
Abstract
Background
Cyanobacteria are considered potential photosynthetic microbial cell factories for biofuel and biochemical production. Ethylene, one of the most important organic chemicals, has been successfully synthesized in cyanobacteria by introducing an exogenous ethylene-forming enzyme (Efe). However, it remains challenging to significantly improve the biosynthetic efficiency of cyanobacterial ethylene. Genetic modification of transcription factors is a powerful strategy for reprogramming cellular metabolism toward target products. In cyanobacteria, nitrogen control A (NtcA), an important global transcription regulator of primary carbon/nitrogen metabolism, is expected to play a crucial role in ethylene biosynthesis.
Results
The partial deletion of ntcA (MH021) enhanced ethylene production by 23%, while ntcA overexpression (MH023) in a single-copy efe recombinant Synechocystis (XX76) reduced ethylene production by 26%. Compared to XX76, the Efe protein content increased 1.5-fold in MH021. This result may be due to the release of the negative regulation of NtcA on promoter P cpcB , which controls efe expression. Glycogen content showed a 23% reduction in MH021, and the ratio of intracellular succinate to 2-oxoglutarate (2-OG) increased 4.8-fold. In a four-copy efe recombinant strain with partially deleted ntcA and a modified tricarboxylic acid (TCA) cycle (MH043), a peak specific ethylene production rate of 2463 ± 219 μL L−1 h−1 OD −1730 was achieved, which is higher than previously reported.
Conclusions
The effects of global transcription factor NtcA on ethylene synthesis in genetically engineered Synechocystis sp. PCC 6803 were evaluated, and the partial deletion of ntcA enhanced ethylene production in both single-copy and multi-copy efe recombinant Synechocystis strains. Increased Efe expression, accelerated TCA cycling, and redirected carbon flux from glycogen probably account for this improvement. The results show great potential for improving ethylene synthetic efficiency in cyanobacteria by modulating global regulation factors.
Similar content being viewed by others

Background
An increased consumption of fossil resources has accelerated the development of alternative routes for producing renewable fuels and chemicals. Photoautotrophic cyanobacteria are promising solar biocatalysts for the production of various target products due to their genetic tractability, fast growth, and high photosynthetic efficiency [1, 2]. Ethylene, a widely used raw material in the chemical industry and in consumer markets, has already been synthesized in model strains Synechococcus elongatus PCC 7942 (hereafter called S. elongatus PCC 7942) [3] and Synechocystis sp. PCC 6803 (hereafter called S. PCC 6803) [4,5,6,7,8] by introducing an extrinsic ethylene-forming enzyme (Efe) pathway [9, 10].
Many efforts have been devoted to constructing stable and efficient ethylene-producing strains in recent years, including (1) optimizing host-preferred codons for efe expression [5, 7], (2) screening more efficient promoters [5, 6], (3) increasing copy numbers of efe [5, 7], (4) modifying ribosome binding sites upstream of efe [8], and (5) modifying related metabolic pathways [5]. In addition to genetic manipulations, a cultivation process for ethylene production was also optimized, considering factors such as light intensity, medium components, and CO2 supply [5, 7]. A volumetric ethylene production rate of 9.7 mL L−1 h−1 was achieved in our previous study by introducing three copies of efe in S. PCC 6803 with blockage of the 2-oxoglutarate decarboxylase (OGDC) and γ-aminobutyrate (GABA) shunts of the tricarboxylic acid (TCA) cycle and expression of 2-OG permease from E. coli; this is the highest cyanobacterial ethylene productivity to our knowledge [5]. Recently, it was found that S. PCC 6803 harbors a complete TCA cycle with a low flux (13% of total fixed carbon) [8, 11] and that an exogenous Efe pathway can turn the bifurcated architecture into a cyclic pathway with an enhanced carbon flux (37% of total fixed carbon) [8]. With an in-depth understanding of the plasticity of cyanobacterial metabolism, a global rebalance of cellular carbon and nitrogen metabolism is important to develop more powerful ethylene photosynthetic cell factories. Global transcription machinery engineering (gTME) [12] has already been successfully applied to regulate primary metabolism toward target products in both prokaryotic and eukaryotic systems [13]. An improvement in cyanobacterial production of polyhydroxybutyrate (PHB, product of phaAB operon) [14] and hydrogen [15] has been reported by overexpression of rre37 (encoding response regulator 37) or sigE (encoding RNA polymerase sigma factor E), suggesting the huge potential of using the gTME method to enhance target metabolite production in cyanobacteria.
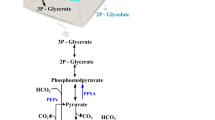
As a key precursor of ethylene biosynthesis, 2-OG is one of the most important indicators of carbon/nitrogen metabolic balance. The 2-OG pool is regulated by global transcription factor nitrogen control A (NtcA), which implies that NtcA should play a crucial role in ethylene production in cyanobacteria. The glutamine synthetase (GS, encoded by glnA)/glutamine oxoglutarate aminotransferase (GOGAT, encoded by glsF or gltB/gltD) [16, 17] cycle is a pathway that represents a connecting step between carbon and nitrogen metabolism in S. PCC 6803 (Fig. 1). GS is a key enzyme in nitrogen assimilation, and it is regulated in multiple ways by NtcA, e.g., NtcA can positively modulate the transcription of glnA [18], negatively regulate the transcription of gifA and gifB (encoding GS inactivating factors IF7 and IF17, respectively) [18, 19], and positively modulate the transcription of nitrogen stress-induced RNA 4 (NsiR4, negative regulator of IF7) [20]. In addition to genes related to nitrogen metabolism, NtcA regulates genes in a variety of other cellular processes (such as carbon metabolism and photosynthesis) as well as several sigma factors [20, 21]. Although it was reported that ntcA can only be partially deleted in S. PCC 6803 [19, 22], a non-completely segregated mutant exhibited abolished activations of ntcA on positive regulons (e.g., glnA, glnB, and glnN promoters) [22] and repressions of ntcA on negative regulons (e.g., gifA and gifB promoters) [19]. On the other hand, overexpression of ntcA leads to wide alterations in primary metabolism and a close to 90% loss of the intracellular 2-OG pool [23]. In addition, NtcA directly acts on sugar catabolism, which is indispensable to metabolism in cyanobacteria, by activating the transcription of sigE [24] and rre37 [25]. These results revealed that genetic modification of ntcA severely disturbed cellular metabolism in S. PCC 6803.

Schema illustrating the involvement of NtcA in carbon and nitrogen metabolism and ethylene production. GS, glutamine synthetase; GOGAT, glutamine oxoglutarate amidotransferase (glutamate synthase); OGDC, 2-oxoglutarate decarboxylase; SSADH, succinic semialdehyde dehydrogenase
In this study, ntcA was chosen as a genetic engineering target to evaluate its effects on ethylene production in S. PCC 6803. We constructed ntcA partial deletion mutants and ntcA overexpression mutants using S. PCC 6803 wild-type (WT) and ethylene-producing Synechocystis recombinants as the parent strains. In addition, we analyzed the Efe protein level, glycogen content, levels of the substrate and the accompanying product of the Efe-catalyzed reaction (2-OG and succinate of TCA cycle, respectively), and ethylene productivity in these strains.
Methods
Chemicals and reagents
The kits used for molecular cloning were from TransGen (China) or Takara (Japan). Taq DNA polymerase and restriction enzymes were obtained from Fermentas (Canada) or Takara (Japan). The antibodies and chemicals used for western blotting were from Sangon Biotech (China). Pure ethylene gas standard was purchased from Heli Gas Co., Ltd (China). The α-ketoglutarate assay kit was ordered from Sigma-Aldrich (USA). Unless otherwise specified, all other chemical reagents were purchased from Sigma-Aldrich (USA).
Strain and plasmid construction
All plasmids, strains, and primers used in this study are listed in Table 1, and the details are described in this section. Clones of the PCR products were confirmed by sequencing.
Plasmids used to knock out ntcA (sll1423) by insertional inactivation
The open reading frame (ORF) of ntcA and its flanking regions were amplified by PCR from S. PCC 6803 genomic DNA using primers ntcA-d1/d4. The amplified PCR fragment was cloned into pMD19-T simple (Takara), resulting in pHM001. Plasmid pHM001 was digested completely with BamHI and BglII to remove the major coding regions of ntcA (465 bp) and was blunted with T4 DNA polymerase. Then, the plasmid was ligated with the kanamycin resistance (Kmr) cassette excised from pRL446 with PvuII, resulting in pHM002. Plasmid pHM002, which harbors the ntcA upstream flanking region (ntcAU) with 125 bp of the 5′ end of ntcA, Kmr cassette, and ntcA downstream-flanking region (ntcAD) with 88 bp of the 3′ end of ntcA, was used to knock out ntcA by insertional inactivation.
Plasmids used to knock out ntcA (sll1423) by deletion
The flanking regions of ntcA (ntcAU 883 bp and ntcAD 905 bp) were amplified by PCR from S. PCC 6803 genomic DNA using primers ntcA-d1/d2 and ntcA-d3/d4, respectively. The Kmr cassette was amplified by PCR from pRL446 using primers kan-1/2. The ntcAU-Kmr-ntcAD cassette was generated by fusion PCR using primers ntcA-d1/d4 and was inserted into pMD19-T simple, resulting in pHM003. Plasmid pHM003, which harbors the ntcA upstream flanking region, Kmr cassette and ntcA downstream-flanking region, was used to knock out ntcA by deletion.
Plasmids used to overexpress ntcA (sll1423) at the phaAB loci
The coding region of ntcA was amplified by PCR from S. PCC 6803 genomic DNA using primers ntcA-x1/x2 and was inserted into the Xcm I-digested pFL-XS BioBrick “T” vector that we previously constructed [5], resulting in pHM004. The DNA fragment containing the promoter region of the cpcB gene (gentamycin resistance (Gmr)-P cpcB cassette) was excised from pXX62 with EcoRI and SpeI and was inserted into the EcoRI and XbaI sites of pHM004 to drive the expression of ntcA, resulting in pHM005. The Gmr-P cpcB -ntcA expression cassette was excised from pHM005 with EcoRI and HindIII, blunted with T4 DNA polymerase, and inserted into the NcoI site (blunted) of pKC104 [26] with the 5′ and 3′ end flanking regions of the phaAB loci, resulting in pHM006. Plasmid pHM006 was used to simultaneously overexpress ntcA and block polyhydroxybutyrate biosynthesis.
Plasmids used to knock out ntcA and express efe at the ntcA locus in multi-copy efe recombinants
The Gmr cassette was excised from pXX58 with EcoRI and HindIII, blunted with T4 DNA polymerase, and inserted into EcoRI-digested and blunted ntcA deletion plasmid pHM003, resulting in pHM008, which was used to disrupt ntcA in strain XX109 (three-copy efe). The P cpcB -efe cassette was excised from pXX55 with EcoRI and XbaI and was inserted into the EcoRI and SpeI sites of pXX58, resulting in pHM009. The Gmr-P cpcB -efe expression cassette was excised from pHM009 with EcoRI and HindIII, blunted with T4 DNA polymerase, and inserted into EcoRI-digested, T4 DNA polymerase-blunted ntcA deletion plasmid pHM003, resulting in pHM010. Plasmid pHM010 was used to introduce another efe copy at the ntcA site.
Generation of cyanobacterial transformants
Transformation of WT and mutant S. PCC 6803 was performed according to the established procedures [27]. Plasmids pHM002 and pHM003 were transformed to S. PCC 6803 to generate the ntcA disruption strains MH013 and MH015, respectively, and pHM006 was transformed to generate the ntcA overexpression strain MH017. Plasmids pHM003 and pHM006 were transformed to the one-copy efe recombinant strain XX76 to generate MH021 and MH023, respectively. Plasmid pHM008 was transformed to the three-copy efe recombinant strain XX109 to generate MH039. Plasmid pHM010 was transformed to XX109 to generate the four-copy efe Synechocystis recombinant MH043. All the Synechocystis mutants constructed here are listed in Table 1, and their genotypes were confirmed by PCR using the corresponding primers listed in Table 1.
Cultivation of E. coli and Synechocystis
Escherichia coli was grown using standard procedures in Luria–Bertani (LB) medium at 37 °C. The Synechocystis strains were grown in standard BG11 medium that was bubbled with air at 30 °C under approximately 50–100 μmol photons m−2 s−1 illumination of white light.
For BG11 solid medium, the culture was supplemented with 1.4% agar, 8 mM TES–NaOH (pH 8.2), and 0.3% Na2S2O3. The culture was supplemented with appropriate antibiotics when necessary (kanamycin, 25 μg mL−1; spectinomycin, 10 μg mL−1; chloramphenicol, 10 μg mL−1; gentamycin, 10 μg mL−1; when two or more antibiotics were used, the concentrations of the antibiotics were reduced by half). Growth and cell densities were measured at OD730 with a T6 UV/VIS spectrophotometer (Persee, China).
Quantification PCR (qPCR) of ntcA copy numbers
The genomic DNA was purified with a Genomic DNA Purification Kit (Sangon Biotech, China). Amplification was performed using SYBR® Premix Ex Taq™ II (Takara) with gene-specific primers (Table 1) and genomic DNA of triplicate technical replicates from duplicate biological cultures. The rnpB (encoding the RNA subunit of ribonuclease P) and rrn16Sa (encoding the 16S ribosomal RNA) genes were used as reference genes. The primers used in qPCR are listed in Table 1: rnpB-f1/f2, with a product size of 246 bp; rrn16Sa-f1/f2, with a product size of 266 bp; and ntcA-f1/f2, with a product size of 242 bp. The final PCR mixture contained primers at a final concentration of 625 nM in a total volume of 20 μL. The two-step cycling conditions were 10 min at 95 °C and 45 cycles at 95 °C for 20 s and 60 °C for 1 min. The relative genomic DNA copy numbers were determined with LightCycler 480 software according to the 2−ΔΔCT method [28]. To correctly calculate the relative genomic DNA copy number, PCR efficiency was determined by a dilution series of the genomic DNA.
Ethylene production assay
Ethylene production was determined as described in our previous report [5]. The quantitative analysis of ethylene production was performed with the help of an ethylene calibration curve (Additional file 1: Figure S1).
Spectrophotometric determination of pigment contents
The Synechocystis strains were inoculated at OD730 ~ 0.1 and were grown photoautotrophically. An appropriate aliquot of culture was centrifuged at the indicated times (the 2nd, 3rd, and 4th days, respectively) and was resuspended in fresh BG11 to OD730 ~ 0.5. Then the whole-cell absorbance spectra (550–750 nm) were recorded on a Beckman Coulter DU-800 UV/VIS spectrophotometer. The pigment contents were calculated based on the absorbance maxima as follows: 630 nm for phycocyanin (PC) and 680 nm for chlorophyll a (Chl a).
Protein extraction and western blotting analysis
Fifty milliliters of cell culture grown for 4 days (OD730 ~ 1.0) was centrifuged, resuspended in 500 μL of 20 mM phosphate buffer (Na2HPO4 and NaH2PO4, pH 7.4), and disrupted with glass beads (0.25–0.30 mm) by vigorous vortexing (45 min in cycles of mixing for 40 s and cooling for 20 s). The supernatant was collected after centrifugation for western blotting analysis, which was performed as described previously [5].
Organic acid measurement
Determination of intracellular succinate
Intracellular succinate was extracted as previously described [8], with minor revisions. Ten milliliters of cell culture grown for 4 days (OD730 ~ 1.0) was filtered onto a sterile nitrocellulose membrane (0.45 μm, Jinteng, China). Then the pellets were scraped from the filter membrane into cold (−80 °C) methanol. After the pellets were thoroughly mixed with chloroform and centrifugation, the supernatant was collected and evaporated by vacuum centrifugation. Next, the extract was re-dissolved in 200 μL of 50% methanol:water (V/V) and was detected by Agilent 1200 High-Performance Liquid Chromatography (USA) using an Aminex HPX-87-H column (300 × 7.8 mm) detected at 210 nm. The column was operated at 60 °C with a mobile phase of 5 mM H2SO4 at a flow rate of 0.5 mL min−1.
Determination of intracellular 2-OG
Intracellular 2-OG was extracted as previously described [18], with minor revisions. Fifty milliliters of cell culture grown for 4 days (OD730 ~ 1.0) was harvested by centrifuging at 10,000g for 5 min at 4 °C. The pellets were resuspended in 500 μL of cold 0.3 M HClO4 and were incubated for 15 min on ice. The resulting lysates were centrifuged at 15,000g for 15 min at 4 °C. Then the supernatant was neutralized by the addition of 37.5 μL of cold 2 M K2CO3. The KClO4 precipitate was removed by centrifugation, and the supernatant containing 2-OG was stored at −80 °C for analysis. The intracellular 2-OG concentration was determined using the α-ketoglutarate assay kit (Sigma-Aldrich, USA).
Glycogen measurement
Glycogen content was measured as described previously [29], with minor modification. To isolate glycogen, a certain amount of cells, equivalent to 10 OD730 units of Synechocystis culture, was collected and rinsed twice with 1 mL of deionized water. After centrifugation, the cells were suspended in 30% (W/V) KOH and were incubated at 95 °C for 2 h. Glycogen was then precipitated by the addition of ice-cold ethanol to a final concentration of 70–75% (V/V) and was subsequently incubated at −20 °C overnight. After centrifugation, the glycogen pellet was washed twice with 70 and 100% (V/V) ethanol and was dried by vacuum centrifugation at 65 °C. The isolated glycogen was resuspended in 100 mM sodium acetate (pH 4.5) and was digested to glucose by glucosidase at 65 °C for 2 h. Soluble glucose from the digests was determined using an SBA-40c biosensor analyzer (Shandong Academy of Sciences, China) equipped with a glucose oxidase-immobilized membrane. To determine dry cell weight (DCW), cells were filtered through sterile nitrocellulose membranes (0.45 μm, Jinteng, China), washed twice with 1 mL of deionized water, and dried at 105 °C for 24 h. Then the content of glycogen was calculated based on the weight of the dried cells equivalent to those were used for glycogen determination.
Results
Inactivation and overexpression of ntcA in S. PCC 6803 and ethylene-producing recombinants
With the aim of evaluating the effect of NtcA on ethylene production, we first inactivated the transcription factor in S. PCC 6803. Two strategies, insertional inactivation and deletion, were employed, and the resultant mutants were denoted as MH013 (Additional file 1: Figure S2) and MH015 (Fig. 2a), respectively. Unlike ntcA of S. elongatus PCC 7942 and Anabaena sp. PCC 7120, which can be completely inactivated (using ammonium instead of NaNO3 in BG11 medium, hereafter referred to as BG11A) [30, 31], we found that both MH013 (ntcA insertional inactivation) (Additional file 1: Figure S2) and MH015 (ntcA deletion) (Fig. 2a) were only partially inactivated. These findings are consistent with previous reports that NtcA is essential for S. PCC 6803 and cannot be fully knocked out when cultured in BG11 or BG11A medium [19, 22]. Next, we constructed MH021 (Additional file 1: Figure S3a, b) by deleting ntcA in XX76 (single-copy efe). For segregation-level analysis, qPCR was performed to measure the relative gene copy number of ntcA in MH015 (ntcA deletion) and MH021 (ntcA deletion in single-copy efe). Our results indicated that MH015 and MH021 exhibited approximately 51 and 34% loss of nctA WT copies relative to their starting strains, S. PCC 6803 and XX76, respectively (Fig. 2c).

Inactivation and overexpression of ntcA in S. PCC 6803 WT and ethylene-producing recombinant. a Schematic representation of the deletion of ntcA. The open reading frame (ORF) of ntcA (sll1423) was replaced by a kanamycin resistance (Kmr) cassette through homologous recombination with plasmid pHM003. DNA fragments were amplified by PCR and were analyzed by agarose gel electrophoresis, showing the partial segregation of ntcA in the mutant strains. PCR products from S. PCC 6803 were generated using the indicated primer pairs. Primer sequences are listed in Table 1. Lanes were loaded with PCR products that were generated with genomic DNA from the indicated strain as a template. The sizes of the PCR products are indicated on the right. b Schematic representation of the overexpression of ntcA. The Gmr-P cpcB -ntcA expression cassette was inserted into the phaAB loci through homologous recombination with plasmid pHM006. DNA fragments were amplified by PCR and were analyzed by agarose gel electrophoresis, which showed the complete replacement of phaAB by the Gmr-P cpcB -ntcA expression cassette. PCR products from S. PCC 6803 were generated using the indicated primer pairs. Primer sequences are listed in Table 1. Lanes were loaded with the PCR products that were generated with genomic DNA from the indicated strains as templates. The sizes of the PCR products are indicated on the right. c Quantitative PCR results of ntcA deletion and overexpression mutants. The copy numbers of ntcA were measured through qPCR. The reference genes for S. PCC 6803 were rnpB and the 16S rRNA gene. The relative ratios of gene copy numbers of ntcA were quantified in the WT strain, ethylene producer XX76, and ntcA mutant strains (MH015, MH021, MH017, and MH023). Data represent the means ± standard deviations from three independent experiments
We also overexpressed ntcA under the control of the P cpcB promoter on the phaAB loci in S. PCC 6803 and XX76, denoted as MH017 (ntcA overexpression) and MH023 (ntcA overexpression in single-copy efe), respectively (Fig. 2b, Additional file 1: Figure S3a, b). Successful integration of the Gmr-P cpcB -ntcA cassette was achieved, and qPCR revealed that the relative gene copy number of ntcA increased by more than twofold in both MH017 and MH023 compared to their respective starting strains (Fig. 2c).
Moreover, we found that the deletion and overexpression mutants were both genetically stable with a similar segregation level after several rounds of subculture.
Ethylene production was enhanced by ntcA inactivation and was reduced by ntcA overexpression
After the generation of ntcA deletion and overexpression mutants, specific growth rates were evaluated in standard BG11 medium. As shown in Fig. 3a, the two starting strains, S. PCC 6803 and XX76 (single-copy efe), showed similar specific growth rates. However, ntcA partial deletion mutant MH015 and ntcA overexpression strain MH017 both showed restrained growth (Fig. 3b). Intriguingly, the specific growth rates of the ethylene producer (MH021) with ntcA partial deletion and the ntcA overexpression recombinant (MH023) recovered in comparison to the growth rates of MH015 and MH017, respectively. Strain MH021 even exhibited a similar specific growth rate to those of S. PCC 6803 and XX76 (Fig. 3a).

Effect of ntcA on cell growth and ethylene productivity. a Specific growth rate of S. PCC 6803 WT, XX76 (single-copy efe), MH015 (ntcA deletion), MH017 (ntcA overexpression), MH021 (ntcA deletion in single-copy efe), and MH023 (ntcA overexpression in single-copy efe). b Growth curves. c Volumetric ethylene production. d Specific ethylene productivity. Data represent the means ± standard deviations from three independent experiments
Ethylene production was also monitored, and the results indicated that all ethylene producers reached their peak production in terms of volumetric ethylene production in the first 4 days (Fig. 3c). Ethylene production in the ntcA overexpression recombinant (MH023) was maximally reduced by 26% compared to that of the baseline strain XX76 (Fig. 3c). In contrast, volumetric ethylene production in the ntcA partial deletion mutant (MH021) was maximally increased by 23% compared to that of XX76 (Fig. 3c). Regarding specific ethylene production, MH021 reached a peak value of 1572 ± 77 μL L−1 h−1 OD −1730 , which was 1.7-fold higher than that of XX76. However, MH023 (ntcA overexpression in single-copy efe) generally showed slightly reduced or unchanged specific ethylene production, despite its 7% lower specific growth rate (Fig. 3a, d). Thus, our results indicate that the deficiency of ntcA is an effective strategy for enhancing ethylene production in S. PCC 6803.
Deficiency of ntcA enhanced the expression of the P cpcB -controlled exogenous efe gene
To explore why the inactivation of ntcA positively affected ethylene production, we first detected the level of Efe in recombinant Synechocystis. From western blotting and subsequent quantitative analysis, we found that strain MH021 (ntcA deletion in single-copy efe) produced 1.5-fold more Efe protein than the starting strain, XX76 (single-copy efe) (Fig. 4a–c). This alteration was in accordance with the 1.7-fold increase in peak specific ethylene production of MH021 (Fig. 4d). In the ntcA overexpression producer MH023, both the Efe level and the peak specific ethylene production were similar to those of XX76.

Efe level and the highest ethylene productivity. a SDS-PAGE showed that equal amounts of proteins were loaded in each lane. b Western blotting detection of Efe in single-copy efe recombinant Synechocystis strains. S. PCC 6803 WT was used as a negative control. c Quantitative analysis of Efe level from the results of two repeats. d Maximal specific productivity. Data in d represent the means ± standard deviations from three independent experiments
As the efe gene was under the control of P cpcB in this study, the increased level of Efe indicated a possible direct or indirect transcriptional regulation of NtcA on P cpcB . Hence, we set up a sequence analysis of P cpcB and found that P cpcB might harbor a putative NtcA binding site (Additional file 1: Figure S4; Additional file 2: Table S1). The 34-bp region containing the putative NtcA binding site is directly upstream of the P cpc560 identified by Zhou et al. [32]. In addition, a broader investigation of P cpcB from other cyanobacterial species indicated a widespread existence of putative NtcA recognition sites (Additional file 2: Table S1). Furthermore, we measured the PC content and PC/Chl a ratio of the Synechocystis strains with whole-cell absorbance spectra. MH015 (ntcA deletion) and MH021 (ntcA deletion in single-copy efe) showed increases at most of the investigated culture stages, while MH017 (ntcA overexpression) and MH023 (ntcA overexpression in single-copy efe) showed decreases in PC content and PC/Chl a ratio relative to S. PCC 6803 and XX76, respectively (Additional file 1: Figure S5a, b). Thus, improved cellular Efe levels and ethylene production are partially related to the release of negative regulation of NtcA on the P cpcB -efe cassette.
NtcA modification mobilized cellular glycogen for ethylene production
Since NtcA was found to regulate sugar metabolism, the glycogen content of S. PCC 6803 and the recombinant strains were determined to explore the relationship between cellular glycogen and ethylene production. We found that the glycogen content of S. PCC 6803 ranged from 6 to 7.4% of the DCW in the period corresponding to robust ethylene synthesis in the recombinant strains (Fig. 5). Compared to S. PCC 6803, the single-copy efe strain XX76 showed a similar pattern of glycogen accumulation. The deletion of ntcA (MH015) caused a 20% loss of glycogen on the 2nd day but rapid glycogen accumulation relative to S. PCC 6803, and 20% more glycogen content was observed on the 4th day (Fig. 5). As expected, MH021 (ntcA deletion in single-copy efe) showed a 14–23% reduction in glycogen content compared to XX76 (Fig. 5). In terms of the overexpression of ntcA, both MH017 (ntcA overexpression) and MH023 (ntcA overexpression in single-copy efe) showed significantly lower glycogen contents relative to their parent strains, S. PCC 6803 and XX76, respectively. Interestingly, the glycogen content of MH023 was much higher than that of MH017 by the 2nd day, and the glycogen accumulation trends in MH017 and MH023 were reversed; the glycogen level in MH017 was lower at the beginning and increased later, while the glycogen level in MH023 was higher at the beginning and decreased later (Fig. 5). The results of glycogen content analysis revealed that the introduction of the Efe pathway did not change the cellular glycogen level. However, the glycogen level was changed by simultaneously introducing the Efe pathway and modifying NtcA. NtcA deficiency might mobilize cellular glycogen for ethylene synthesis considering the enhanced ethylene production in MH021.

Glycogen levels of S. PCC 6803 WT, MH015 (ntcA deletion), MH017 (ntcA overexpression), XX76 (single-copy efe), MH021 (ntcA deletion in single-copy efe), and MH023 (ntcA overexpression in single-copy efe). Data represent the means ± standard deviations from three independent experiments
NtcA inactivation affected the intracellular pool of succinate and 2-OG
Succinate and 2-OG, two intermediates of the TCA cycle, are directly related to the Efe reaction. To investigate the metabolic effect of ntcA deletion on the unusual TCA cycle, the intracellular levels of succinate and 2-OG in the Synechocystis strains were monitored on the 4th day. We found that the pool of 2-OG (1.62 ± 0.26 nmol mL−1 OD −1730 ) was over 6 times larger than that of succinate (0.22 ± 0.05 nmol mL−1 OD −1730 ) under standard culture conditions in S. PCC 6803 WT (Fig. 6). These results confirmed the reported metabolic gap between these two metabolites, even with functional OGDC and GABA shunts [11]. In ethylene producer XX76 (single-copy efe), the levels of succinate and 2-OG both increased, and the ratio of succinate to 2-OG (RatioSuc/2-OG) increased by 73% compared to that of S. PCC 6803 (Fig. 6), indicating enhanced TCA cycling, as previously noted [8]. In MH015 (ntcA deletion), the level of intracellular 2-OG decreased while succinate increased, and the RatioSuc/2-OG increased threefold compared to that of S. PCC 6803. Deleting ntcA in XX76 (MH021) caused a dramatic enlargement of the intracellular pool of succinate and a slight increase in 2-OG, with the RatioSuc/2-OG being enlarged by 9-, 4.8-, and 1.5-fold compared to that of S. PCC 6803, XX76, and MH015, respectively. These results indicated that the flux from 2-OG to succinate dramatically increased by the introduction of Efe when coupled with the partial deletion of ntcA and that this genetic modification reprogrammed the metabolic network of the TCA cycle.

Intracellular levels of succinate and 2-OG in S. PCC 6803 WT, MH015 (ntcA deletion), MH017 (ntcA overexpression), XX76 (single-copy efe), MH021 (ntcA deletion in single-copy efe), and MH023 (ntcA overexpression in single-copy efe). Data represent the means ± standard deviations from at least two independent experiments
NtcA inactivation in combination with TCA cycle modification enhanced cellular ethylene production in recombinant Synechocystis
In our previous study, we constructed Synechocystis recombinant strain XX109 with three efe copies. In this recombinant, the ogdc (sll1981) and ssadh (slr0370) genes were inactivated by inserting the Kmr-P cpcB -efe and Cmr-P cpcB -efe cassettes, respectively [5]. Theoretically, this strain should get an Efe-dominated TCA cycle with primarily cyclic architecture because the OGDC and GABA pathways were both blocked.
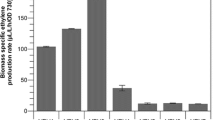
To further improve ethylene production, we constructed strains MH039 and MH043 by replacing ntcA with a gentamycin cassette (Gmr) or a Gmr-P cpcB -efe cassette, respectively, based on the three-copy efe strain XX109 (Additional file 1: Figure S3c, d). Quantitative PCR analysis revealed that MH039 (ntcA deletion in three-copy efe) lost approximately 56% and that MH043 (ntcA deletion in four-copy efe) lost 30% of the nctA WT copies (Additional file 1: Figure S6). Compared to XX109, MH039 showed a similar specific growth rate, and MH043 showed a slightly improved specific growth rate (Fig. 7a). In addition, specific ethylene production was enhanced in these strains (Fig. 7b). A maximal specific ethylene production rate of 2463 ± 219 μL L−1 h−1 OD −1730 was achieved in MH043 (Fig. 7b), which was higher than the previously reported rate.

Effect of the deletion of ntcA on ethylene productivity and Efe level in multi-copy efe recombinant Synechocystis strains (XX109, MH039, and MH043). a Specific growth rate. b Maximal specific productivity. c Western blotting detection of Efe. d Quantitative analysis of Efe level from the results of two repeats. Data in a and b represent the means ± standard deviations from three independent experiments
The Efe level (Fig. 7c, d) and PC content (PC/Chl a ratio as well) (Additional file 1: Figure S5c, d) both increased in MH039 (ntcA deletion in three-copy efe) and MH043 (ntcA deletion in four-copy efe) compared to that in XX109. The results confirmed the possible negative regulation of NtcA on the P cpcB -efe cassette, even when the OGDC and GABA shunts of the TCA cycle were blocked. However, despite harboring four efe copies, MH043 exhibited a similar Efe level to that of MH039.
Discussion
Here, we enhanced ethylene production by the partial deletion of ntcA and analyzed the corresponding physiological and metabolic effects in Synechocystis strains. NtcA was the target for genetic modification based on its essential role in integrating the signals of nitrogen and carbon balance (Fig. 1). We found that ntcA deletion and overexpression mutants both showed a stable genotype (Fig. 2; Additional file 1: Figures S3, S6). Compared to previous studies, the lower segregation level of ntcA observed here might be due to different nitrogen resources. Furthermore, cell growth of the non-segregated ntcA mutant was not seriously restrained. Recently, it has been found that ethylene can modulate several important aspects of Synechocystis biology by the functional ethylene receptor (SynEtr1, encoded by slr1212) [33]. NtcA has also been shown to control transcription at a genome-wide level and widely affect the primary metabolism of S. PCC 6803. Consequently, the recovered growth of MH021 (ntcA deletion in single-copy efe) and MH023 (ntcA overexpression in single-copy efe) might relate to the combined effect of ethylene modulation and ntcA modification. On the other hand, the stable genotype, in combination with the recovered physiological feature, demonstrated the feasibility of improving ethylene production in S. PCC 6803.
Previous studies have revealed that the modification of transcription regulators can cause alterations in primary metabolism and can enhance the production of native biological products in S. PCC 6803 [25, 34, 35]. Thus, it is reasonable that ethylene production was affected by the deletion or overexpression of transcription factor NtcA. We first suspected that the enhanced ethylene production in MH021 (ntcA deletion in single-copy efe) (Fig. 3c, d) was related to only the cellular metabolic reprogramming imposed by ntcA deletion. However, Efe level detection (Fig. 4 and 7) and PC content analysis (Additional file 1: Figure S5) implied the possible native regulation of NtcA on P cpcB . Moreover, we found a putative NtcA binding site between 560 bp and 594 bp upstream of the initiation codon of the cpcB gene (Additional file 1: Figure S4; Additional file 2: Table S1). In fact, it has been reported that the transcription factor binding sites (TFBSs) on P cpcB are crucial for its promoter strength in S. PCC 6803; however, the NtcA site was not included (only 560 bp upstream of the initiation codon was analyzed) [32]. NtcA belongs to the CRP (cAMP receptor protein) family and binds as a homodimer to the conserved palindromic sequence GTAN8TAC [20, 21]. As NtcA promoters were thought to serve as regulatory points to coordinate photosynthesis and nitrogen uptake [21], the genes involved in various stages of photosynthesis (such as allophycocyanin apc, PC cpc) were supposed to be new members of the NtcA regulons [21]. The possible regulation of NtcA on the P cpcB -efe cassette found in this study is a good example of this hypothesis.
Glycogen is the main carbohydrate storage compound that can divert a significant portion of fixed carbon in S. PCC 6803 [36, 37]. A rapid accumulation of glycogen usually occurs when cyanobacteria face stress conditions [37]. In this study, we observed rapid glycogen accumulation in the ntcA mutant MH015, but a lower level of glycogen (maximal loss of 23%) and a slow glycogen accumulation rate were observed in MH021 (ntcA deletion in single-copy efe) under standard culture conditions (Fig. 5). Thus, it is plausible that the Efe reaction can draw many carbons from glycogen when the ntcA transcription factor is deleted. Lower glycogen content results from enhanced catabolism and/or attenuated anabolism. It was reported that the key genes involved in glycogen catabolic pathways, such as glycolysis and the oxidative pentose phosphate (OPP) pathway, were regulated by several transcription regulators and sigma factors, including NtcA [22], Rre37 [25, 35, 38], and SigE [25, 34, 35]. The coding genes of such regulators (including NtcA itself) were among the regulons of NtcA [21, 39, 40]. Thus, under the regulation of NtcA, the intermediates involved in glycogen catabolic pathways can be converted to acetyl-CoA and malate, which are inputted into the TCA cycle. In agreement, a sharply increased RatioSuc/2-OG was found in MH021 (Fig. 6), which indicated a remodeled metabolic network for the TCA cycle. The results were also supported by identification of the predominantly cyclic architecture of the TCA cycle in recombinant Synechocystis with an efficient Efe pathway. In addition, the positive regulation of NtcA on the GS-GOGAT cycle of nitrogen metabolism might also bring carbon flux to the Efe reaction.
As previously found, ethylene production in cyanobacteria is a complex process involving multiple genetic and environmental factors [4,5,6,7,8]. The highest specific ethylene production in the multi-copy efe strain MH043 indicated robust metabolism in the host cells with blockage of both the OGDC and GABA pathways. Furthermore, carbon flux redistribution probably occurred, considering the alterations in glycogen content and organic acid pools. The four-copy efe strain MH043 showed a similar Efe level to that of MH039, which might suggest the upper limit of Efe protein expression in both strains.
Conclusions
Based on the plasticity of the cyanobacterial metabolic network and inspired by the notion of gTME, the global nitrogen control transcription factor NtcA was chosen as the target for genetic modification to enhance ethylene production. We found that ntcA deficiency enhanced ethylene production in recombinant Synechocystis, resulting in reduced glycogen content and a drastic increase in the intracellular level of succinate. P cpcB might be a new member of the NtcA regulons, and it exhibited a promising application prospect as a super-strong and tunable promoter in cyanobacterial biotechnology. In combination with the deletion of ntcA and the blockage of TCA cycle bypasses, a peak specific ethylene production rate of 2463 ± 219 μL L−1 h−1 OD −1730 was achieved in multi-copy efe recombinants, which is a significant increase in ethylene productivity by photoautotrophic microorganisms. The strains developed in this study and the gTME strategy can be further exploited to construct highly efficient photoautotrophic biocatalysts for ethylene production.
Abbreviations
- Efe:
-
ethylene-forming enzyme
- gTME:
-
global transcription machinery engineering
- 2-OG:
-
2-Oxoglutarate
- TCA cycle:
-
tricarboxylic acid cycle
- OGDC:
-
2-Oxoglutarate decarboxylase
- GABA:
-
γ-Aminobutyrate
- PHB:
-
polyhydroxybutyrate
- GS:
-
glutamine synthetase
- GOGAT:
-
glutamine oxoglutarate aminotransferase
- WT:
-
wild type
- ORF:
-
open reading frame
- PCR:
-
polymerase chain reaction
- qPCR:
-
quantitative real-time PCR
- PC:
-
phycocyanin
- Chl a:
-
chlorophyll a
- DCW:
-
dry cell weight
- OD:
-
optical density
- RatioSuc/2-OG :
-
ratio of succinate to 2-OG
- TFBSs:
-
transcription factor binding sites
- RP:
-
cAMP receptor protein
- OPP:
-
oxidative pentose phosphate pathway
References
Lu X. A perspective: photosynthetic production of fatty acid-based biofuels in genetically engineered cyanobacteria. Biotechnol Adv. 2010;28:742–6.
Quintana N, Van der Kooy F, Van de Rhee MD, Voshol GP, Verpoorte R. Renewable energy from cyanobacteria: energy production optimization by metabolic pathway engineering. Appl Microbiol Biotechnol. 2011;91:471–90.
Sakai M, Ogawa T, Matsuoka M, Fukuda H. Photosynthetic conversion of carbon dioxide to ethylene by the recombinant cyanobacterium, Synechococcus sp. PCC 7942, which harbors a gene for the ethylene-forming enzyme of Pseudomonas syringae. J Ferment Bioeng. 1997;84:434–43.
Zavrel T, Knoop H, Steuer R, Jones PR, Cerveny J, Trtilek M. A quantitative evaluation of ethylene production in the recombinant cyanobacterium Synechocystis sp PCC 6803 harboring the ethylene-forming enzyme by membrane inlet mass spectrometry. Bioresour Technol. 2016;202:142–51.
Zhu T, Xie X, Li Z, Tan X, Lu X. Enhancing photosynthetic production of ethylene in genetically engineered Synechocystis sp. PCC 6803. Green Chem. 2015;17:421–34.
Guerrero F, Carbonell V, Cossu M, Correddu D, Jones PR. Ethylene synthesis and regulated expression of recombinant protein in Synechocystis sp. PCC 6803. PLoS ONE. 2012;7:e50470.
Ungerer J, Tao L, Davis M, Ghirardi M, Maness P-C, Yu J. Sustained photosynthetic conversion of CO2 to ethylene in recombinant cyanobacterium Synechocystis 6803. Energ Environ Sci. 2012;5:8998–9006.
Xiong W, Morgan JA, Ungerer J, Wang B, Maness P-C, Yu J. The plasticity of cyanobacterial metabolism supports direct CO2 conversion to ethylene. Nat Plants. 2015;1:15053.
Fukuda H, Ogawa T, Tazaki M, Nagahama K, Fujii T, Tanase S, Morino Y. Two reactions are simultaneously catalyzed by a single enzyme: the arginine-dependent simultaneous formation of two products, ethylene and succinate, from 2-oxoglutarate by an enzyme from Pseudomonas syringae. Biochem Biophys Res Commun. 1992;188:483–9.
Martinez S, Hausinger RP. Biochemical and spectroscopic characterization of the non-heme Fe(II)- and 2-oxoglutarate-dependent ethylene-forming enzyme from Pseudomonas syringae pv. phaseolicola PK2. Biochemistry. 2016;55:5989–99.
Xiong W, Brune D, Vermaas WF. The gamma-aminobutyric acid shunt contributes to closing the tricarboxylic acid cycle in Synechocystis sp. PCC 6803. Mol Microbiol. 2014;93:786–96.
Alper H, Stephanopoulos G. Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab Eng. 2007;9:258–67.
Lanza AM, Alper HS. Global strain engineering by mutant transcription factors. Methods Mol Biol. 2011;765:253–74.
Osanai T, Oikawa A, Numata K, Kuwahara A, Iijima H, Doi K, Saito K, Hirai MY. Pathway-level acceleration of glycogen catabolism by a response regulator in the cyanobacterium Synechocystis species PCC 6803. Plant Physiol. 2014;164:1831–41.
Osanai T, Kuwahara A, Iijima H, Toyooka K, Sato M, Tanaka K, Ikeuchi M, Saito K, Hirai MY. Pleiotropic effect of sigE over-expression on cell morphology, photosynthesis and hydrogen production in Synechocystis sp. PCC 6803. Plant J. 2013;76:456–65.
Navarro F, Chavez S, Candau P, Florencio FJ. Existence of two ferredoxin-glutamate synthases in the cyanobacterium Synechocystis sp. PCC 6803. Isolation and insertional inactivation of gltB and gltS genes. Plant Mol Biol. 1995;27:753–67.
Suzuki A, Knaff DB. Glutamate synthase: structural, mechanistic and regulatory properties, and role in the amino acid metabolism. Photosynth Res. 2005;83:191–217.
Muro-Pastor MI, Reyes JC, Florencio FJ. Cyanobacteria perceive nitrogen status by sensing intracellular 2-oxoglutarate levels. J Biol Chem. 2001;276:38320–8.
Garcia-Dominguez M, Reyes JC, Florencio FJ. NtcA represses transcription of gifA and gifB, genes that encode inhibitors of glutamine synthetase type I from Synechocystis sp. PCC 6803. Mol Microbiol. 2000;35:1192–201.
Klahn S, Schaal C, Georg J, Baumgartner D, Knippen G, Hagemann M, Muro-Pastor AM, Hess WR. The sRNA NsiR4 is involved in nitrogen assimilation control in cyanobacteria by targeting glutamine synthetase inactivating factor IF7. Proc Natl Acad Sci USA. 2015;112:E6243–52.
Su Z, Olman V, Mao F, Xu Y. Comparative genomics analysis of NtcA regulons in cyanobacteria: regulation of nitrogen assimilation and its coupling to photosynthesis. Nucleic Acids Res. 2005;33:5156–71.
Osanai T, Imamura S, Asayama M, Shirai M, Suzuki I, Murata N, Tanaka K. Nitrogen induction of sugar catabolic gene expression in Synechocystis sp. PCC 6803. DNA Res. 2006;13:185–95.
Osanai T, Oikawa A, Iijima H, Kuwahara A, Asayama M, Tanaka K, Ikeuchi M, Saito K, Hirai MY. Metabolomic analysis reveals rewiring of Synechocystis sp. PCC 6803 primary metabolism by ntcA overexpression. Environ Microbiol. 2014;16:3304–17.
Osanai T, Azuma M, Tanaka K. Sugar catabolism regulated by light- and nitrogen-status in the cyanobacterium Synechocystis sp PCC 6803. Photochem Photobiol Sci. 2007;6:508–14.
Azuma M, Osanai T, Hirai MY, Tanaka K. A response regulator Rre37 and an RNA polymerase sigma factor SigE represent two parallel pathways to activate sugar catabolism in a cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol. 2011;52:404–12.
Tan XM, Liang FY, Cai K, Lu XF. Application of the FLP/FRT recombination system in cyanobacteria for construction of markerless mutants. Appl Microbiol Biotechnol. 2013;97:6373–82.
Williams JGK. Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis 6803. In: LesterPacker ANG, editor. Methods enzymol, vol. 167. Cambridge: Academic Press; 1988. p. 766–78.
Lee C, Kim J, Shin SG, Hwang S. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J Biotechnol. 2006;123:273–80.
Grundel M, Scheunemann R, Lockau W, Zilliges Y. Impaired glycogen synthesis causes metabolic overflow reactions and affects stress responses in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology. 2012;158:3032–43.
Espinosa J, Forchhammer K, Contreras A. Role of the Synechococcus PCC 7942 nitrogen regulator protein PipX in NtcA-controlled processes. Microbiology. 2007;153:711–8.
Wei TF, Ramasubramanian TS, Golden JW. Anabaena sp. strain PCC 7120 ntcA gene required for growth on nitrate and heterocyst development. J Bacteriol. 1994;176:4473–82.
Zhou J, Zhang H, Meng H, Zhu Y, Bao G, Zhang Y, Li Y, Ma Y. Discovery of a super-strong promoter enables efficient production of heterologous proteins in cyanobacteria. Sci Rep. 2014;4:4500.
Lacey RF, Binder BM. Ethylene regulates the physiology of the cyanobacterium Synechocystis sp. PCC 6803 via an ethylene receptor. Plant Physiol. 2016;171:2798–809.
Osanai T, Oikawa A, Azuma M, Tanaka K, Saito K, Hirai MY, Ikeuchi M. Genetic engineering of group 2 sigma factor SigE widely activates expressions of sugar catabolic genes in Synechocystis species PCC 6803. J Biol Chem. 2011;286:30962–71.
Nakaya Y, Iijima H, Takanobu J, Watanabe A, Hirai MY, Osanai T. One day of nitrogen starvation reveals the effect of sigE and rre37 overexpression on the expression of genes related to carbon and nitrogen metabolism in Synechocystis sp. PCC 6803. J Biosci Bioeng. 2015;120:128–34.
Carrieri D, Paddock T, Maness P-C, Seibert M, Yu J. Photo-catalytic conversion of carbon dioxide to organic acids by a recombinant cyanobacterium incapable of glycogen storage. Energ Environ Sci. 2012;5:9457.
Damrow R, Maldener I, Zilliges Y. The multiple functions of common microbial carbon polymers, glycogen and PHB, during stress responses in the non-diazotrophic cyanobacterium Synechocystis sp. PCC 6803. Front Microbiol. 2016;7:966.
Tabei Y, Okada K, Tsuzuki M. Sll1330 controls the expression of glycolytic genes in Synechocystis sp. PCC 6803. Biochem Biophys Res Commun. 2007;355:1045–50.
Muro-Pastor AM, Herrero A, Flores E. Nitrogen-regulated group 2 sigma factor from Synechocystis sp. strain PCC 6803 involved in survival under nitrogen stress. J Bacteriol. 2001;183:1090–5.
Joseph A, Aikawa S, Sasaki K, Teramura H, Hasunuma T, Matsuda F, Osanai T, Hirai MY, Kondo A. Rre37 stimulates accumulation of 2-oxoglutarate and glycogen under nitrogen starvation in Synechocystis sp. PCC 6803. FEBS Lett. 2014;588:466–71.
Authors’ contributions
HM carried out strain constructions and evaluations, participated in data analysis, and helped to draft the manuscript. XX carried out strain constructions and evaluations, participated in data analysis, and helped to draft the manuscript. TZ designed the research, analyzed the data, and drafted the manuscript. XL supervised the project, participated in data analysis, and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Prof. Xudong Xu for kindly providing plasmid pRL446 and strain S. PCC 6803.
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
The data supporting our findings can be found in this manuscript and in the additional files provided. The authors are willing to provide any additional data and materials related to this research that may be requested for research purposes.
Consent for publication
The authors agree to publish in the journal.
Funding
This work was supported by the Excellent Youth Award of Shandong Natural Science Foundation (JQ201306 to X. Lu), the Shandong Taishan Scholarship (X. Lu), and the Qingdao Science and Technology Innovation Leader Foundation [15-10-3-15-(31)-zch to X. Lu].
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
13068_2017_832_MOESM1_ESM.docx
Additional file 1: Figure S1. Ethylene calibration curve. Figure S2. Schematic representation of the construction of MH013 (ntcA insertional inactivation). Partial coding region of ntcA on the genome of S. PCC 6803 was replaced by a kanamycin resistance (Kmr) cassette through homologous recombination with plasmid pHM002. DNA fragments amplified by PCR and analyzed by agarose gel electrophoresis showing the partial segregation of ntcA in the mutant strain. PCR products from WT and mutants S. PCC 6803 were generated using the indicated primer pairs. Primer sequences are listed in Table 1. Lanes were loaded with PCR products that were generated with genomic DNA from the indicated strains as template. The sizes of the PCR products are indicated on the right. Figure S3. Construction of MH021 (ntcA deletion in single-copy efe), MH023 (ntcA overexpression in single-copy efe), MH039 (ntcA deletion in three-copy efe) and MH043 (ntcA deletion in four-copy efe). (a) and (c) Schematic representation. (b) and (d) Genotypic confirmation by PCR and agarose gel electrophoresis. PCR products from WT and mutants S. PCC 6803 were generated using the indicated primer pairs. Primer sequences are listed in Table 1. Lanes were loaded with PCR products that were generated with genomic DNA from the indicated strains as templates. The DNA marker with relevant sizes (in bp) was indicated on the left. Figure S4. Schematic representation of putative NtcA site on P cpcB of S. PCC 6803. Figure S5. Effect of ntcA deletion or overexpression on pigment contents. (a) and (c) Absorbance spectra for the 2nd, 3rd and 4th day. Peaks of PC and Chl a are indicated. Groups of spectra were shifted along the y-axis for better viewing. (b) and (d) PC/Chl a ratios as a function of time. XX76 (single-copy efe), MH015 (ntcA deletion), MH017 (ntcA overexpression), MH021 (ntcA deletion in single-copy efe), MH023 (ntcA overexpression in single-copy efe), XX109 (three-copy efe), MH039 (ntcA deletion in three-copy efe), MH043 (ntcA deletion in four-copy efe). Figure S6. Quantitative PCR results of ntcA deletion in MH039 (ntcA deletion in three-copy efe) and MH043 (ntcA deletion in four-copy efe). The copy numbers of ntcA were measured through qPCR. The reference genes for S. PCC 6803 were rnpB and the 16S rRNA gene. The relative ratio of gene copy numbers of ntcA were quantified in XX109 (three-copy efe), MH039 and MH043. Data represent the means ± standard deviations from at least two independent experiments.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mo, H., Xie, X., Zhu, T. et al. Effects of global transcription factor NtcA on photosynthetic production of ethylene in recombinant Synechocystis sp. PCC 6803. Biotechnol Biofuels 10, 145 (2017). https://doi.org/10.1186/s13068-017-0832-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-017-0832-y