
Abstract
Background
2-Aminothiazoles are significant class of organic medicinal compounds utilized as starting material for the synthesis of diverse range of heterocyclic analogues with promising therapeutic roles as antibacterial, antifungal, anti-HIV, antioxidant, antitumor, anthelmintic, anti-inflammatory & analgesic agents.
Experimental
Eight compounds 1a, 2a–2g were synthesized and characterized by FTIR and NMR (1H and 13C). Evaluation of antibacterial potential against multi-drug resistant clinical isolates was performed and minimum inhibitory concentration (MIC) values were determined. Antifungal activity was also performed. Protein–ligand interactions of compounds with target enzyme were evaluated through docking studies.
Results
Resistance profiling of bacterical clinical isolates (MDRs) depicted that some standard drugs used were not active against these MDRs while our synthesized compounds showed good MIC values. Among all the synthesized compounds, 2a and 2b showed significant antibacterial potential towards gram-positive Staphylococcus epidermidis and gram-negative Pseudomonas aeruginosa at MIC 250 µg/mL and 375 µg/mL respectively. Likewise, compound 2d and 2g exhibited inhibitory potential against gram-positive Staphylococcus aureus and gram-negative Escherichia coli at MIC values of 250 and 375 µg/mL respectively. Compound 2b showed maximum antifungal potential against Candida glabrata (ATCC 62934) with a zone of inhibition 21.0 mm as compared to the reference drug nystatin which showed lesser antifungal potential with a zone of inhibition of 19.1 mm. Candida albicans (ATCC 60387) showed maximum sensitivity to compound 2a with a zone of inhibition 20.0 mm. Its antifungal activity is more in comparison to reference drug nystatin with exhibited the zone of inhibition of 19.3 mm. Designed compounds were docked with the target enzyme UDP-N-acetylmuramate/l-alanine ligase. The compound 2b showed highest binding affinity (− 7.6 kcal/mol).
Conclusions
The synthesized compounds showed moderate to significant antibacterial and antifungal potential. It is clear from the binding affinities that compounds having hydroxyl group substituted on benzene ring possess strong binding affinity as compared to other analogues. These designed compounds could be considered to act as antagonists against target UDP-N-acetylmuramate/l-alanine ligase.
Similar content being viewed by others
Introduction
Schiff bases have gained remarkable place in medicinal chemistry because of their diverse therapeutic roles as antibacterial [1, 2], antifungal [3,4,5,6], anti-HIV [7], antioxidant, antitumor, anthelmintic [8], anti-inflammatory & analgesic agents [9]. Thiazole nucleus is present in both natural and synthetic products with notable pharmacological and therapeutic activities [10]. Thiamin also called as vitamin B1, also contains thiazole nucleus. Thiamin functions in human body as co-enzyme in metabolic pathways of carbohydrates and amino acids [11]. Synthesis of 2-amino-6-methylbenzothiazoles Schiff bases with antibacterial activity comparable to that of ampicillin, are reported [12]. 2-amino-4-substituted thiazoles are already reported for their anthelmintic, anti-leukotrienes, anticonvulsant, antimalarial and fungicidal properties [13].
The registered drugs Mirabegron (Anticholinergic agent) and Cefdinir (Antibacterial agent) belong to aminothiazole analogues [14, 15]. Since it is well known that 2-aminothiazoles are significant class of organic medicinal compounds utilized as starting material for the synthesis of diverse range of heterocyclic analogues. Therefore, the design and synthesis of ethyl-2-aminothiazole-4-carboxylate Schiff bases was targeted in this study.
Designing of evident drugs can be achieved through virtual screening and molecular docking strategies. Likewise, interpretation of probable drug specificity with target enzyme or protein can be assessed [16]. In the present study the selected uridine diphosphate-N-acetylmuramate/l-alanine ligase was selected as antimicrobial target for docking [17]. It is the bacterial enzyme that catalyzes the reaction of peptidoglycan synthesis by first amino acid addition to peptidoglycan sugar moiety. Peptidoglycan is the key element for bacterial cell wall [18]. By targeting this ligase enzyme, the bacterial cell integrity can be dismantled ultimately resulting in bacterial cell death.
Experimental
Molecular docking
Ligand designing
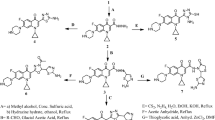
Eight ligands were designed using ChemSketch 12.0 (https://www.acdlabs.com/resources/freeware/). Analogues of ethyl -2-aminothiazole-4-carboxylate were designed by altering the amino group as illustrated in Fig. 1. Four aldehydes and two ketones were used to design the compounds form 2a–2h. Molecular properties were determined using ChemSketch Tool (https://www.acdlabs.com/resources/freeware/). Chemo-informatics, ADME properties and drug likeliness were determined by following Lipinski rule while associated parameters of designed ligands were predicted by Molinspiration (https://www.molinspiration.com/) and pkCSM online tool [19, 20]. The toxicity profile was obtained using TOXTREE and pkCSM online tool [21].

General scheme for the synthesis of ethyl 2-aminothiazole-4-carboxylate and its derivatives
Pocket identification and docking of designed compounds
Pocket identification was accomplished via Dogsitescorer [22]. Crystal structure of bacterial target enzyme UDP-N-acetylmuramate/l-alanine ligase was taken from databank (https://www.rcsb.org/) (PDB ID: 1GQQ). Enzyme-Ligand docking was accomplished with PyRx [23], via AutoDock VINA module [24]. Docking coordinates of x = 42.396, y = 47.393 and z = 84.654 with dimensions x = 47.187, y = 23.629 and z = 38.776 were used according to the best binding site predicted by DogSiteScorer. Open Babel GUI and Discovery Studio 2017 R2 Client (https://www.3dsbiovia.com/) were utilized for preliminary procedures and elaboration of enzyme ligand interactions respectively [25].
Chemistry
Ethyl bromopyruvate (technical grade, 90%), thiourea (ACS reagent) benzaldehyde (ReagentPlus® 99%), salicylaldehyde (reagent grade, 98%), 3-hydroxybenzaldehyde (AR ≥ 99%), 4-hydroxybenzaldehyde (Analytical standard), 4-hydroxy-3-methoxybenzaldehyde (ReagentPlus® 99%), acetophenone (Analytical standard), 2′-hydroxyacetophenone (ReagentPlus® 99%),. Ethanol (absolute, ACS reagent) and glacial acetic acid (100%, anhydrous for analysis ACS, ISO reagent) were used purchased from Sigma Aldrich and Merck. Synthesized compounds were purified by recrystallization in appropriate solvents and examined through thin layer chromatography (Merck Silica gel 60 F254). Melting points were determined by using digital Gallenkamp model MPD BM 3.5 apparatus. Characterization of synthesized compounds was made through spectrophotometric analysis; FT-IR (Thermoscientific NICOLET IS10 spectrophotometer), 1H & 13C NMR (Bruker AM-300 and AM-100 spectrophotometer) using DMSO and CDCl3 respectively. Elemental analysis values were recorded on Model ANALYST 2000 CHNS, Perkin Elmer Analyzer. Multiskan™ GO Microplate Spectrophotometer was used to quantify synthesized compounds for accuracy, precision and sensitivity [26].
General procedure for the preparation of ethyl 2-aminothiazole-4-carboxylate (1a)
Ethyl bromopyruvate (2 mol) and of thiourea (3 mol) in 100 mL of ethanol (99.9%) was refluxed for 24 h. Progress of the reaction was monitored by TLC (petroleum ether: ethyl acetate, 1:3). After completion, the reaction mixture was cooled to room temperature, concentrated and poured into ice cold water and basified (pH 10) with 2 M NaOH resulting in separation of off-white precipitation that was recrystallized using ethanol.
Yield 70%, m.p. 175–177 °C, Rf 0.71 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1690 (C=O ester), 3000 (C–H), 3300–3150 (N–H amine), 1566 (C=C); 1H-NMR (Cholroform, δ ppm): δ = 4.34 (q, 2H, CH2), 1.38 (t, 3H, CH3), 7.41 (s, 1H, Thiazole), 5.85 (s, 2H, NH2); Elemental analysis: C6H8N2O2S, Calculated: C 41.81%, H 4.65%, N 16.26% O 18.59% S 18.59%; Found C 41.79%, H 4.64%, N 16.26% O 18.58% S 18.57%.
General procedure for the synthesis of ethyl 2-aminothiazole-4-carboxylate derivatives
Ethyl 2-aminothiazole-4-carboxylate (0.05 mol) and aldehyde/ketone (0.05 mol) in absolute ethanol (30 mL) were dissolved and few drops of glacial acetic acid were added. Reaction mixture was stirred and refluxed for 12 h. Progress of reaction was monitored by TLC. After cooling the excess solvent was evaporated by using rotary evaporator. The residue was dissolved in ethyl acetate, crystal growth was observed after few days [27, 28]. Physical data of the synthesized compounds is given in Table 1.
Ethyl 2-{[(E)-phenylmethylidene]amino}-1,3-thiazole-4-carboxylate (2a)
Yield 50%, m.p. 160–162 °C, Rf 0.61 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1687 (C=O ester), 3023 (C–H), 1513 (C=C), 1616 (C=N);1H NMR (DMSO, δ ppm): δ = 4.25 (q 2H CH2), 1.26 (t, 3H, CH3), 7.44 (s, 1H, Thiazole), 7.02 (m, 5H, Ar), 9.47 (s, 1H, H–C=N); 13C NMR (CDCl3, d ppm): 166.3 (C5), 165.2 (C12), 162.2 (C7), 143.7 (C2), 134.7 (C13), 130.7 (C18), 129.5 (C14), 129.5 (C15), 127.8 (C16), 127.8 (C17), 122.5 (C1), 61.3 (C10), 13.8 (C11); Elemental analysis: C13H12N2O2S, Calculated: C 59.93%, H 4.61%, N 10.76%; Found C 59.91%, H 4.60%, N 10.76%.
Ethyl 2-{[(E)-(2-hydroxyphenyl)methylidene]amino}-1,3-thiazole-4-carboxylate (2b)
Yield 56%, m.p. 198–199 °C, Rf 0.64 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1687 (C=O ester), 2975 (C–H), 1535 (C=C), 1617 (C=N), 3261 (OH); 1H NMR (DMSO, δ ppm): δ = 4.20 (q, 2H, CH2), 1.24 (t, 3H, CH3), 7.45 (s,1H, Thiazole), 7.40 (m, 4H, Ar), 9.76 (s,1H, H–C=N), 7.26 (s, 1H, Ar–OH); 13C NMR (CDCl3, d ppm): 165.1 (C5), 164.1 (C12), 162.9 (C7), 161.1 (C14), 143.1 (C2), 133.7 (C18), 133.5 (C15), 122.8 (C1), 120.8 (C17), 117.8 (C13), 116.9 (C16), 61.1 (C10), 14.4 (C11); elemental analysis: C13H12N2O3S, calculated: C 56.458%, H 4.343%, N 11.58%; found C 56.45%, H 4.34%, N 11.58%.
Ethyl 2-{[(E)-(3-hydroxyphenyl)methylidene]amino}-1,3-thiazole-4-carboxylate (2c)
Yield 54%, m.p. 195–197 °C, Rf 0.66 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1 1690 (C=O ester), 2983 (C–H), 1502 (C=C), 1614 (C=N), 3259 (OH); 1H NMR (DMSO, δ ppm): δ = 4.21 (q, 2H, CH2), 1.24 (t, 3H, CH3), 7.43 (s, 1H, Thiazole), 7.30 (m, 4H, Ar), 8.89 (s,1H, H–C=N); 13C NMR (CDCl3, d ppm): 166.8 (C5), 164.9 (C12), 161.9 (C7), 158.9 (C16), 142.4 (C2), 135.7 (C13), 129.5 (C17), 121.9 (C15), 121.3 (C1), 116.3 (C18), 115.6 (C14), 61.9 (C10), 14.1 (C11); elemental analysis: C13H12N2O3S, calculated: C 56.458%, H 4.343%, N 11.58%; found C 56.45%, H 4.34%, N 11.57%.
Ethyl 2-{[(E)-(4-hydroxyphenyl)methylidene]amino}-1,3-thiazole-4-carboxylate (2d)
Yield 60%, m.p. 200–202 °C, Rf 0.69 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1688 (C=O ester), 2953 (C–H), 1517 (C=C), 1612 (C=N), 3250 (OH); 1H NMR (DMSO, δ ppm): δ = 4.25 (q, 2H, CH2), 1.25 (t, 3H, CH3), 7.42 (s, 1H, Thiazole), 7.20 (m, 4H, Ar), 9.48 (s, 1H, H–C=N); 13C NMR (CDCl3, d ppm): 167.1 (C5), 163.8 (C12), 161.9 (C7), 157.3 (C18), 142.4 (C2), 131.5 (C14), 131.5 (C15), 127.7 (C13), 121.2 (C1), 117.8 (C16), 117.8 (C17), 61.6 (C10), 14.2 (C11); elemental analysis: C13H12N2O3S, calculated: C 56.458%, H 4.343%, N 11.58%; found C 56.45%, H 4.33%, N 11.56%.
Ethyl 2-{[(E)-(4-hydroxy-3-methoxyphenyl)methylidene]amino}-1,3-thiazole-4-carboxylate (2e)
Yield 50%, m.p. 196–198 °C, Rf 0.61 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1696 (C=O ester), 3030 (C–H), 1506 (C=C), 1619 (C=N), 3210 (OH); 1H NMR (DMSO, δ ppm): δ = 4.22 (q, 2H, CH2),1.26 (t, 3H, CH3), 7.45 (s, 1H, Thiazole), 7.01 (m, 3H, Ar), 8.40 (s, 1H, H–C=N), 3.37 (s, 3H, OCH3), 7.25 (s, 1H, Ar–OH); 13C NMR (CDCl3, d ppm): 165.9 (C5), 165.1 (C12), 162.9 (C7), 147.8 (C16), 146.2 (C18), 142.7 (C2), 125.7 (C13), 124.2 (C15), 121.2 (C1), 115.1 (C17), 112.8 (C14), 61.9 (C10), 57.5 (C21), 14.2 (C11); Elemental analysis: C14H14N2O4S, Calculated: C 54.842%, H 4.57%, N 9.14%; Found C 54.84%, H 4.57%, N 9.14%.
Ethyl 2-{[(1E)-1-phenylethylidene]amino}-1,3-thiazole-4-carboxylate (2f)
Yield 52%, m.p. 190–192 °C, Rf 0.59 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1690 (C=O ester), 2985 (C–H), 1553 (C=C), 1617 (C=N); 1H NMR (DMSO, δ ppm): δ = 4.25 (q, 2H, CH2), 1.26 (t, 3H, CH3), 7.64 (s, 1H, Thiazole), 7.14 (m, 5H, Ar), 3.38 (s, 3H, CH3–C=N); 13C NMR (CDCl3, d ppm): 175.1 (C12), 168.8 (C5), 162.2 (C7), 142.7 (C2), 136.5 (C13), 131.7 (C18), 129.1 (C16), 129.1 (C17), 128.5 (C14), 128.5 (C15), 124.1 (C1), 61.9 (C10), 18.2 (C19), 14.3 (C11); elemental analysis: C14H14N2O2S, calculated: C 61.24%, H 5.103%, N 10.206%; found C 61.24%, H 5.10%, N 10.21%.
Ethyl 2-{[(1E)-1-(2-hydroxyphenyl)ethylidene]amino}-1,3-thiazole-4-carboxylate (2g)
Yield 62%, m.p. 164–165 °C, Rf 0.60 (petroleum ether: ethyl acetate, 1:3); IR (KBr) cm−1: 1685 (C=O ester), 2990 (C–H), 1570 (C=C), 1615 (C=N), 3437 (OH); 1H NMR (DMSO, δ ppm): δ = 4.21 (q, 2H, CH2), 1.23 (t, 3H, CH3), 7.45 (s, 1H, Thiazole), 7.25 (m, 4H, Ar), 3.34 (s, 3H, CH3–C=N); 13C NMR (CDCl3, d ppm): 178.9 (C12), 168.1 (C5), 160.9 (C7), 158.7 (C14), 141.7 (C2), 133.7 (C18), 129.9 (C15), 124.1 (C1), 123.7 (C13), 117.8 (C17), 114.2 (C16), 61.1 (C10), 19.5 (C19), 14.9 (C11); elemental analysis: C14H14N2O3S, calculated: C 57.89%, H 4.82%, N 9.65%; found C 57.87%, H 4.82%, N 9.64%.
Antimicrobial assay
Resistance profiling
Analysis of resistance pattern of pathogenic microbe (resistant, intermediate, susceptible), antibiotic sensitivity assay was performed. Fresh cultures of bacterial strains were made on nutrient agar and incubated for 24 h at 37 °C. The following day isolated colonies from bacterial cultures were picked and dissolved in 1 mL of PBS (autoclaved normal saline) and inoculum turbidity was then confirmed with 0.5% McFarland standard [29].
Dried Muller Hinton (MH) agar plates were used for resistance profiling. With the help of sterilized syringe antibiotic discs of known concentration were placed on MH agar plates namely Cefepime, Ciprofloxacin, Imipenem, Cefoxitin, Ampicillin, Aztreonam, Tetracycline, Ceftazidime, Minocyclin, Gentamycin, Co-trimoxazole, Colistin, Clindamycin, Vancomycin, Doxycycline, Erythromycin and Chloramphenicol. Later these plates were incubated for 24 h at 37 °C. Afterwards, resistance pattern of particular strain was studied based upon the zone of inhibition measurement following the CLSI 2017 guideline [30, 31].
Quantification by nanophotometer
For quantification purpose Multiskan™ GO Microplate Spectrophotometer was used. The standard stock solution of 2 mg/mL was prepared for each active compound by adding 0.02 g in 10 mL distilled water. From this stock solution various working dilutions ranging from 40 to 2000 μg/mL were prepared in triplicate. Wave scan of the dilutions was performed between 200–900 nm and λmax value was obtained from absorbance spectra. Afterwards, particular active compound λmax was used to measure optical density of dilutions and standard curve was plotted.
Antibacterial activity and minimum inhibitory concentration
Antimicrobial activity was assessed by broth dilution method. Clinical isolates were obtained from Microbiology and Public Health Laboratory culture collection, COMSATS University, Islamabad. Staphylococcus epidermidis (MDR) and Staphylococcus aureus (MDR) were the gram-positive and Escherichia coli (MDR) and Pseudomonas aeruginosa (MDR) were the gram-negative strains selected for this study.
Broth dilution method was performed for antibacterial activity of synthesized compounds against selected clinical microbes. MIC was calculated by broth dilution method, stock solution of 2 mg/mL for each active compound with DMSO 20% as solvent was prepared which was further consumed for preparing working dilutions ranged from 40 to 1000 μg/mL. The selected model bacterial strains were subjected to these active compounds dilutions separately and OD values were obtained at 595 nm [32].
Antifungal assay
Agar well diffusion method was utilized for antifungal assay using nystatin as positive control while DMSO (20%) was employed as a negative control. Sabouraud dextrose agar (SDA) and nutrient broth were prepared and sterilized by autoclaving at 121 °C for 15 min. Sterile agar plates were prepared by pouring the sterile agar in disposable sterile plates and incubated for 24 h for sterility check at 28 °C after congealing. The fungal strains Candida albicans (ATCC 60387) and Candida glabrata (ATCC 62934) were refreshed by inoculation in nutrient broth followed by 24 h incubation at 28 °C. Lawn of both fungal strains were made on nutrient agar plates and wells were made by employing sterile borer (6 mm). 50 μL of the synthesized compounds, nystatin and DMSO (20%) were poured through micropipette into individual well. The concentration of synthesized compounds, positives and negative controls used were 10 mg/mL and 750 μg/mL respectively. Plates were sealed by using paraffin film (Parafilm M) and incubated for 3–7 days at 28 °C to be examined for zone of inhibition that reflects the antifungal potential [33].
Result and discussion
Chemistry
Ethyl 2-aminothiazole-4-carboxylate was synthesized by reacting ethyl bromopyruvate and thiourea. Ethyl 2-aminothiazole-4-carboxylate (1a) was collected as off-white precipitates. Schiff bases 2a–2g were synthesized by reacting Ethyl 2-aminothiazole-4-carboxylate (1a) with different aldehydes and ketones as shown in Fig. 1.
Purity of all the synthesized compounds was ensured by recrystallization in appropriate solvents and checked by thin layer chromatography plates using ethyl acetate: petroleum ether (3:1) solvent system. Single spot yielded by each synthesized compound was obtained.
Synthesis of compound 1a was confirmed through FTIR and 1H NMR spectral data. Strong peak of C=O (ester) at 1690 cm−1, NH2 (amine) 3300–3150 cm−1 and C–H 3000 cm−1 stretch was observed in FTIR spectrum. 1H NMR spectrum showed singlet of amine and thiazole proton at 5.85 ppm and 7.41 ppm. Moreover, quartet and triplet of CH2 and CH3 was observed 4.34 ppm and 1.38 ppm respectively.
Confirmation of synthesized compounds was done by FTIR and 1H NMR spectral data. FTIR spectral data showed strong peak of C=O (ester) in case of each compound from 2a to 2e at cm−1: 1687, 1687, 1690, 1688 and 1696 respectively. Absence of amine peak and appearance of C=N (imine) confirmed the synthesis of Schiff bases in case of each derivative. FTIR spectral data showed strong peak of C=N (imine) in case of each compound from 2a to 2e at cm−1: 1616, 1617, 1614, 1612 and 1619 respectively. C–H stretch was observed at cm−1: 3023, 2975, 2983, 2953 and 3030 for compounds 2a–2e respectively. 1H NMR spectral data showed singlet of imine in case of each synthesized compound from 2a to 2e; ppm: 9.47, 9.76, 8.89, 9.48 and 8.42 respectively. Moreover, quartet of CH2 of synthesized compounds from 2a to 2e was observed a ppm: 4.25, 4.20, 4.21, 4.25 and 4.20 respectively. While triplet of CH3 in 1H NMR spectrum was observed in case of each compound from 2a to 2e at ppm: 1.26, 1.24, 1.24, 1.25 and 1.24 respectively.
Strong peak of C=O (ester) 1690 cm−1, C=N (imine) 1617 cm−1 and C–H stretch 2985 cm−1 were observed in FTIR spectrum of compound 2f. Singlets of CH3–C=N imine at 3.38 ppm and thiazole proton at 7.64 ppm were observed. Moreover, quartet and triplet of CH2 and CH3 were observed at 4.25 ppm and 1.26 ppm respectively. While in compound 2g peak of C=O (ester) was observed at 1685 cm−1. C=N (imine) 1615 cm−1 and C–H stretch 2990 cm−1 were also perceived in FTIR spectrum of compound 2g. 1H NMR spectral data showed singlets of CH3–C=N imine at 3.34 ppm and thiazole proton at 7.45 ppm were observed. Moreover, quartet and triplet of CH2 and CH3 were observed at 4.21 ppm and 1.23 ppm respectively.
Antibacterial assay of synthesized compounds
All experiments were conducted in the boundaries of ethical principles and there was no involvement of human or animal samples in this project.
Selected clinical gram-negative and gram-positive bacteria were subjected to antibiotic disks diffusion by Kirby Bauer assay and their pattern of resistance were determined. The resistance pattern of synthesized derivatives is given in Table 2.
Quantification
Spectrophotometric analysis was utilized for the quantification of compounds 2a, 2b, 2d and 2g. After performing wave scan the values obtained for λmax were 2a at 320 nm, 2b at 420 nm, 2d at 310 nm and 2g at 325 nm. λmax values were used to measure dilutions optical density (OD). These OD values were used to plot the standard curve and coefficient of determination (R2) value was calculated. The curve was plotted between OD values on y-axis while the concentration values on horizontal x-axis. From regression analysis, the R2 values obtained for compound 2a, 2b, 2d and 2g were 0.976, 0.999, 0.951 and 0.997 respectively. Values closer to 1 indicated the fitness of the data against the regression line. The graphs plotted are given in Fig. 2.

Optical Density (OD). Figure 1 illustrates the quantification of compounds by UV visible spectrophotometer. Linear line indicates the fitness of data against regression line
Minimum inhibitory concentration (MIC)
All the synthesized compounds manifested mild to moderate antibacterial potential. The lowest concentration of the drug exhibiting bacteriostatic action is stated to be the Minimum inhibitory concentration (MIC) of the drug. The MIC of synthesized derivatives is given in Table 3. Compound 2a and 2b showed quite good MIC of 250 µg/mL against gram-positive Staphylococcus epidermidis (MDR). Likewise compound 2d and 2g exhibited MIC of 250 µg/mL against gram-positive bacteria S. aureus (MDR). Compounds 2d and 2g exhibited MIC of 375 µg/mL against E. coli (gram-negative) (MDR). While compounds 2a and 2b showed MIC of 375 µg/mL against P. aeruginosa (gram-negative) (MDR).
Antifungal activity
Compounds 2a, 2b and 2d showed maximam antifungal potential against Candida glabrata (ATCC 62934) with a zone of inhibition (mm) of 13.0, 21.0 and 13.1 respectively. Candida albicans (ATCC 60387) showed sensitivity to compounds 2a, 2b 2d, 2f and 2g with a zone of inhibition (mm) of 20.0, 13.8, 19.1, 15.4 and 14.9 respectively (Table 4).
Insilico studies
Best compounds are selected computationally by analyzing them for their chemo-informatics and ADMET properties as given in Tables 5 and 6. According to literature, established qualifying range for log P value is (− 0.4 to 5.6). All compounds log p value lie within the limits. Molar refractivity should lie between (40 to 130) while molecular weight limits are (160 to 480). Moreover, the required number of atoms of compounds should lie between (20 to 70). Number of rotatable bonds should be less than 10. All compounds have 4 rotatable bonds except the compounds 2e and 2h that are having 5 rotatable bonds. All the compounds are fulfilling the set criteria for above parameters. Moreover, the drug likeliness following the Lipinski rule is also fulfilled. That’s why these compounds can be used as drug candidates.
Toxicity profile was determined by using TOXTREE and pkCSM online tools. All the compounds employed were having a therapeutically safe profile excluding compound 2h, which holds genotoxic potential. Therefore, compound 2h was not utilized in the docking step. Table 7 contains the toxicity profile of designed compounds.
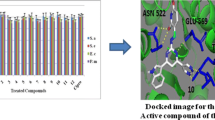
Our designed compounds were docked with the target enzyme UDP-N-acetylmuramate/l-alanine ligase and binding affinities were determined. The binding pocket of target enzyme was identified through Dogsitescorer. All the compounds docked with the biggest pocket of the enzyme target located on chain B. The volume and surface of the pocket is 963.07 [Å3] and 1100.44 [Å2] respectively. While the pocket possesses quite good drug score of 0.77. Figure 3 illustrates the druggable pocket of UDP-N-acetylmuramate/l-alanine ligase target enzyme.

a depicts the ligand binding pocket of UDP-N-acetylmuramate/l-alanine ligase target protein while b is showing the enlarged view of chain B ligand binding pocket
Results of docking of compounds are displayed (Table 8) in terms of free binding energy of ligand also stated as binding affinity (kcal/mol), root mean square deviation (RMSD) upper bound along with values of RMSD lower bound of all the docked ligands as well. Strength of binding interactions between ligand and receptor is termed as affinity. Calculation of RMSD values is based on the best binding pose and it employs movable heavy atoms only. Among the compounds 2a, 2b, 2d and 2g; 2b shown the strongest binding affinity of − 7.6 kcal/mol against UDP-N-acetylmuramate/l-alanine ligase. While compounds 2a, 2d and 2g exhibited binding affinity of − 6.8, − 7.3, − 7.1 and − 6.9 kcal/mol respectively. It is inferred from the binding affinities that the compounds having hydroxyl gyoup substituted on benzene ring possess strong binding affinity as compared to others. These designed compounds could be considered as antagonist lead molecules for target UDP-N-acetylmuramate/l-alanine ligase. The enzyme-ligand binding interactions of compounds 2b, 2c, 2d, and 2g with target UDP-N-acetylmuramate/l-alanine ligase are shown in Figs. 4, 5, 6, 7 respectively.

Interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2a. a Two dimensional diagram of protein–ligand interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2a. b Three dimensional view of compound 2a docking with UDP-N-acetylmuramate/l-alanine ligase, illustrates the pocket amino acids and bond distances of interacting groups of protein receptor and the ligand

Interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2b. a Two dimensional diagram of protein–ligand interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2b. b Three dimensional view of compound 2b docking with UDP-N-acetylmuramate/l-alanine ligase, illustrates the pocket amino acids and bond distances of interacting groups of protein receptor and the ligand

Interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2d. a Two dimensional diagram of protein–ligand interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2d. b Three dimensional view of compound 2d docking with UDP-N-acetylmuramate/l-alanine ligase, illustrates the pocket amino acids and bond distances of interacting groups of protein receptor and the ligand

Interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2g. a two dimensional diagram of protein–ligand interaction of UDP-N-acetylmuramate/l-alanine ligase with compound 2g. b Three dimensional view of compound 2g docking with UDP-N-acetylmuramate/l-alanine ligase, illustrates the pocket amino acids and bond distances of interacting groups of protein receptor and the ligand
Conclusion
The aim of the study is to design and synthesize novel Schiff base derivatives of ethyl-2-aminothiazole-4-carboxylate. Characterization was done by FTIR, 1H NMR and 13C NMR spectral data and quantification is done through Multiskan™ GO Microplate Spectrophotometer. Synthesized derivatives are screened for their antibacterial potential against Multi drug resistant (MDR) clinical isolates. Moreover, synthesized derivatives also exhibited good antifungal activity against ATCC fungal strains. It is anticipated that these synthesized compounds are promising potent antibacterial therapeutic agents. In future it is aimed to develop a pharmacophore model from these compounds to have a best lead molecule for the UDP-N-acetylmuramate/l-alanine ligase target enzyme.
Availability of data and materials
All the relevant data supporting the conclusions of this article is included in the article.
Abbreviations
- ATCC:
-
American Type Culture Collection
- CDCl3 :
-
deuterated chloroform
- DMSO:
-
dimethyl sulfoxide
- FTIR:
-
Fourier transform infrared
- MIC:
-
minimum inhibitory concentration
- NMR:
-
nuclear magnetic resonance
- SDA:
-
sabouraud dextrose agar
- TLC:
-
thin layer chromatography
References
Ajani OO, Obafemi CA, Nwinyi OC, Akinpelu DA (2010) Microwave assisted synthesis and antimicrobial activity of 2-quinoxalinone-3-hydrazone derivatives. Bioorg Med Chem 18(1):214–221
Ashraf MA, Mahmood K, Wajid A, Maah MJ, Yusoff I (2011) Synthesis, characterization and biological activity of Schiff bases. IPCBEE. 10:1–7
Karegoudar P, Karthikeyan MS, Prasad DJ, Mahalinga M, Holla BS, Kumari NS (2008) Synthesis of some novel 2, 4-disubstituted thiazoles as possible antimicrobial agents. Eur J Med Chem 43(2):261–267
Turan-Zitouni G, Kaplancıklı ZA, Yıldız MT, Chevallet P, Kaya D (2005) Synthesis and antimicrobial activity of 4-phenyl/cyclohexyl-5-(1-phenoxyethyl)-3-[N-(2-thiazolyl) acetamido] thio-4H-1, 2, 4-triazole derivatives. Eur J Med Chem 40(6):607–613
Rezanka T, Sobotka M, Spízek J, Sigler K (2006) Pharmacologically active sulfur-containing compounds. Anti Infect Agents Med Chem Form Curr Med Chem Anti Infect Agents 5(2):187–224
Costa M, Costa-Rodrigues J, Fernandes MH, Barros P, Vasconcelos V, Martins R (2012) Marine cyanobacteria compounds with anticancer properties: a review on the implication of apoptosis. Marine Drugs 10(10):2181–2207
Wang MW, Wei S, Faccio R, Takeshita S, Tebas P, Powderly WG, Teitelbaum SL, Ross FP (2004) The HIV protease inhibitor ritonavir blocks osteoclastogenesis and function by impairing RANKL-induced signaling. J Clin Investig 114(2):206–213
Morgan JB, Mahdi F, Liu Y, Coothankandaswamy V, Jekabsons MB, Johnson TA, Sashidhara KV, Crews P, Nagle DG, Zhou YD (2010) The marine sponge metabolite mycothiazole: a novel prototype mitochondrial complex I inhibitor. Bioorg Med Chem 18(16):5988–5994
Siddiqui N, Arya SK, Ahsan W, Azad B. Int J Drug Dev Res | October–December 2011| vol. 3| Issue 4| ISSN 0975-9344|. http://www.ijddr.in. Covered in Official Product of Elsevier, The Netherlands© 2010 IJDDR. Int. J. Drug Dev Res. 2011;3(4):156–64
de Souza MV (2005) Synthesis and biological activity of natural thiazoles: an important class of heterocyclic compounds. J Sulfur Chem 26(4–5):429–449
Siddiqui N, Arshad MF, Ahsan W, Alam MS (2009) Thiazoles: a valuable insight into the recent advances and biological activities. Int J Pharm Sci Drug Res. 1(3):136–143
Kajal A, Bala S, Kamboj S, Sharma N, Saini V (2013) Schiff bases: a versatile pharmacophore. J Catal 27:2013
Khalifa ME (2018) Recent developments and biological activities of 2-aminothiazole derivatives. Acta Chim Slov 65(1):1–22
Sacco E, Bientinesi R (2013) Mirabegron, a novel, non-antimuscarinic drug for the overactive bladder: an up-to-dated review. World J Obstet Gynecol 2(4):65–73
Guay DR (2002) Cefdinir: an advanced-generation, broad-spectrum oral cephalosporin. Clin Ther 24(4):473–489
Stroganov OV, Novikov FN, Stroylov VS, Kulkov V, Chilov GG (2008) Lead finder: an approach to improve accuracy of protein–ligand docking, binding energy estimation, and virtual screening. J Chem Inf Model 48(12):2371–2385
Jin H, Emanuele JJ, Fairman R, Robertson JG, Hail ME, Ho HT, Falk PJ, Villafranca JJ (1996) Structural studies of Escherichia coli UDP-N-acetylmuramate: l-alanine ligase. Biochemistry 35(5):1423–1431
Alderwick LJ, Harrison J, Lloyd GS, Birch HL (2015) The Mycobacterial cell wall—peptidoglycan and arabinogalactan. Cold Spring Harb Perspect Med 5(8):a021113
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1(64):4–17
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Patlewicz G, Jeliazkova N, Safford RJ, Worth AP, Aleksiev B (2008) An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ Res 19(5–6):495–524
Fährrolfes R, Bietz S, Flachsenberg F, Meyder A, Nittinger E, Otto T, Volkamer A, Rarey M (2017) Proteins Plus: a web portal for structure analysis of macromolecules. Nucleic Acids Res 45(W1):W337–W343
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Chemical biology. Humana Press, New York, pp 243–250
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Chem Inform 3(1):33
Niaz T, Shabbir S, Noor T, Abbasi R, Raza ZA, Imran M (2018) Polyelectrolyte multicomponent colloidosomes loaded with nisin Z for enhanced antimicrobial activity against foodborne resistant pathogens. Front Microbiol 15(8):2700
Hussain Z, Yousif E, Ahmed A, Altaie A (2014) Synthesis and characterization of Schiff’s bases of sulfamethoxazole. Org Med Chem Lett 4(1):1
Aziz HJ, Ali HH (2010) Synthesis of a new series of schiff bases using both traditional and the ultrasonic techniques. Tikrit J Pure Sci 15(3):70–74
Reller LB, Weinstein M, Jorgensen JH, Ferraro MJ (2009) Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin Infect Dis 49(11):1749–1755
Yarbrough ML, Lainhart W, Burnham CA (2018) Epidemiology, clinical characteristics, and antimicrobial susceptibility profiles of human clinical isolates of Staphylococcus intermedius group. J Clin Microbiol 56(3):e01788-17
Peng Z, Jin D, Kim HB, Stratton CW, Wu B, Tang YW, Sun X (2017) Update on antimicrobial resistance in Clostridium difficile: resistance mechanisms and antimicrobial susceptibility testing. J Clin Microbiol 55(7):1998–2008
Jamil B, Habib H, Abbasi SA, Ihsan A, Nasir H, Imran M (2016) Development of cefotaxime impregnated chitosan as nano-antibiotics: De novo strategy to combat biofilm forming multi-drug resistant pathogens. Front Microbiol 18(7):330
Sen A, Batra A (2012) Evaluation of antimicrobial activity of different solvent extracts of medicinal plant: melia azedarach L. Int J Curr Pharm Res. 4(2):67–73
Acknowledgements
We are thankful to Dr. Muhammad Imran Assistant Professor, Department of Biosciences, Comsats University Islamabad for his valuable support and suggestions.
Funding
Not applicable. (No specific funding or grant).
Author information
Authors and Affiliations
Contributions
Authors HN and RZP carried out study design, supervised the work and refined the manuscript for publication. SE1 did experimental work in synthesis of compounds, molecular docking and antifungal assay and prepared the manuscript. SE2 and SS performed anti-microbial evaluation including resistant profiling, quantification and minimum inhibitory concentration assays and prepared the related manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ejaz, S., Nadeem, H., Paracha, R.Z. et al. Designing, synthesis and characterization of 2-aminothiazole-4-carboxylate Schiff bases; antimicrobial evaluation against multidrug resistant strains and molecular docking. BMC Chemistry 13, 115 (2019). https://doi.org/10.1186/s13065-019-0631-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0631-6