regeneration medium in the process of electroporation, sufficient numbers of transformants were obtained, although the efficiency which was less than 102 CFU/µg of plasmid DNA was still lower than that of pBBR1-rfp. Transformants identified by PCR and sequencing were cultured in LB with 2 mg/mL arabinose and 200 µg/mL kanamycin for 120–168 h, after which they were streaked onto LB plates with arabinose and kanamycin at the same concentrations. 48 single colonies were picked randomly and red fluorescence was detected with a microplate reader, while C5rfp and C5 were selected as positive and negative controls, respectively. The result indicated that none of the rfp genes was disrupted (Table 2), which therefore showed that CRISPR-Cas systems and the native NHEJ mechanism alone were not able to edit the target gene in R. eutropha.
To introduce a heterologous NHEJ mechanism into R. eutropha, the ligD and ku70 genes from M. tuberculosis H37Rv were cloned together with the constitutive promoter BBa_J23119 into both pBBR1-Cas9 and pBBR1-Cpf1, to construct the plasmids pBBR1-Cas9-ligD-ku and pBBR1-Cpf1-ligD-ku [32]. The two plasmids were individually transferred into C5rfp and the resulting strain was induced under the conditions described above. However, while 48 single colonies were obtained, none of them had disruptions of the rfp genes (Table 2), which indicated that no editing events occurred with the CRISPR-Cas and heterologous NHEJ systems.
Because NHEJ was not able to repair the DSB induced by CRISPR-Cas9, HR system for R. eutropha was investigated. Homologous arms of ~ 500 bp were cloned into both pBBR1-Cas9 and pBBR1-Cpf1 to construct the plasmids pBBR1-Cas9-rfpF-rfpR (Additional file 2) and pBBR1-Cpf1-rfpF-rfpR, respectively, which were individually transferred into C5rfp. After induction with 2 mg/mL arabinose for 168 h, the rfp genes in all the 23 randomly selected colonies were successfully edited using pBBR1-Cas9-rfpF-rfpR (Table 2, Additional file 1: Figure S1), while no editing occurred in the strain containing pBBR1-Cpf1-rfpF-rfpR (Table 2). Phage-derived recombinases λ-Red [19] and recET [25] were used to enhance the editing efficiency in E. coli and Corynebacterium glutamicum. In this study, the editing efficiency was 100%, even though none of the heterologous recombinases was expressed, which indicated that R. eutropha possesses an efficient endogenous homologous recombination system that supports successful genome editing. Although the CRISPR-Cpf1 system did not work, this is the first report of CRISPR-Cas9-based genome editing in R. eutropha. A similar situation was observed in C. glutamicum [25, 26], whereby the cas9 gene from Streptococcus pyogenes did not function, while a cas9 codon-optimized for actinomycetes and cpf1 from Francisella tularensis worked well.
To cure the editing plasmid pBBR1-Cas9-rfpF-rfpR after the editing process, the edited strain was cultured in LB medium without antibiotics for 24 h, after which randomly picked colonies were investigated for resistance against kanamycin. It was found that 98 of 100 colonies lost resistance against kanamycin, which indicated that almost all the cells were cleared of the plasmids (Additional file 1: Figure S2). The high curing efficiency may be due to the instability of the large pBBR1-Cas9-rfpF-rfpR plasmid, which has a low copy number.
In addition, four other R. eutropha genes (H16_A1814, H16_A1334, H16_A1437 and H16_B0204) were selected for editing using the method described above. A schematic illustration is shown in Fig. 3. Induction time was investigated; although the editing efficiency of H16_A1814 was less than 50% when induction time was 96 h, it increased with the extension of induction time, which reached 78.3% when induction time was extended to 168 h (Table 3). Longer induction time was not investigated, because the activity of the strains decreased sharply. As a result, about 2 weeks were required to edit a single gene, which was faster than the conventional method with an average of 2–3 weeks. All genes in our study were precisely edited as designed, with editing efficiencies ranging from 78.3 to 100% (Table 4, Additional file 1: Figure S3), which was also higher than that of the conventional method with an efficiency lower than 50%.

Schematic illustration of the genome editing method for R. eutropha using electroporation-based CRISPR-Cas9 technique. The editing plasmid for genome was transferred into R. eutropha via electroporation, while 10 mg/mL fructose was added to the regeneration medium in the process of electroporation. Transformants were induced by 2 mg/mL l-arabinose for 96–168 h, then streaked on LB plate with 2 mg/mL l-arabinose and 200 µg/mL kanamycin. Before curing the plasmid, edited strains were identified by PCR and sequencing
It was found that genes difficult to be disrupted via the conventional methods may be deleted by the CRISPR-Cas9 method. Gene H16_B0204 failed to be knocked out in our previous work by the conventional pK18mobsacB method, even after several attempts. However, it was successfully knocked out with an efficiency of 95.6% using the CRISPR-Cas9 technique (Table 4, Additional file 1: Figure S3).
Fragment insertion and multigene editing were not performed in this study due to the large size of the editing plasmid, but this shortcoming will be solved by using two compatible broad-host-range plasmids in our future study. Since the plasmid system we used in this research is derived from the broad-host-range plasmid pBBR1MCS2, this method may be applied to other strains or species with minor modifications.
Conclusions
In summary, an efficient and convenient genome editing method that utilizes the CRISPR-Cas9 system was developed in R. eutropha for the first time. Compared to conventional methods, this novel technique significantly increases the efficiency and decreases the time required to manipulate the genome of this important facultative chemolithoautotrophic microbe. The novel technique will facilitate more advanced researches and applications of R. eutropha for PHA production and CO2 conversion.
Abbreviations
- PHAs:
-
polyhydroxyalkanoates
- RM:
-
restriction modification
- HR:
-
homologous recombination
- NHEJ:
-
non-homologous end joining
- CBB:
-
Calvin–Benson–Bassham
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- rfp :
-
red fluorescent protein
References
Anne P, Florian FW, Frank R, Bernhard K, Heiko L, Rainer C, Thomas E, Christian E, Markus P, Edward S, et al. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat Biotechnol. 2006;24(10):1257–62.
Ryu HW, Hahn SK, Chang YK, Chang HN. Production of poly(3-hydroxybutyrate) by high cell density fed-batch culture of Alcaligenes eutrophus with phosphate limitation. Biotechnol Bioeng. 1997;55(1):29–33.
Li H, Opgenorth PH, Wernick DG, Rogers S, Wu TY, Higashide W, Malati P, Huo YX, Cho KM, Liao JC. Integrated electromicrobial conversion of CO2 to higher alcohols. Science. 2012;335:1596.
Liu C, Colón BC, Ziesack M, Silver PA, Nocera DG. Water splitting-biosynthetic system with CO2 reduction efficiencies exceeding photosynthesis. Science. 2016;352(6290):1210–3.
Müller J, MacEachran D, Burd H, Sathitsuksanoh N, Bi C, Yeh YC, Lee TS, Hillson NJ, Chhabra SR, Singer SW, Beller HR. Engineering of Ralstonia eutropha H16 for autotrophic and heterotrophic production of methyl ketones. Appl Environ Microbiol. 2014;79:4433–9.
Bi C, Su P, Müller J, Yeh YC, Chhabra SR, Beller HR, Singer SW, Hillson NJ. Development of a broad-host synthetic biology toolbox for Ralstonia eutropha and its application to engineering hydrocarbon biofuel production. Microb Cell Fact. 2013;12:107.
Grousseau E, Lu J, Gorret N, Guillouet SE, Sinskey AJ. Isopropanol production with engineered Cupriavidus necator as bioproduction platform. Appl Microbiol Biotechnol. 2014;98:4277–90.
Tanaka K, Ishizaki A. Production of poly(d-3-hydroxybutyrate) from CO2, H2, and O2 by high cell density autotrophic cultivation of Alcaligenes eutrophus. Biotechnol Bioeng. 1995;45:268–75.
Tanaka K, Ishizaki A. Production of poly-d-3-hydroxybutyric acid from carbon dioxide by a two-stage culture method employing Alcaligenes eutrophus ATCC 17697T. J Ferment Bioeng. 1994;77(4):425–7.
Park JM, Jang Y-S, Kim TY, Lee SY. Development of a gene knockout system for Ralstonia eutropha H16 based on the broad-host-range vector expressing a mobile group II intron. FEMS Microbiol Lett. 2010;309:193–200.
Park HC, Lim KJ, Park JS, Lee YH, Huh TL. High frequency transformation of Alcaligenes eutrophus producing poly-β-hydroxybutyric acid by electropotation. Biotechnol Tech. 1995;9:31–4.
Shimizu R, Chou K, Orita I, Suzuki Y, Nakamura S, Fukui T. Detection of phase-dependent transcriptomic changes and Rubisco-mediated CO2 fixation into poly (3-hydroxybutyrate) under heterotrophic condition in Ralstonia eutropha H16 based on RNA-seq and gene deletion analyses. BMC Microbiol. 2013;13:169.
Schgfer A, Tauch A, Jsger W, Kalinowski J, Thierbachb G, Piihler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutumicum. Gene. 1994;145:69–73.
Simon R, Priefer U, Puher A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol. 1983;1(9):784–91.
Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1077–84.
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31(3):233–9.
Selle K, Barrangou R. Harnessing CRISPR–Cas systems for bacterial genome editing. Trends Microbiol. 2015;23(4):225–32.
Luo ML, Leenay RT, Beisel CL. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol Bioeng. 2016;113(5):930–43.
Zhao D, Yuan S, Xiong B, Sun H, Ye L, Li J, Zhang X, Bi C. Development of a fast and easy method for Escherichia coli genome editing with CRISPR/Cas9. Microb Cell Fact. 2016;15:205.
Cobb RE, Wang Y, Zhao H. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol. 2015;4(6):723–8.
Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol. 2015;4(9):1020–9.
Oh JH, van Pijkeren JP. CRISPR–Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res. 2014;42(17):e131.
Huang H, Chai C, Li N, Rowe P, Minton NP, Yang S, Jiang W, Gu Y. CRISPR/Cas9-based efficient genome editing in Clostridium ljungdahlii, an autotrophic gas-fermenting bacterium. ACS Synth Biol. 2016;5(12):1355–61.
Altenbuchner J. Editing of the Bacillus subtilis genome by the CRISPR-Cas9 system. Appl Environ Microbiol. 2016;82(17):5421–7.
Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY. CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng. 2017;42:157–67.
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y, et al. CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun. 2017;8:15179.
Hillson NJ, Rosengarten RD, Keasling JD. j5 DNA assembly design automation software. ACS Synth Biol. 2012;1(1):14–21.
Peccoud J, Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4(5):e5553.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–5.
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166:175–6.
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz S, Joung J, Jvd Oost, Regev A, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–71.
Su T, Liu F, Gu P, Jin H, Chang Y, Wang Q, Liang Q, Qi Q. A CRISPR-Cas9 assisted non-homologous end-joining strategy for one-step engineering of bacterial genome. Sci Rep. 2016;6:37895.
Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:234–6.
Grant SGN, Jessee J, Bloom FR, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA. 1990;87:4645–9.
Raberg M, Peplinski K, Heiss S, Ehrenreich A, Voigt B, Doring C, Bomeke M, Hecker M, Steinbuchel A. Proteomic and transcriptomic elucidation of the mutant Ralstonia eutropha G+1 with regard to glucose utilization. Appl Environ Microbiol. 2011;77(6):2058–70.
Weller GR, Kysela B, Roy R, Tonkin LM, Scanlan E, Della M, Devine SK, Day JP, Wilkinson A, di Fagagna FD, et al. Identification of a DNA nonhomologous end-joining complex in bacteria. Science. 2002;297(5587):1686–9.
Authors’ contributions
CB, XZ and BX conceived the experiments. BX, ZL, LL and DZ performed the experiments. CB, XZ and BX analyzed the results and wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This research was financially supported by the Key Deployment Project of the Chinese Academy of Sciences (ZDRW-ZS-2016-3), the National High Technology Research and Development Program of China (2015AA020202) and Tianjin Key Technology R&D program of Tianjin Municipal Science and Technology Commission (14ZCZDSY00067).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Figure S1.
rfp editing identified by agarose gel electrophoresis and sequencing. Figure S2. pBBR1-Cas9-rfpF-rfpR clearance. Figure S3. Four genes edited by CRISPR-Cas9. Table S1. Putative restriction endonuclease genes in R. eutropha H16. Table S2. Genes related to putative NHEJ in R. eutropha. Table S3. List of plasmids used in this study. Table S4. List of main primers used in this study.
Additional file 2.
Profile and sequence of rfp editing plasmid pBBR1-Cas9-rfpF-rfpR.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xiong, B., Li, Z., Liu, L. et al. Genome editing of Ralstonia eutropha using an electroporation-based CRISPR-Cas9 technique. Biotechnol Biofuels 11, 172 (2018). https://doi.org/10.1186/s13068-018-1170-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-018-1170-4