
Abstract
Background
Cellulose, an abundant and renewable polysaccharides, constitutes the largest resource for bioconversion of biofuels. Plant polysaccharides hydrolysis is catalyzed by cellulases, which include endoglucanases, exoglucanases, and β-glucosidases. Converting cellulose and hemicellulose to short chains of oligosaccharides by endo-/exoglucanases is the key step for biofuel transformation. Intriguingly, β-glucanases with transglycosylation activity not only can relieve product inhibition of glucan hydrolysis but also has potential application as biocatalysts for functional materials.
Results
Here, a metagenomic fosmid library was constructed from a paddy soil for cellulase screening. One purified clone showing carboxymethylcellulase activity was isolated, and the complete β-glucanase gene (umcel9y-1) was cloned and overexpressed in Escherichia coli. Phylogenetic analysis indicated that β-glucanase Umcel9y-1 belonged to the theme C of glycoside hydrolase family 9. Amino acids sequence showed 58.4 % similarity between Umcel9y-1 and its closest characterized reference, cellulase Cel01. Biological characterization showed that Umcel9y-1 was an efficient endoglucanase and also exhibited high activities of exoglucanase and transglycosylation. The transglycosylation products of Umcel9y-1 including sophorose, laminaribiose, and gentiobiose, and transglycosylation was detected under all activated conditions. The order of catalytic efficiency for polysaccharides, cellooligosaccharides, and aryl-β-glycosides was p-nitrophenol-D-cellobioside, barley glucan, cellopentaose, cellotetraose, cellotriose, hydroxyethylcellulose, cellohexose, laminarin, and carboxymethylcellulose, respectively. The barley glucan was the optimal polysaccharides for Umcel9y-1 with K m and K cat/K m values of 13.700 mM and 239.152 s−1 mM−1, respectively.
Conclusion
Biological characterizations of recombinant Umcel9y-1 showed that the versatile β-glucanase had efficient endoglucanase activity to barley glucan and also exhibited high activities of exoglucanase and transglycosylation. The optimum conditions of recombinant Umcel9y-1 was pH 6.5–7.0 at 37 °C with predominant halotolerance and high-thermal stability. These results indicate that the novel metagenomic-derived β-glucanase may be a potent candidate for industrial applications.
Similar content being viewed by others

Background
Cellulose fibers are composed of bundles of linear polymers of D-glucose linked by β-1, 4-glycosyl bonds [1]. Cellulose is the most abundant and renewable polysaccharide and constitutes one-third of all existing plant cell-wall material, which is the largest potential resource for bioconversion of ethanol and other biofuels, feedstock chemicals, and pharmaceuticals [2–4]. Cellulases catalyze cellulolysis through hydrolyzing β-1, 4-glycosidic bonds in cellulose, and mainly consisted of three types of enzymes: endoglucanases (EC3.2.1.4), exoglucanase (or cellobiohydrolase, EC3.2.1.91), and β-glucosidase (EC3.2.1.21) [5]. Endoglucanases randomly hydrolyze internal β-1, 4-glycosidic bonds to decrease the length of cellulose chain, exoglucanases split off cellobiose from the shortened cellulose chains, and β-glucosidases degrade cellobiose to glucose [5].
Nowadays, some cellulases from fungi and bacteria have been technically employed in cellulose biotransformation, including polysaccharide degradation, textile processing, juice and wine fermentation, animal feed and fuel production, and other industries [6–8]. Recently, cellulases (including β-glucanase) with transglycosylation activity are attracted as substitute of glycosyltransferases for the synthesis of stereo- and regiospecific glycosides (or oligosaccharides) which are considered as functional materials, nutraceuticals, or pharmaceuticals [9, 10]. Previously, glycosyltransferases were the primary choice as the biocatalyst for glycosidic synthesis, but the industrial application processes come to a standstill for years. The major difficulties are the expensive nucleotide sugar precursors, narrow substrate specificity, and low enzyme availability. Comparatively, cellulases or β-glucanases are more abundant, commercially available, and exhibit a broad acceptor-substrate specificity with simple substrates [10, 11]. Biochemical characterizations of cellulases require intensive efforts but are likely to generate new opportunities for the use of renewable resource as biofuels, food and drug additives, and other agricultural products [6–12].
Metagenomic approach as a molecular biological access to retrieve uncultured microbial resource has been widely employed to isolate novel genes and prospect for important biocatalysts [13]. Under the development of metagenomic technology, considerable functional bioenzymes have been isolated from uncultured microorganisms in soils and other environmental samples. The cellulases are one of the most important group of functional bioenzymes targeted by metagenomic technology [10, 13–16].
The need for more suitable biocatalysts has reinforced the scientific research for novel cellulase, especially from the uncultured microbial resources. In this study, a metagenomic fosmid library was constructed from a paddy soil and a novel β-glucanase Umcel9y-1 was successfully screened. The full length gene of Umcel9y-1 was cloned and overexpressed in Escherichia coli BL21 (DE3). Biological study revealed that Umcel9y-1 was an efficient endoglucanase with versatile activities (i.e., exoglucanase and transglycosylation), and the potential industrial values of Umcel9y-1 were evaluated.
Methods
Sample collection and metagenomic library construction
Paddy soil was collected from Liaoning province (41°07′03”N 122°03′09”E) of China in October 2010. The total microbial DNA was extracted using SoilMaster™ DNA Extraction Kit (Epicentre, Madison, WI) according to the manufacturer instructions. The metagenomic library was constructed using CopyControl™ Fosmid Library Production Kit (Epicentre, Madison, WI) according to the manufacturer’s instructions. DNA products were examined by agarose electrophoresis, and the fragments within the sizes of 25–35 kb were recovered for an end-repair reaction, and ligated into pEpiFOS-1 fosmid vector prepared in the kit. In vitro packaging was performed with a MaxPlax lambda packaging extract kit (Epicentre, Madison, WI). Finally, the products were infected into E. coli EPI 300 (Epicentre, Madison, WI). The quality of the library was tested by NotI (Promega, Madison, WI) digestion of the prepared fosmids plasmids, and colony counting was carried out by automatic bacteria counter (Shineso Science & Technology Co., LTD, China).
Cellulase screening and gene cloning
The metagenomic library was screened using the substrate carboxymethylcellulose (CMC, Sigma, St. Louis, MO, USA) for CMC hydrolysis genes. The transformed E. coli EPI 300 was inoculated on CMCase screening agar containing 1.0 % tryptone, 0.5 % yeast extract, 1.0 % NaCl, 0.5 % CMC, 100 µg/mL chloramphenicol, and 0.2 mM isopropy-β-D-thiogalactoside (IPTG). The screening agar was incubated at 37 °C for 24 h. After incubation, the plates were stained with 0.2 % Congo-red for 20 min [17]. The CMCase positive clones were screened out by a hydrolysis zone around the bacterial colony. CMCase positive plasmids were enriched with fosmid autoinduction solution and purified by FosmidMAX™ DNA purification Kit (Epicentre, Madison, WI). For subcloning, the plasmid DNA of CMCase positive clone was digested by NotI, then recovered for further digestion by Sau3AI (Promega, Madison, WI), and ligated into a pBlueScript SK(+) vector (Stratagene, La Jolla, CA). The subclone of CMCase positive transformant was screened further, and the insert DNA sequencing was performed on an Applied Biosystems DNA sequencer, model ABI PRISM 377 (Invitrogen, Shanghai).
Sequence analysis and secondary structure prediction
Nucleotide sequence translation, and the possible ORF, signal peptide, theoretical pI, and Mw predictions were performed at online programs (ExPASy and SignalP 3.0) [18, 19]. The conserved region analysis and comparison of sequence identity to the related cellulase genes were performed by BlastX (http://www.ncbi.nlm.nih.gov). Sequences of complete cellulase proteins retrieved from the GenBank database were aligned using the CLUSTAL_X software and the alignments were corrected manually [20]. Phylogenetic analysis was performed by neighbor-joining algorithm using MEGA v5.0, and the topology of the phylogenetic tree was assessed by the bootstrap analysis based on 1000 replications. The circular dichroism (CD) spectroscopy was carried out to estimate the secondary structure of the purified recombinant protein. The CD spectra were determined at room temperature with a JASCO J-810 spectrometer (JASCO Japan), and the protein concentration was 10 µM in Tris–HCl buffer [21]. The secondary structure of the protein was predicted by the online program Protein Homology/analog Y Recognition Engine v2.0 and the online tool K2D2 [21, 22].
Umcel9y-1 gene amplification, overexpression, and purification
Umcel9y-1 gene of the screened subclone was amplified by PCR using the LA PCR™ Kit Ver.2.1 (Takara, Dalian). A primer pair of 5′-GACACCCATGGGCAGCAGCCATCATCATCATCATCAC-3′ (forward primer) and 5′-GTGTCCATATGTCACATTGTTGGAAGCAA-3′ (reverse primer) was employed in the PCR amplification, and the restriction sites of NcoI and NdeI were introduced and underlined. The PCR conditions were 1 min at 94 °C, followed by 30 cycles of 10 s at 95 °C, 35 s at 58 °C, and 5 min at 72 °C. The PCR fragments was first ligated into pUC57 (Sangon, Shanghai), then excised by NcoI and NdeI, ligated into pET-15b(+) vector (Novagen, San Diego, CA), and transformed into the E. coli BL21 CodonPlus™ (DE3) strain. The transformant cells were incubated in Luria-Bertani medium (containing 50.0 µg/mL kanamycin) at 15, 22, and 28 °C for appropriate temperature assay. Until the cell density of OD600 reached 0.6, the broth was induced by adding 0.1 mM IPTG and followed by additional 5 h incubation. His-tagged recombinant protein in cell disruption (precipitant) was purified by affinity chromatography with nickel-nitrilotriacetic acid agarose resin (Ni–NTA, Qiagen, CA). After sample loading, His-tagged recombinant protein was purified by washing buffer (2 M Urea, 50 mM Tris, 2 mM DTT, 10.0–50.0 mM imidazole, pH 8.0), and collected by elusion buffer (2 M Urea, 50 mM Tris, 2 mM DTT, 500 mM imidazole, and pH 8.0). Then, the purified protein was de-His-tagged by TEV protease at 37 °C. The de-His-tag protein was further purified by affinity chromatography to remove the hydrolyzed His-tags, and the Ni2+ ion from Ni-NTA resin was eliminated by dialysis. The purity of recombinant protein was determined by SDS-PAGE using ChemiDoc™ XRS+ system (BioRad, CA), and the protein concentration was estimated by the solution absorbance at 280 nm using a molar extinction coefficient [23].
Substrate specificity and kinetic analysis
Cellulase activity was assessed by measuring the amount of reducing sugars released from CMC (or other substrates) at 575 nm using dinitrosalicylic acid (DNS) reagent [12]. Enzymatic reaction was carried out in 2 mL of mixtures (in phosphate buffer, pH 7.0) containing 1.2 mL cellulase solution (0.12 mg/mL) and 0.8 mL 2.5 % CMC (w/v) at optimum temperature for 30 min. One unit (U) of hydrolysis activity was defined as the amount of enzyme to release 1 µmol of reducing sugar per minute. The substrate specificity of recombinant enzyme was assayed according to the standard methods in phosphate buffer (pH 7.0) at 37 °C [15, 16], and all of the substrates were obtained from Sigma-Aldrich. Other than CMC, the polysaccharides of hydroxyethyl cellulose (HEC), laminarin from Eisenia bicyclis, β-D-glucan from barley, xylan from oat spelt and microcrystalline cellulose (MCC), the cellooligosaccharides of cellobiose (G2), cellotriose (G3), cellotetraose (G4), cellopentaose (G5) and cellohexose (G6), and the aryl-β-glycosides of p-nitrophenol-β-D-cellobioside (pNPCel), p-nitrophenyl-β-D-glucopyranoside (pNPGlc), p-nitrophenyl-β-D-galactopyranoside (pNPGal), p-nitrophenyl-β-D-xylopyranoside (pNPXyl), and p-nitrophenyl-β-D-fucopyranoside (pNPFuc) were selected and determined for substrate specificity. The substrate specificity was assayed under the optimal conditions at a final concentration of 1.0 % substrate (w/v) for 120 min (except 8 h for MCC and 15 min for barley glucan). The specific activities were determined under the optimal condition for 30 min at the substrate concentrations of 40.0 mg/mL for CMC; 20.0 mg/mL for xylan, pNPXyl, pNPGlc, pNPCel, and laminarin; and 15.0 mg/mL for HEC and oligosaccharides. The specific activity of barley glucan was assayed under the optimal condition for 15 min at the substrate concentration of 10.0 mg/mL, and MCC was assayed under the optimal condition for 4 h at the substrate concentration of 20.0 mg/mL. To determine the kinetic parameters of recombinant enzyme, reactions were carried out under the optimal condition for 30 min (except 15 min for barley glucan) at appropriate substrate concentrations (4.0–20.0 mg/mL for CMC, HEC, pNPCel, laminarin and oligosaccharides, and 1.0–10.0 mg/mL for β-D-glucan). The reaction rate versus the substrate concentration was plotted, and the kinetic constants K m and K cat were calculated by a nonlinear regression of the Michaelis-Menten equation with GraphPad PRISM version 5.0 (GraphPad Software, La Jolla, CA).
Biological characterization of recombinant Umcel9y-1
The pH range was determined with appropriate buffers (Citrate buffer, pH 3.0–6.0; Phosphate buffer, pH 6.0–8.0; Tris-HCl buffer, pH 8.0–9.0; and glycine-NaOH buffer, pH 9.0–10.0), and the temperature range was measured from 20 to 90 °C at the interval of 10 °C using the method for enzymatic activity assay [15]. The enzymatic stability at various temperatures and pH were performed as described previously. The recombinant Umcel9y-1 was incubated at various temperatures for 0–90 min and at various pH for 0–96 h, then the residual activities were determined [24]. Effects of metal ions, EDTA, and chemical agents on the enzymatic activity were tested by appropriate concentrations. The final concentration of the divalent metal ions and EDTA were 1.0 mM, respectively, and the concentration of each chemical agent for the reaction mixtures was 10 % (v/v). The halotolerance was determined by measuring residual activity under optimal condition following pre-incubation of recombinant Umcel9y-1 in 1.0–4.0 M NaCl or KCl for 10 days. The enzymatic activity of recombinant Umcel9y-1 in high-salt concentrations was determined by the enzymatic activity assay with a final concentration of 1–4 M NaCl or KCl in reaction buffers [15]. Influences of monosaccharides on recombinant Umcel9y-1 activity (kinetic parameters) toward barley glucan and cellooligosaccharide were evaluated as previously with 100 mM additive to substrate barley glucan, and 200 or 1000 mM additive to cellopentaose [10].
Substrate hydrolysis products and transglycosylation assay
The hydrolysis products of barley glucan and cellooligosaccharides (G6) by recombinant Umcel9y-1 were detected by high-performance liquid chromatography (HPLC) as described previously [10, 25]. Briefly, hydrolysis was carried out in 1 mL of mixture (in phosphate buffer, pH 7.0) containing 0.6 mL cellulase solution (0.12 mg/mL) and 0.4 mL substrate (50 mg/mL). The hydrolysis was carried out for 24 h on barley glucan and 30 h on G6 at 37 °C. Transglycosylation activity of recombinant Umcel9y-1 was determined in 20 µL mixtures (phosphate buffer, pH 7.0) containing 10 mM pNPGlc, 100 mM glucose, and 0.05 µg recombinant protein. Other than the standard mixture, the reaction system contain 1000 mM glucose was further evaluated. Transglycosylation products were analyzed using a high-performance anion exchange chromatograph with pulsed amperometric detection (HPAE-PAD; Dionex ICS-3000, Sunnyvale CA) as described as Uchiyama et al. [10].
Statistical analysis
Unless specified otherwise, all assays in this study were performed in triplicate. Data are presented as mean ± SD. Statistic analyses were assessed by Student’s t test. The p values <0.05 were considered statistically significant.
Results
Fosmid library construction and CMCase screening
A fosmid library constructed using the purified metagenomic DNAs contained approximate 2.5 × 104 clones. The sizes of the inserted DNA fragments from 20 randomly selected clones were 18–33 kb with an average size of 22 kb. Many positive clones exhibiting CMCase activity were screened. One positive clone, designated as WP-2, was selected for further characterization due to its high CMCase activity.
Sequence analysis of CMCase
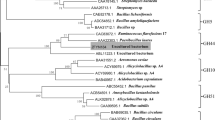
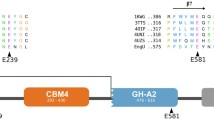
The CMCase positive subclone (the recombinant plasmid designated as pumcel9y-1) with an inserted DNA fragment of about 2.7 kb was sequenced and identified by BlastX. A complete gene sequence (2043 bp) of a CMCase was obtained (GenBank accession number: KR780677/ALA62876). The CMCase gene umcel9y-1 was expected to encode a protein of 680 amino acid residues (Additional file 1: Figure S1) and was identified as endoglucanase Umcel9y-1 consisting of an immunoglobulin-like domain (Cel-N) at the N-terminal (Amino acids 76–161th: pfam 02927) and an endoglucanase catalytic domain (Amino acids 166–621th: pfam 00759) (Additional file 2: Figure S2). Signal peptide analysis indicated that Umcel9y-1 is a non-secreted protein. The theoretical pI and Mw of Umcel9y-1 were evaluated to be 4.74 and 73296.9 Da, respectively. Phylogenetic analysis of glycosyl hydrolase family 9 (GH9) indicated that Umcel9y-1 was closely (58.4 % amino acids identity) related to the characterized endoglucanase Cel01. Although Umcel9y-1 and Cel01 were the closest related proteins and formed a separated branch within the phylogenetic tree, the two proteins belonged to different themes of GH9 based on their domain organization (Additional file 3: Figure S3). Umcel9y-1 belongs to theme C which contains a family 9 catalytic domain and an immunoglobulin-like domain, but the Cel01 belongs to theme D which has an additional family 4 CBM9 (Additional file 2: Figure S2), and different themes of GH9 proteins indicate different enzymatic activities [25]. The multiple alignment of the amino acid sequence of Umcel9y-1 with highly homologous GH9 enzymes is shown in Fig. 1. The two regions containing putative catalytic residues Asp234, Asp237, and Glu607 are well conserved among GH9 enzymes. The putative catalytic residues Asp234 and Asp237 act as nucleophile and Glu607 is proton donor during substrate hydrolysis [26]. The secondary structure of Umcel9y-1 predicted by the online program Protein Homology/analog Y Recognition Engine v2.0 (Phyre2) contained 29.4 % α-helix and 8.8 % β-strand. However, 23.0 % α-helix and 23.1 % β-strand were obtained from K2D2 database, which calculated by the protein spectrum of circular dichroism spectroscopy (Additional file 4: Figure S4). The distance is large between Umcel9y-1 to the closest spectrum in K2D2 database, suggesting Umcel9y-1 might be a relatively novel protein compare to the database [27].

Multiple alignments of Umcel9y-1 with seven highly homologous GHF9 proteins from different microorganisms. The identical residues are shown in white with a black background, and conservative changes are shown in black with a gray background. The residues of catalytic site and active pocket, and the secondary structure elements of CbhA from Clostridium thermocellum are indicated as the symbols in figure. The amino acid sequences used were as follows: Umcel9y-1 (ALA62876), Cel01 (AFG25775), CbhA (1UT9_A), Man5A (BAI52927), unnamed cellulase (YP_001612873), CelA (AAD01959), unnamed cellulase (S03818), unnamed cellulase (WP_014546521)
Recombinant production of Umcel9y-1
His-tagged Umcel9y-1 protein was efficiently expressed as a soluble protein fraction in cells of E. coli BL21 CodonPlus™ (DE3) from 15–28 °C. Umcel9y-1 expression production indicated that the optimum overexpression condition in E. coli BL21 is at 28 °C with 0.1 mM IPTG (Fig. 2a). His-tagged Umcel9y-1 was purified to almost homogeneity from the soluble protein fraction by affinity chromatography (Fig. 2b). The purified fusion Umcel9y-1 was de-His-tagged by TEV protease, and the recombinant Umcel9y-1 (de-His-tag protein) contained only one additional glycine residue on N-terminal of Umcel9y-1 (Additional file 5: Figure S5). After de-His-tagged, the recombinant Umcel9y-1 was purified again, and the purity was 90.8 % as determined by SDS-PAGE (Fig. 2c). The final yield of the expression was 2.8 mg pure protein per liter of E. coli culture, and the purification yield was about 42.5 %. The apparent Mw of recombinant Umcel9y-1 protein was about 70 kDa, and the fusion Umcel9y-1 was little bigger determined by SDS-PAGE (Fig. 2c). The apparent Mws of the recombinant Umcel9y-1 and the fusion protein in SDS-PAGE were consistent with that of prediction at ExPASy online program.

Overexpression and purification of recombinant Umcel9y-1 in Escherichia coli BL21(DE3). a SDS-PAGE analysis showing the Umcel9y-1 overexpression in Escherichia coli BL21(DE3). M, Markers; (1) Control without IPTG at 28 °C (supernatant); (2) Control without IPTG at 28 °C (precipitant); (3) 15 °C overnight (supernatant); (4) 15 °C overnight (precipitant); (5) 22 °C overnight (supernatant); (6) 22 °C overnight (precipitant); (7) 28 °C overnight (supernatant); (8) 28 °C overnight (precipitant). b SDS-PAGE analysis showing the purification of recombinant Umcel9y-1 by affinity chromatography. M, Marker; (1) Precipitant before loading; (2) Precipitant flow through; (3) Whole cell disruption before loading; (4) Washing through with 10 mM imidazole; (5) Washing through with 20 mM imidazole; (6) Washing through with 50 mM imidazole; (7–8) Purified eluate with 500 mM imidazole. c SDS-PAGE analysis showing the apparent Mw of purified recombinant Umcel9y-1 protein. M, Marker; (1–2) De-His-tag Umcel9y-1; 3, Fusion Umcel9y-1 with His-tag; 4, Repurified de-His-tag Umcel9y-1
Hydrolytic properties of recombinant Umcel9y-1
Substrate specificity assays showed that CMC, HEC, laminarin, barley glucan, pNPCel, and oligosaccharides (G3-G6) could be hydrolyzed, and MCC was hydrolyzed weakly, but cellobiose, Xylan, pNPXyl, pNPGlc, pNPGal, or pNPFuc was not hydrolyzed. Recombinant Umcel9y-1 exhibited the highest activity toward barley glucan (2372.9 ± 184.3 U/mg) followed by G3, pNPCel, and HEC. The enzymatic activity toward MCC was very low (2.18 ± 0.21 U/mg), and CMC was not the optimal substrate for Umcel9y-1 (Fig. 3e). Substrate specificity assays displayed a wide range of β-(1,4)-, β-cellobiose, β-(1,3)-/β-(1,6)-, and β-(1,3)-/β-(1,4)-/β-(1,6)- linked polysaccharides and amorphous cellulose were hydrolyzed by recombinant Umcel9y-1. Umcel9y-1 exhibits extraordinarily efficient activity on barley glucan and shows high hydrolytic activities to pNPCel and CMC, but no hydrolytic activity was observed for pNPGlc, these results indicate that the enzyme possesses endo-/exoglucanases activities, but not β-glucosidase activity.

Biochemical characteristics of purified recombinant Umcel9y-1. a Temperature range and optimal temperature; b Thermal stability; c pH range and optimal pH; d pH stability; e Specific activity for different substrates
The steady-state kinetic parameters of recombinant Umcel9y-1 were determined and compared for the various hydrolysable substrates (Table 1). The enzyme showed the most efficient hydrolytic abilities to pNPCel and barley glucan, followed by cellopentaose, cellotetraose, and cellotriose. Other than barley glucan, HEC was the most hydrolysable polysaccharide for Umcel9y-1. Barley glucan was the optimal polysaccharide substrate and cellopentaose was the optimal cellooligosaccharides substrate. The substrate pNPCel was the only efficiently degraded aryl-β-glycosides in this assays, indicating high exoglucanases activity. The steady-state kinetic parameters further proved that CMC is not the optimal substrate for Umcel9y-1. The V max value of recombinant Umcel9y-1 for CMC was also much lower than that of barley glucan. Comparing to other characterized endoglucanases from organisms and environmental samples, Umcel9y-1 showed prominent catalytic efficiency for barley glucan [15, 16, 24, 25, 28, 29].
Biochemical characteristics of recombinant Umcel9y-1
Recombinant Umcel9y-1 showed the maximum activity at 37 °C (Fig. 3a). The temperature stability indicated that Umcel9y-1 was stable from 30–50 °C. More than 90 % original activity was retained after 90 min incubation at 37 °C, and 68.4 % activity was observed after 90 min incubation at 50 °C (Fig. 3b). It was most active at pH range of 5.0–8.0 and the maximum activity was observed on pH 6.5–7.5 (Fig. 3c). When pH was lower than 4.0 or higher than 9.0, the activity was almost lost. The pH stability indicated that recombinant Umcel9y-1 was most stable from 6.5–7.5. Approximately 80 % original activity was retained after 24 h storage at pH 7.0–7.5, and approximately 70 % original activity was observed after 24 h storage at pH 6.5. After 48 h storage at 6.5–7.5, 50 % original activity was retained and the residual activity was stable until 96 h (Fig. 3d).
Halotolerance results indicated that Umcel9y-1 was highly salt tolerant, retaining more than 70 % activity to the control after 10 days of pre-incubation with 4 M NaCl or 4 M KCl. More than 80 % activity was observed after 10 days of pre-incubation with 1M NaCl or 1M KCl (Fig. 4a, b). Recombinant Umcel9y-1 was also active in high-salt reaction systems, with 79.9 and 86.2 % activities to control in 1 M NaCl and 1 M KCl, respectively. At concentrations of 2M NaCl or KCl, the relative activities were still higher than 70 % (Fig. 4c). The halotolerance of Umcel9y-1 was even higher than that of the well-known halotolerant protein Cel5G [15].

Effects of high-salt concentrations, divalent cations, and chemical agents on enzymatic activity of recombinant Umcel9y-1. a NaCl tolerance; b KCl tolerance; c Catalytic activity in high-salt systems; d Divalent cations and chemical agents influences. Data are expressed as mean ± standard deviation. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control (t test)
The activity of recombinant Umcel9y-1 was improved significantly by divalent cations of Mn2+, Ca2+, Mg2+, or Co2+ but was intensively inhibited by Zn2+, Cu2+, or EDTA. The relative activity increased to 288.8 % (Mn2+), 183.6 % (Ca2+), 165.2 % (Mg2+), and 148.8 % (Co2+) of the original by 1.0 mM corresponding divalent cation, but the activity was reduced to 26.9 and 5.0 % of the original when 1.0 mM Zn2+ and Cu2+ was applied, respectively. The enzymatic activity almost disappeared with the addition of 1.0 mM EDTA and was inhibited intensively with the addition of 10 % of ethanol (EtOH), ethyl acetate, N, N-Dimethyl formamide (DMF), or dimethyl sulfoxide (DMSO) (Fig. 4d).
The effects of 100 mM monosaccharides on recombinant Umcel9y-1 activity were evaluated for substrate barley glucan, and two concentrations (200 mM and 1000 mM) of monosaccharides were evaluated for substrate cellopentaose. The addition of 100 mM monosaccharides generally decreased k cat/K m for barley glucan with exception of D-xylose. The most significant change of k cat/K m on barley glucan was observed on 100 mM D-glucose, by which the inhibition was 2.3-fold. However, the addition of monosaccharides generally increased k cat/K m for cellopentaose with exception of 200 mM D-galactose, and the degree of stimulation was positively correlated to the concentration of monosaccharide additive. The most significant change of k cat/K m on cellopentaose was observed with 1000 mM D-xylose, by which the improvement was 9.5-fold (Table 2). In this study, the Umcel9y-1 hydrolysis activity on barley glucan was exclusively inhibited by 100 mM monosaccharides, and the activity on cellopentaose was significantly increased by 200–1000 mM D-arabinose and 1000 mM D-xylose, but was significantly inhibited by 200 mM D-mannose (Fig. 5). The decrease of hydrolysis activity for barley glucan of this study should be attributed to product inhibition [10], but the monosaccharide stimulations on cellopentaose hydrolysis suggesting that these monosaccharides may induce transglycosylation activity of Umcel9y-1.

Effects of various monosaccharides on recombinant Umcel9y-1 activity toward barley glucan and cellopentaose. Data are expressed as mean ± standard deviation. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control (t test)
Substrate hydrolysis products and transglycosylation activity
In the polysaccharide and oligosaccharide hydrolysis assays, G3 was the predominant product released from barley glucan, but G2 was predominant product released from G6 by recombinant Umcel9y-1, as determined by HPLC analysis (Fig. 6). Barley glucan hydrolysis initially yielded G4, G3, G2, and D-glucose, but G2 and G4 began to decrease after 1 h and reached steady concentrations at 4 h later; G3 and G1 was continuous increase until 6th h, then D-glucose decreased quickly and almost disappeared at the time point of the 24th h (Fig. 6a). As described above, β-cellobiose (G2) could not be hydrolyzed by Umcel9y-1, and D-glucose is also impossible to be degraded, but the products of G2 and D-glucose were consumed or even disappeared during the hydrolysis course, these results indicated that the transglycosylation may occurred during the hydrolyzation. During the process of G6 hydrolysis, the product G3 was further degraded to G2 and G1, and G3 was disappeared after 12 h incubation. Meanwhile, G1 began to decrease at the time point of 12th h, this also may attributed to transglycosylation (Fig. 6b).

Hydrolysis properties of barley glucan and cellohexose by recombinant Umcel9y-1 monitored by HPLC. a Time course of barley glucan hydrolysis; b Time course of cellohexose hydrolysis; c Chromatogram for G1 to G6 standard mixture; d Chromatogram for hydrolysis products of barley glucan; e Chromatogram for hydrolysis products of cellohexose
Transglycosylation activity was determined using 10 mM pNPGlc and 100 mM (or 1.0 M) D-glucose as substrates. The results indicated that the recombinant Umcel9y-1 with 10 mM pNPGlc plus 100 mM D-glucose resulted in transglycosylation products of gentiobiose laminaribiose and sophorose. When D-glucose concentration elevated to 1.0 M as additive, only gentiobiose and laminaribiose were detected as the products, and sophorose was difficult to be detected by HPAE-PAD at high-D-glucose concentrations (Fig. 7). Transglycosylation product or pNPGlc hydrolysis product pNP was not detected, when recombinant Umcel9y-1 incubated with 10 mM pNPGlc in absence of D-glucose.

HPAE-PAD analyses of the transglycosylation products of recombinant Umcel9y-1. Chromatograms are given for standard glucodisaccharides (a), transglycosylation products with 100 mM D-glucose (b), and transglycosylation products with 1000 mM D-glucose (c)
Discussion
The paddy soil is rich of plant biomass (i.e., straw and root) which can be degraded and utilized by cellulose-degrading microorganisms. Unfortunately, only a small fraction of environmental microorganisms (<5 %) can be isolated by laboratory conditions, and thus soil metagenomes represent an unlimited resource for novel biocatalysts exploitation [30]. Metagenomic-derived GH9 glucanase have been isolated from compost and grassland soils [31, 32], aquatic community [33], elephant dung [34], and enrichment culture of alkaline lakes [35], but never from paddy soil. In this study, an efficient and versatile β-glucanase Umcel9y-1 was cloned from the metagenome of paddy soil and characterized for potential industrial development.
Substrate specificity suggested that Umcel9y-1 can hydrolyze CMC and pNPCel that are indicative of endo- and exoglucanase activities, and the enzyme also exhibits transglycosylation activity in presence of D-glucose. Amino acid sequence analysis indicated that Umcel9y-1 belongs to theme C of GH9 (Fig. 1, Figure S2), but phylogenetic analysis showed Umcel9y-1 was separated from theme C and showed closest relatedness to theme D (Additional file 3: Figure S3) [25]. Therefore, the specific modular structure may be one of the key factor for the efficient and multiple activities. Analyses of the X-ray crystal structure of Umcel9y-1 are currently under way. The resulting three-dimensional model may provide clues for glucosyl-binding subsites and offer an explanation for the efficient endo-/exoglucanase and transglycosylation activity. Umcel9y-1 as the theme C protein of GH9 consisted of a Cel-N-term domain and a GH9 catalytic domain, but without carbohydrate-binding modules (CBMs). However, CBMs in cellulases are considered to be responsible for the association of catalytic modules to insoluble carbohydrates. So, GH9 cellulases of themes B and D consisting of CBMs often exhibited efficient activities toward crystalline cellulose, which was not observed on Umcel9y-1 [25, 36].
Among the tested biochemical characteristics, the prominent advantages of Umcel9y-1 are the halotolerant and thermal stability. It is more halotolerant than other salt-tolerant cellulases from saline environment [14, 15, 33, 35]. The stability and activity of Umcel9y-1 in high-salt concentrations are remarkable for an endoglucanase from non-halophilic environmental community (Fig. 4). Umcel9y-1 was highly active at 20–50 °C and the activity was relatively stable at 50 °C. The optimal temperatures of metagenomic-derived cellulases Cel01, Cel5G, and Cel5A also ranged 45–50 °C with rapid inactivation at 60 °C [14, 15, 31]. The relative activity of Umcel9y-1 was greatly enhanced by addition of Mg2+, Mn2+, or Ca2+ but was inhibited intensively by other divalent cations, chelating agent, or chemical agents. This result indicated that divalent cations (especially Mn2+) are required for enzyme activation. However, the divalent cations (e.g., Ca2+, Co2+, Mg2+, and Zn2+), chelating agents. or other chemical agents (e.g., EtOH, SDS, and DMSO) showed limited influences on the other endoglucanases (e.g., Cel01, Cel5A, Cel9D, and Cel5G) [14, 15, 25, 31].
Another prominent advantage of Umcel9y-1 is the excellent catalytic efficiency on barley glucan. The k cat/K m value for barley glucan (239.14 s−1 mM−1) is higher than that of most reported cellulases from bacteria, fungi and metagenomes (Additional File 6: Table S1). For example, the K cat/K m value of CelA obtained from Alicyclobacillus acidocaldarius was 2.3 s−1 mM−1 [24]; the K cat/K m value of Cel9D obtained from Fibrobacter succinogenes was 0.0097–22.8 s−1 mM−1 for different substrates [25]; the K cat/K m values of EngD and EngB-CBD that modified from endoglucanase EngB of Clostridium cellulovorans were 25.3–42.1 s−1mM−1 [28], and the K cat/K m value of a endoglucanase from Arachniotus citrinus were 40.0–132.0 s−1 mM−1 at different temperatures [29]. The high K cat/K m indicate that Umcel9y-1 showed prominent catalytic efficiency for barley glucan hydrolysis than other cellulases from bacteria, actinomycetes, fungi, and metagenomes to their optimal substrates.
Different from the most other glucanases, transglycosylation activity was observed for Umcel9y-1 during polysaccharide and oligosaccharide hydrolysis. In the hydrolysis process of barley glucan and cellohexose, the end product of D-glucose was consumed as substrate of transglycosylation reactions to produce G3 and G2, respectively. Other than the minor components of G2 and G4, G3 is the unique predominant hydrolysis product from barley glucan, and Umcel9y-1 might be considered as a potent biocatalyst for G3 production. Moreover, transglycosylation activity can also relieve the product inhibition of cellulases [10], and some monosaccharides even improved the hydrolysis efficiency of Umcel9y-1 toward cellopentaose, but the activation results were not observed for barley glucan.
Conclusions
In this study, an efficient and versatile β-glucanase gene was cloned from the metagenome of paddy soil, and its enzymatic activities were characterized for the potential industrial applications (e.g., β-glucan saccharification, G3 production, or as glycosyltransferases substitute). The recombinant Umcel9y-1 possesses some interesting characteristics (e.g., easiness for expression and purification, high halotolerance, and prominent catalytic ability to barley glucan) and efficient activities of exo-gluancase and transglycosylation, indicating that it is a novel metagenome-derived cellulase, a potent candidate for industrial applications (Addditional file 6: Table S1).
Abbreviations
- CMC:
-
carboxymethylcellulose
- IPTG:
-
isopropy-β-D-thiogalactoside
- DNS:
-
dinitrosalicylic acid
- HEC:
-
hydroxyethyl cellulose
- MCC:
-
microcrystalline cellulose
- G2:
-
cellobiose
- G3:
-
cellotriose
- G4:
-
cellotetraose
- G5:
-
cellopentaose
- G6:
-
cellohexose
- pNPCel:
-
p-nitrophenol-β-D-cellobioside
- pNPGlc:
-
p-nitrophenyl-β-D-glucopyranoside
- pNPGal:
-
p-nitrophenyl-β-D-galactopyranoside
- pNPXyl:
-
p-nitrophenyl-β-D-xylopyranoside
- pNPFuc:
-
p-nitrophenyl-β-D-fucopyranoside
- HPLC:
-
high-performance liquid chromatography
- HPAE-PAD:
-
high-performance anion exchange chromatograph with pulsed amperometric detection
- GH9:
-
glycosyl hydrolase family 9
References
Ding SY, Himmel ME. The maize primary cell wall microfibril: a new model derived from direct visualization. J Agric Food Chem. 2006;54(3):597–606.
Detroy RW, St Julian G. Biomass conversion: fermentation chemicals and fuels. Crit Rev Microbiol. 1983;10(3):203–28.
Merino ST, Cherry J. Progress and challenges in enzyme development for biomass utilization. Adv Biochem Eng Biotechnol. 2007;108:95–120.
Varel VH, Yen JT. Microbial perspective on fiber utilization by swine. J Anim Sci. 1997;75(10):2715–22.
Wang H, Squina F, Segato F, Mort A, Lee D, Pappan K, Prade R. High-temperature enzymatic breakdown of cellulose. Appl Environ Microbiol. 2011;77(10):5199–206.
Bhat MK. Cellulases and related enzymes in biotechnology. Biotechnol Adv. 2000;18(5):355–83.
Ando S, Ishida H, Kosugi Y, Ishikawa K. Hyperthermostable endoglucanase from Pyrococcus horikoshii. Appl Environ Microbiol. 2002;68(1):430–3.
Lynd LR, Weimer PJ, Zyl WH, Pretorius IS. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol R. 2002;66(3):506–77.
Bhatia Y, Mishra S, Bisaria VS. Microbial β-glucosidases: cloning, properties, and applications. Crit Rev Biotechnol. 2002;22(4):375–407.
Uchiyama T, Miyazaki K, Yaoi K. Characterization of a novel β-glucosidase from a compost microbial metagenome with strong transglycosylation activity. J Biol Chem. 2013;288(25):18325–34.
Vladimir K, Joachim T. Glycosylation employing bio-systems: from enzymes to whole cells. Chem Soc Rev. 1997;26(6):463–73.
Brunecky R, Alahuhta M, Xu Q, Donohoe BS, Crowley MF, Kataeva IA, et al. Revealing nature’s cellulase diversity: the digestion mechanism of Caldicellulosiruptor bescii CelA. Science. 2013;342(6165):1513–6.
Voget S, Leggewie C, Uesbeck A, Raasch C, Jaeger KE, Streit WR. Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol. 2003;69(10):6235–42.
Voget S, Steele HL, Streit WR. Characterization of a metagenome-derived halotolerant cellulase. J Biotechnol. 2006;126(1):26–36.
Liu J, Liu WD, Zhao XL, Shen WJ, Cao H, Cui ZL. Cloning and functional characterization of a novel endo-beta-1,4-glucanase gene from a soil-derived metagenomic library. Appl Microbiol Biotechnol. 2011;89(4):1083–92.
Feng Y, Duan CJ, Pang H, Mo XC, Wu CF, Yu Y, et al. Cloning and identification of novel cellulase genes from uncultured microorganisms in rabbit cecum and characterization of the expressed cellulases. Appl Microbiol Biotechnol. 2007;75(2):319–28.
Teather RM, Wood PJ. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl Environ Microbiol. 1982;43(4):777–80.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–8.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: signalP 3.0. J Mol Biol. 2004;340(4):783–95.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–82.
Miao Y, Chen L, Wang C, Wang Y, Zheng Q, Gao C, et al. Expression, purification and antimicrobial activity of puroindoline A protein and its mutants. Amino Acids. 2012;43(4):1689–96.
Kelley LA, Sternberg MJ. Protein structure prediction on the web: a case study using the phyre server. Nat Protoc. 2009;4(3):363–71.
Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4(11):2411–23.
Eckert K, Zielinski F, Leggio L, Schneider E. Gene cloning, sequencing, and characterization of a family 9 endoglucanase (CelA) with an unusual pattern of activity from the thermoacidophile Alicyclobacillus acidocaldarius ATCC27009. Appl Microbiol Biotechnol. 2002;60(4):428–36.
Qi M, Jun HS, Forsberg CW. Cel9D, an Atypical 1, 4-β-D-Glucan Glucohydrolase from Fibrobacter succinogenes: characteristics, Catalytic Residues, and Synergistic Interactions with Other Cellulases. J Bioact. 2008;190(6):1976–84.
Parsiegla G, Belaïch A, Belaïch JP, Haser R. Crystal structure of the cellulase Cel9 M enlightens structure/function relationships of the variable catalytic modules in glycoside hydrolases. Biochemistry. 2002;41(37):11134–42.
Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2012;80(2):374–81.
Murashima K, Kosugi A, Doi RH. Thermostabilization of cellulosomal endoglucanase EngB from Clostridium cellulovorans by in vitro DNA recombination with non-cellulosomal endoglucanase EngD. Mol Microbiol. 2002;45(3):617–26.
Saleem M, Rashid MH, Jabbar A, Perveen R, Khalid AM, Rajoka MI. Kinetic and thermodynamic properties of an immobilized endoglucanase from Arachniotus citrinus. Process Biochem. 2005;40(2):849–55.
Ward DM, Weller R, Bateson MM. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature. 1990;345(6270):63–5.
Nacke H, Engelhaupt M, Brady S, Fischer C, Tautzt J, Daniel R. Identification and characterization of novel cellulolytic and hemicellulolytic genes and enzymes derived from German grassland soil metagenomes. Biotechnol Lett. 2012;34(4):663–75.
Pang H, Zhang P, Duan CJ, Mo XC, Tang JL, Feng JX. Identification of cellulase genes from the metagenomes of compost soils and functional characterization of one novel endoglucanase. Curr Microbiol. 2009;58(4):404–8.
Pottkämper J, Barthen P, Ilmberger N, Schwaneberg U, Schenk A, Schulte M, Ignatiev N, WR S. Applying metagenomics for the identification of bacterial cellulases that are stable in ionic liquids. Green Chem. 2009;11(4):957–65.
Wang F, Li F, Chen G, Liu W. Isolation and characterization of novel cellulase genes from uncultured microorganisms in different environmental niches. Microbiol Res. 2009;164(6):650–7.
Grant S, Sorokin DY, Grant WD, Jones BE, Heaphy S. A phylogenetic analysis of Wadi el Natrun soda lake cellulase enrichment cultures and identification of cellulase genes from these cultures. Extremophiles. 2004;8(5):421–9.
Schubot FD, Kataeva IA, Chang J, Shah AK, Ljungdahl LG, Rose JP, et al. Structural basis for the exocellulase activity of the cellobiohydrolase CbhA from Clostridium thermocellum. Biochemistry. 2004;43(5):1163–70.
Authors’ contributions
YZ, XW, WW, and ZZ designed experiments. XW, JX, and WWA constructed fosmid library, gene isolation, and enzyme assay. WW, JX, and QW performed biological characterizations. YZ, JX, ZX, WW, CW, and HJ analyzed data. YZ, XW, CW, ZX, ZZ, and HJ wrote and reviewed the manuscript. YZ, XW, and CW supervised the research. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by grants from International Cooperation Project of Ministry of Science and Technology (2013DFA31450), Natural Science Foundation for Distinguished Young Scholars of Anhui Province (1608085J08) and Startup Foundation for Advanced Talents of Anhui Agricultural University.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
13068_2016_449_MOESM1_ESM.tif
Additional file 1: Figure S1. Nucleotide sequence and putative amino acids of the subclone insert DNA fragment of umcel9y-1 gene. The immunoglobulin (Ig)-like domain was underlined and the GH9 catalytic domain was shadowed.
13068_2016_449_MOESM2_ESM.tif
Additional file 2: Figure S2. Module structures of Umcel9y-1 and the two closest GH9 cellulases. Abbreviations: SP, signal peptide; CBM, carbohydrate-binding module; Cel-N-term, cellulase N-terminal module; GH9, family 9 glycoside hydrolase module. The cellulases are Umcel9y-1 from a uncultured bacterium of paddy soil, Cel01 from a uncultured bacterium of grassland soil and an unnamed cellulase from Sorangium cellulosum So ce56.
13068_2016_449_MOESM3_ESM.tif
Additional file 3: Figure S3. Neighbor-joining phylogenetic tree based on complete amino acid sequences of endogluancases, showing the phylogenetic position of Umcel9y-1 in GH9 family. Bootstrap values are shown as percentages of 1000 replicates, and only the bootstrap values above 50 % are shown. GenBank accession numbers are shown in parentheses. Bar, 2 substitutions per 10 animo acid positions.
13068_2016_449_MOESM5_ESM.tif
Additional file 5: Figure S5. Putative amino acid sequence of recombinant Umcel9y-1 expressed by Escherichia coli BL21 in the vector of pET-15b(+). His-tag in front of recombinant Umcel9y-1 was indicated as red font, TEV protease site was underlined, and the recombinant Umcel9y-1 was shadowed, in which one additional amino acid ‘Glycine’ (G with blue font) was included after de-His-tagged.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Y., Wang, X., Wei, W. et al. A novel efficient β-glucanase from a paddy soil microbial metagenome with versatile activities. Biotechnol Biofuels 9, 36 (2016). https://doi.org/10.1186/s13068-016-0449-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-016-0449-6