
Abstract
Background
Typically, oxazolinyl metal complexes are synthesized in two steps, where the free ligand is prepared by the condensation reaction between a functionalized nitrile and an amino alcohol in the presence of a Lewis or Brønsted acid catalyst, followed by a further reaction with metal salts to obtain the corresponding metal complexes. Very often, the yield afforded by the two-step procedure is not high, and very few oxazolinyl zinc complexes have been prepared by this route. Given that metal-oxazoline complexes often contain Lewis acidic metals, it is conceivable that the two steps may be telescoped.
Results
A series of novel chiral organozinc complexes 1–15 were assembled in a single step, All crystalline compounds were fully characterized, including the report of 15 X-ray crystal structures, including a wide structural diversity.
Conclusions
A series of novel chiral organozinc complexes were assembled in a single step, from nitriles, chiral D/L amino alcohols, and a stoichiometric amount of ZnCl2, with moderate to high yields (20–90%).

A series of novel chiral organozinc complexes can be assembled in moderate to high yields (20–90%), from a mixture of nitriles, chiral amino alcohols and ZnCl2. All crystalline compounds were fully characterized, including the report of 15 X-ray crystal structures, including a wide structural diversity.
Similar content being viewed by others
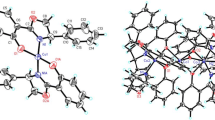


Background
Chiral oxazolines constitute an important class of ‘privileged’ ligands in asymmetric catalysis [1,2,3,4,3]. Chiral zinc complexes containing these ligands exhibit a broad range of catalytic activities, including the asymmetric Mukaiyama-aldol reactions of α-ketoesters [4], the Henry reaction [5], isoselective ring-opening polymerization of rac-lactide [6], and asymmetric co-polymerisation of cyclohexene oxide with CO2 [7]. More recently, a chiral boxmi-Zn catalyst has been reported to be highly effective for the enantioselective alkylation of oxindoles and α-ketoesters, thought to proceed through an usual radical pathway [8].
Typically, oxazolinyl metal complexes are synthesized in two steps, where the free ligand is prepared by the condensation reaction between a functionalized nitrile and an amino alcohol in the presence of a Lewis or Brønsted acid catalyst, followed by a further reaction with metal salts to obtain the corresponding metal complexes (Scheme 1) [9, 10]. Very often, the yield afforded by the two-step procedure is not high, and very few oxazolinyl zinc complexes have been prepared by this route. Given that metal-oxazoline complexes often contain Lewis acidic metals, it is conceivable that the two steps may be telescoped. Herein, we will report a simple, one-pot procedure for the preparation of oxazolinyl-zinc complexes by the atom-efficient assembly of three reactive components: a nitrile, an amino alcohol and a zinc salt. In all cases, the complexes were isolated, purified and characterized; their structures were further confirmed by X-ray crystallography.

Two-step synthesis of oxazolinyl-metal complexes
Results and discussion
The one-pot procedure was initially tested by refluxing a mixture of 1-piperidinepropionitrile with 2–3 eq of amino alcohol in the presence of ZnCl2 (1–2.5 eq) in chlorobenzene. Following the reaction, excess ZnCl2 can be removed by an aqueous wash, and the metal complexes were isolated and purified by column chromatography. During the preliminary work, it became quickly apparent that the reaction outcome is highly dependent upon the amount of ZnCl2 used (Scheme 2): Using 1.1 eq of the metal salt, the desired amino-oxazolidinyl complex 1 can be obtained from l-leucinol, but only in a low yield (25%). While the use of an excess (2.5 eq) of the zinc salt with l-valinol led to the formation of the bis-oxazolidinyl zinc complex 2, containing two monodentate ligands.

Effect of reaction stoichiometry (metal precursor) ZnCl2
The nature of the side chain (R1) also influenced the reaction outcome: using l-phenylalaninol with ZnCl2 (1.6 eq) led to the cleavage of the propionitrile to give the unsymmetrical diamine complex 3 in a very good yield (86%). Similarly, addition of the 1.5 eq of ZnCl2 to 1-morpholinepropionitrile (X=O) and d-phenylglycinol furnished complex 4 in 90% yield. Interestingly, using 1-(2-cyanoethyl)-4-methylpiperazine (Z=NMe) as a precursor with 2.5 eq of the ZnCl2 led only to the formation of the zwitterionic piperazine-complex 5, irrespective of the amino alcohol used.
The formation of complexes 2–5 indicates that the propionitrile precursors are unstable under the reaction conditions in the presence of excess ZnCl2, which can decompose into acetonitrile (affording 2) or the parent cyclic amines (3–5). With this in mind, a number of nitrile precursors were chosen which are more robust against degradation under the reaction conditions. Consequently, a number of aromatic nitrile precursors containing additional N-donors were examined as precursors in these 3-component reactions. In these reactions, the amount of ZnCl2 was carefully optimizedto ensure a specific outcome. The use of 3-aminobenzonitrile and D-leucinol in the presence of 0.44 eq of ZnCl2 led to the formation of complex 6 containing two monodentate ligands coordinating via the oxazoline nitrogen (Scheme 3). The use of 2-cyanopyridine with 1.2 eq of ZnCl2, on the other hand, led to different outcomes with different amino alcohols: the formation of a bis-chelated complex 7 was obtained with l-phenylalaninol, while the mono-chelated complex 8 was obtained from d-valinol. This result highlights the importance of the sidechain present in the amino alcohol precursor; presumably, the sterically bulky isopropyl group prevented the formation of the bis-chelate complex.

Zinc complexes derived from 3-aminobenzonitrile and 2-cyanopyridine
It was anticipated that oxazolines derived from 1, 2-dicyanobezene will provide C2-symmetricalbis-oxazolines that form 7-membered chelate rings, which can only form a 1:1 adduct with zinc dichloride. Indeed, the condensation of isophthalonitrile with d-phenylglycinol (0.56 eq) afforded the predicted mono-chelated complex 9 [11] in a good yield (Scheme 4). However, the presence of a slight excess of l-valinol (0.72 eq) caused the condensation of three amino alcohols in complex 10.

Complexes derives from isophathalonitrile
Condensation of l-leucinol and phenyl glycinol with tetracyanoethylene in the presence of 0.42 eq of ZnCl2 provided neutral bis[bis(oxazoline)]zinc (II) complexes 11 and 12, respectively, in good yields (Scheme 5). The formation of these methylene-bis(oxazoline) structures indicates disproportionation-rearrangement of the tetracyanoethylene precursor (to tricyanomethane), although the precise mechanism of this is unclear. During the preparation of this manuscript, the synthesis complex 12 (by a different route) was reported by Kögel et al. [12] Interestingly, compound 12 was reported to display intense Cotton effect as a result of exciton coupling. Indeed, a comparison of their X-ray crystal structures revealed that the isobutyl-substituted complex 11 possesses a fairly symmetrical tetrahedral coordination environment; while, in contrast, complex 12 is highly distorted (See Figs. 11 and 12 in Additional file 1). We speculate this may be due to the favourable intramolecular π-interaction between one of the phenyl substituent with the semicorrin structure of the adjacent ligand within 3.5 Å, effectively bringing the two chiral chromophores into close proximity to facilitate exciton coupling [13].

Neutral zinc complexes derived from tetracyanoethylene
In the final part of this study, 2-hydroxy-6-methylnicotinonitrile was employed as a precursor, to test the utility of the one-pot methodology in assembling complex multinuclear structures. Condensation product with valinol furnished the binuclear zwitterionic complex 13 (Scheme 6). Presumably, the formation of higher aggregates is prevented by the sterically demanding isopropyl substituent.
Highly symmetrical tetramers 14 and 15 (Scheme 6) were formed when leucinol or phenylalaninol were used as precursors in the presence of 1.5 eq of ZnCl2 (See Figs. 14 and 15 in Additional file 1). A six-membered N, O-chelate is formed preferentially at each metal centre, and the pendant pyridine acting as a bridging donor ligand to another metal centre. With each zinc occupying a corner of a square grid, the planar N,O,N-ligands are oriented perpendicularly to one another with diagonal Zn···Zn distance of ca. 6 Å.

Multinuclear zinc complexes
The X-ray crystal structures of all the complexes are determined and reported in the supporting information. In all cases, a distorted tetrahedral geometry is found at the zinc(II), and the C=N double bond character of the oxazolindinyl ligand is largely retained in the metal complexes.
General remarks
Unless otherwise stated, all chemical reagents were purchased from Acros, Aldrich, or Fluka USA. Flash column chromatography was performed using Merck silica gel (60, particle size 0.02–0.03 mm). 1H and 13C NMR spectra were recoZrded using Bruker AM-500 or AM-600 spectrometers. Chemical shifts are reported in ppm (δ) with the solvent relative to tetramethylsilane (TMS) employed as the internal standard (residual CHCl3, δH 7.26 ppm; CDCl3, δc 77 ppm). The following abbreviations were used to δ designate multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet. Infrared spectra were recorded on a Mattson Galaxy Series FTIR 3000 spectrometer; peaks are reported in cm−1. Elemental analyses were obtained on Elemental Analyzer AE-3000. High-resolution mass spectra (HRMS) were obtained on a Micro GCT-MS equipped with an EI ion source. Optical rotations were measured on a WZZ-1 automatic polarimeter with a 2-cm cell, recorded at the sodium d-line.
The procedure for the synthesis of the complexes 1–15
1-[2-(4-isobutyl-4,5-dihydro-oxazol-2-yl)-ethyl]-piperidine zinc(II) dichloride, 1
A dry 100 mL Schlenk flask was purged with N2 and charged with anhydrous ZnCl2 (2.515 g, 18.45 mmol), 3-piperidin 1-yl propionitrile (2.462 g, 17.81 mmol) and l-leucinol (4.824 g, 41.16 mmol). 40 mL of chlorobenzene was added, and the reaction mixture was refluxed for 72 h. After cooling to room temperature, the solvent was removed under reduced pressure, and the residue was dissolved in 15 mL of H2O and extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were evaporated to give a crude red oil, which was purified by column chromatography (petroleum ether/CH2Cl2, 4/1) to afford the title compound as colourless crystals in 25% yield, m.p. 50–52 °C \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +67.5° (c = 0.02, MeOH); δH (600 MHz, CDCl3, 27 °C) 4.52–4.56 (m, 1H), 4.12–4.16 (m, 1H), 4.01–4.03 (m, 1H), 2.89–2.92 (m, 1H), 2.73–2.75 (m, 1H), 2.59–2.64 (m, 3H), 2.31 (t, J = 12.4 Hz, 1H), 1.62–1.78 (m, 6H), 1.43–1.44 (m, 2H), 1.29–1.35 (m, 2H), 1.15–1.20 (m, 1H), 0.84–0.90 (m, 6H); δC (150 MHz, DMSO-d6), 170.0, 73.7 (×2), 65.5, 62.9 (×2), 54.7, 46.1, 44.4, 43.3, 25.1, 24.0, 23.4, 22.6. νmax(cm−1) 3274, 2954, 2869, 1648, 1587, 1468, 1387, 1368, 1319, 1283, 1169, 1076 1041, 979, 956, 949, 904, 864, 839, 780, 607, 493. Found C: 45.36, H: 7.19, N: 7.75%; C14H26Cl2N2OZn requires C: 44.88, H: 7.00, N: 7.48%.
Bis-[4-isopropyl-2-methyl-4,5-dihydro-oxazole]zinc(II) dichloride, 2
Prepared using the same procedure described above for complex 1, from a mixture of ZnCl2 (5.401 g, 39.63 mmol), 3-piperidin1-yl propionitrile (2.321 g, 16.79 mmol) and l-valinol (5.319 g, 51.56 mmol) in chlorobenzene (80 mL). The product was obtained as colourless crystals in 65% yield after column chromatography (petroleum ether/CH2Cl2, 2/1), m.p. 60–62 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +9.97° (c = 0.35, MeOH); δH (600 MHz, CDCl3, 27 °C) 3.64–3.71 (m, 6H), 2.03 (s, 6H), 1.85–1.88 (m, 2H), 0.92 (d, J = 6.8 Hz, 6H), 0.96 (d, J = 7.8 Hz, 6 H); δC (150 MHz, CDCl3), 171.2(×2), 63.7, 63.6, 57.1, 53.5, 31.7, 28.9, 23.4, 22.9, 19.3, 19.0, 18.8, 17.9; νmax(cm−1) 3436, 3284, 3145, 2954, 2864, 1725, 1659, 1585, 1458, 1420, 1393, 1319, 1278, 1280, 1219, 1092, 1038, 974, 952, 905, 765. Found C: 43.49, H: 7.19, N: 7.23%; C14H26Cl2N2O2Zn requires C: 43.04, H: 6.71, N: 7.17%.
α-Phenyl-1-hexahydropyridyl ethylamine zinc(II), 3
Prepared using the same procedure described above, using anhydrous ZnCl2 (3.5002 g, 25.68 mmol), 3-piperidin1-yl propionitrile (2.4590 g, 17.79 mmol), and l-phenylalaninol (5.5420 g, 40.40 mmol) in 80 mL of dry chlorobenzene. The product was obtained as colorless crystals after column chromatography (petroleum ether/dichloromethane, 1/2) in 86% yield, m.p: 168–172 °C; \( \left[ {{\text{a}}} \right]_{\text{D}^{25}} \) = −29.30º (c = 0.016, CH3OH): δH (600 MHz, CDCl3, 27 °C) 7.32–7.43 (m, 5H), 4.23–4.32 (m, 1H), 3.42–3.64 (m, 3H), 2.96–3.05 (m, 2H), 2.61–2.65 (m, 1H), 2.19–2.41 (t, J = 912.4 Hz, 1H), 1.62–1.78 (m, 6H), 1.43–2.41 (m, 3H), 1.69–1.98 (m, 6H), 1.24–1.31 (m, 1H), δC (150 MHz, DMSO-d6), 169.2, 141.8, 128.9, 128.4, 127.4, 127.3, 127.1 65.2, 65.1, 55.4, 55.2, 53.8, 51.9, 25.3, 23.2(×2); νmax: 3447, 3027,2943, 2860, 1648, 1603, 1496, 1455, 1132, 1043, 1060,1040, 1030, 762, 705. Elemental analysis: Found C: C:47.20%, H, 6.05%, N, 7.60%; C14H22Cl2N2Zn requires C: 47.42, H: 6.25, N: 7.90%.
2-Morpholin-4-(R)-yl-1-phenyl-ethylamine zinc(II) dichloride complex, 4
Prepared as described above, from a mixture of anhydrous ZnCl2 (1.780 g, 13.06 mmol), N-cyanoacetylmorpholine (1.501 g, 9.74 mmol), d-phenylglycinol (4.097 g, 29.87 mmol) and dry chlorobenzene (40 mL). The reaction mixture was refluxed for 60 h. The product was purified by column chromatography (petroleum ether/CH2Cl2, 1/100) to afford the title compound as colourless crystals in 90% yield, m.p. 196–198 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = −42.43° (c = 0.13, THF); δH (500 MHz, DMSO-d6, 27 °C), 7.48 (d, J = 8.8 Hz, 2H), 7.36 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.3 Hz, 1H), 4.95 (br s, 2H), 4.14 (t, J = 11.9 Hz, 1H), 3.88-3.91 (m, 2H), 3.78–3.81 (m, 2H), 2.97–3.00 (m, 2H), 2.67–2.82 (m, 4H); δC (125 MHz, DMSO-d6) 139.9, 128.5 (×2), 127.9, 127.0 (×2), 65.6, 65.2 (×2), 54.6 (×2), 51.2; νmax(cm−1) 3435, 3271, 3228, 3145, 2974, 2928, 2904, 2865, 1591, 1499, 1458, 1447, 1290, 1263, 1146, 1126, 1094, 1072, 1060, 1037, 988, 899, 874, 749, 694. Found C: 42.39, H: 5.24, N: 8.05%; C12H18N2Cl2OZn requires C: 42.07, H: 5.30, N: 8.18%.
(1-Methyl-piperazine)zinc(II) trichloride, 5
Prepared using the same procedure described for complex 5, from a mixture of anhydrous ZnCl2 (5.008 g, 36.75 mmol), 1-(2-cyanoethyl)-4-methylpiperazine (2.313 g, 15.09 mmol) and d-phenylglycinol (10.696 g, 77.97 mmol) in 40 mL of dry chlorobenzene. The product was recrystallized from ethanol/CH2Cl2, to furnish colourless crystals in 56% yield; m.p. 148–152 °C; δH (600 MHz, CDCl3 and DMSO-d6, 27 °C) 4.09–4.12 (m, 1H), 3.64–3.67 (m, 1H), 3.56–3.59 (m, 1H), 2.86–2.88 (m, 4H), 2.37–2.40 (m, 3H), 2.16 (s, 3H); δC (150 MHz, CDCl3 and DMSO-d6) 62.1, 54.7, 51.4, 44.0, 42.5. νmax(cm−1) 3491, 3455, 3189, 3006, 2956, 2771, 1585, 1458, 1387, 1128, 1099, 1058, 1035, 998, 976, 870, 701. Found: C: 22.20, H: 4.56, N: 10.10%; C5H13Cl3N2Zn requires C: 22.01, H: 4.80, N: 10.27%.
2-[4R-4,5-dihydro-4-(1′,1′-dimethylethyl)-3-oxazolinyl]aniline zinc(II) dichloride, 6
Prepared using the same procedure described for complex 1, from a mixture of anhydrous ZnCl2 (3.002 g, 22.02 mmol), 3-amino-benzonitrile (6.702 g, 56.73 mmol), and d-leucinol (10.008 g, 85.40 mmol) in 80 mL of dry chlorobenzene. The reaction mixture was refluxed for 72 h. After evaporation, the residue was dissolved in 15 mL of H2O and extracted with CH2Cl2 (3 × 10 mL). The organic layer was evaporated under vacuum, and the red oily residue was purified by column chromatography over silica gel (petroleum ether/CH2Cl2, 1/4), yield: 90%; m.p. 168–170 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = −54.9° (c = 0.0364, EtOH). δH (600 MHz, CDCl3 and DMSO-d6, 27 °C) 7.77–7.90 (m, 1H), 6.98–7.19 (m, 5H), 6.63–6.76 (m, 2H), 5.19–5.33 (m, 1H), 4.26–4.58 (m, 4H), 3.92–3.93 (m, 1H), 3.21–3.25 (m, 4H), 1.70–1.82 (m, 4H), 1.35–1.44 (m, 2H), 0.89–0.96 (m, 12H); δC (150 MHz, CDCl3 and DMSO-d6) 167.0, 161.3, 146.9, 146.4, 134.2, 126.9, 115.0, 113.9, 113.0, 112.8, 111.5(×2), 109.9(×2), 70.9, 63.0, 46.5, 45.3, 43.7, 39.5, 23.5, 23.2, 21.5, 21.1, 21.0, 20.3; νmax(cm−1) 3353, 2957, 2928, 2870, 1625, 1498, 1467, 1386, 1333, 1290, 1171, 1135, 1108, 996, 966, 948, 882, 797, 750, 688, 576, 537. Found C: 54.65, H: 6.24, N: 10.16%; C26H36N4Cl2O2Zn requires C: 54.51, H: 6.33, N: 9.78%.
Bis-[2-(4R-benzyl-4,5-dihydro-oxazol-2-yl)-pyridine]zinc(II) tetrachlorozincate, 7
Prepared using the same procedure described for compound 1, using anhydrous ZnCl2 (3.340 g, 24.51 mmol), 2-cyanopyridine (2.095 g, 20.13 mmol) and l-phenylalaninol (3.992 g, 26.40 mmol) in 40 mL of dry chlorobenzene, and the reaction mixture was refluxed for 60 h. The product was extracted into CH2Cl2 as described above, and purified by column chromatography (petroleum ether/CH2Cl2, 1/4) to give colourless crystals in 80% yield; m.p. 134–136 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +51.4° (c = 0.0272, MeOH); δH (600 MHz, DMSO-d6, 27 °C) 8.78–8.81 (m, 2H), 7.95–8.03 (m, 4H), 7.63–7.68 (m, 2H), 7.22–7.30 (m, 10H), 4.64–4.67 (m, 4H), 4.49–4.51 (m, 2H), 3.30–3.39 (m, 2H), 3.35 (s, 2H), 2.82–2.86 (m, 2H); δC (150 MHz, DMSO-d6) 163.8, 148.5, 137.7, 135.5, 128.2, 127.6, 126.7, 125.7, 122.4, 73.6, 64.5, 39.2; νmax(cm−1) 3493, 3061, 3027, 2955, 2920, 2853, 1660, 1590, 1571, 1492, 1469, 1452, 1440, 1404, 1388, 1325, 1293, 1244, 1223, 1154, 1143, 1088, 1045, 1014, 947, 847, 801, 746, 703, 681, 632; Found C: 57.43, H: 5.03, N: 8.67%; C30H30Cl2N4O3Zn2 requires C: 57.12, H: 4.79, N: 8.88%.
[2-(4S-isopropyl-4,5-dihydro-oxazol-2-yl)-pyridine] zinc (II) dichloride, 8
Prepared using the procedure described above for compound 1, refluxing a mixture of anhydrous ZnCl2 (3.423 g, 25.12 mmol), 2-cyanopyridine (2.128 g, 20.44 mmol), and l-valinol (3.386 g, 32.82 mmol) in 40 mL of dry chlorobenzene for 60 h. The product was purified by column chromatography (petroleum ether/CH2Cl2, 1/8). Colourless crystals were obtained in 85% yield; m.p. 178–180 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +23.1° (c = 0.17, MeOH); δH (600 MHz,CDCl3, 27 °C) 8.78–8.80 (m, 1H), 8.20–8.23 (m, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.86–7.88 (m, 1H), 4.96 (t, J = 9.5 Hz, 1H), 4.65 (t, J = 8.9 Hz, 1H), 4.42–4.45 (m, 1H), 2.10–2.14 (m, 1H), 1.04–1.14 (m, 6H); δC (150 MHz, CDCl3) 166.1, 149.8, 141.2, 140.2, 129.7, 124.1, 75.4, 69.3, 31.6, 18.4, 17.8; νmax(cm−1) 3223, 3188, 2962, 2875, 1662, 1587, 1470, 1392, 1372, 1320, 1251, 1129, 1046, 878, 836, 791, 752, 690, 539. Found C: 40.81, H: 3.85, N: 8.47%; C11H14Cl2N2OZn requires C: 40.46, H: 4.32, N: 8.58%.
[1, 2-bis–(4R-phenyl-4,5-dihydro-oxazol-2-yl)phenyl] zinc(II) dichloride, 9
Prepared using the same procedure described above for compound 1, using anhydrous ZnCl2 (2.590 g, 19.01 mmol), isophthalonitrile (3.353 g, 26.17 mmol), and d-phenylglycinol (8.492 g, 61.91 mmol) in 80 mL of dry chlorobenzene, and refluxing for 72 h. The product was purified by column chromatography (petroleum ether/CH2Cl2, 1/8). Yield = 86%; m.p. >250 °C (dec), \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = −54.9° (c = 0.0364, EtOH).δH (600 MHz, CDCl3, 27 °C) 7.77–7.79 (m, 2H), 7.55–7.56 (m, 2H), 7.18–7.28 (m, 10H), 5.28 (t, J = 9.2 Hz, 2H), 4.68 (t, J = 9.2 Hz, 2H), 4.10 (t, J = 8.4 Hz, 2H),δC (150 MHz, CDCl3) 163.5, 140.3, 129.4 (×2), 128.4, 127.0 (×2), 126.0,125.3 (×2), 73.9, 68.3. νmax(cm−1) 3447, 3058, 2965, 2907, 1650, 1639, 1592, 1495,1473, 1455, 1379, 1363, 1318, 1308, 1278, 1238, 1207, 1153, 1120, 1067, 1020, 991, 945, 760, 704, 648, 594, 556. Found C: 56.92, H: 3.92, N: 5.41%; C24H20Cl2N2O2Zn requires C: 57.11, H: 3.99, N: 5.55%.
2-{(4S-isopropyl-4,5-dihydro-oxazol-2-yl)-phenyl-4,5-dihydro-imidazol-1-yl}-3-methyl-butan-1-ol zinc(II), 10
Prepared using the same procedure described for compound 1, refluxing a mixture of anhydrous ZnCl2 (4.000 g, 29.35 mmol), isophthalonitrile (6.700 g, 52.29 mmol), and l-valinol (16.000 g, 15.51 mmol) in 80 mL of dry chlorobenzene for 72 h. The product was purified by column chromatography (petroleum ether/CH2Cl2, 1/4). Yield: 90%; m.p. >250 °C (dec), \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +34.4° (c = 0.0436, CHCl3),δH (600 MHz, DMSO-d6, 27 °C), 7.78–7.81 (m, 2H), 7.69–7.71 (m, 1H), 7.63–7.66 (m, 1H), 4.90–4.93 (m, 1H), 4.65 (t, J = 9.4 Hz, 1H), 4.51–4.55 (m, 1H), 4.45 (t, J = 7.8 Hz, 2H), 4.27 (t, J = 5.0 Hz, 1H), 3.75 (d, J = 11.2 Hz, 1H), 3.62–3.65 (m 2H), 3.47–3.50 (m, 1H), 2.21–2.24 (m, 1H), 1.70–1.74 (m, 1H), 0.94–0.99 (m, 8H), 0.85–0.86 (m, 4H), 0.72 (d, J = 6.6 Hz, 3H), 0.59 (d, J = 6.6 Hz, 3H); δC (150 MHz, CDCl3 and DMSO-d6) 165.6, 163.1, 130.2, 129.4, 128.7 (×2), 125.4, 123.5, 68.4, 67.7, 64.2, 61.1, 57.5, 42.5, 29.9, 29.4, 27.0, 25.1, 17.9, 17.5, 16.9, 16.2, 13.9, 13.0. Found C: 53.55, H: 6.87, N: 7.78%; C23H35N3Cl2O2Zn requires C: 52.94, H: 6.76, N: 8.05%. νmax(cm−1) 3436, 2961, 2923, 2874, 1635, 1604, 1571, 1520, 1464, 1377, 1317, 1300, 1138, 1074, 1047, 1026, 946, 784, 766.
Bis-[(4S-isobutyl-4,5-dihydro-oxazol-2-yl)-acetonitrile] zinc(II), 11 [11]
Prepared using the procedure described above for compound 1, by refluxing a mixture of anhydrous ZnCl2 (0.450 g, 3.30 mmol), tetracyanoethylene (1.000 g, 7.81 mmol), and l-leucinol (4.029 g, 34.38 mmol) in 40 mL of dry chlorobenzene for 60 h. The product was obtained in 88% yield as colourless crystals after column chromatography (petroleum ether/dichlormethane, 4/1). m.p. > 220 °C (dec); \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +166.33° (c = 0.30, CH2Cl2): δH (500 MHz, CDCl3, 27 °C) 4.60 (t, J = 7.3 Hz, 4H), 3.94–4.05 (m, 8H), 1.29–1.72 (m, 12H), 0.89–0.93 (m, 24H); δC (125 MHz, CDCl3) 170.1, 118.3, 73.0, 61.6, 45.6, 25.0, 22.3, 21.8. νmax(cm−1) 3439, 2955, 2927, 2871, 2201, 1611, 1530, 1430, 1386, 1368, 1342, 1281, 1260, 1239 1218, 1133, 1068, 1048, 951, 746. Found: C: 59.32, H: 7.46, N: 13.77%; C32H48N6O4Zn requires C: 59.48, H: 7.49, N: 13.01%.
Bis-[(4S-phenyl-4,5-dihydro-oxazol-2-yl)-acetonitrile] zinc(II), 12 [12]
Prepared using the procedure described above for compound 1, by refluxing a mixture of anhydrous ZnCl2 (0.450 g, 3.30 mmol), tetracyanoethylene (1.000 g, 7.81 mmol), and L-phenylglycinol (10.089 g, 7.35 mmol) in 40 mL of dry chlorobenzene for 60 h. The product was obtained in 86% yield as colourless crystals after column chromatography (petroleum ether/CH2Cl2, 2/1) m.p. > 220 °C (dec), \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +306.6° (c = 0.17, CH2Cl2). δH (500 MHz, CDCl3, 27 °C) 7.22–7.26 (m, 12H), 6.82 (d, J = 6.9 Hz, 8H), 4.50–4.60 (m, 8H), 3.95 (t, J = 7.2 Hz, 4H); δC (125 MHz, CDCl3) 171.3, 138.6, 129.3 (×2), 129.1, 126.8 (×2), 118.5, 74.6, 67.4; νmax(cm−1) 3032, 2903, 2202, 1608, 1526, 1429, 1455, 1362, 1264, 1220, 1075, 1051,911, 734, 701. Found C: 65.99, H: 4.20, N: 11.28%; C40H32N6O4Zn requires C: 66.17, H: 4.44, N: 11.57%.
[3-(4S-isopropyl-4,5-dihydro-oxazol-2-yl)-6-methyl-2-ol]zinc(II) chloride dimer, 13
Prepared using the procedure described above for compound 1, by refluxing a mixture of anhydrous ZnCl2 (3.502 g, 25.70 mmol), 2-hydro-6-methyl-nicotinonitrile (2.002 g, 14.92 mmol) and l-valinol (8.025 g, 77.79 mmol) in 40 mL of dry chlorobenzene for 60 h. The product was obtained in 80% yield as colourless crystals after column chromatography (petroleum ether/CH2Cl2, 4/1) m.p. 168–170 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = +162.8° (c = 0.181, MeOH); δH (600 MHz, CDCl3 and DMSO-d6, 27 °C) 12.36(br s, 1H), 8.27 (d, J = 7.7 Hz, 2H), 6.57 (d, J = 7.7 Hz, 2H), 4.56–4.58 (m, 2H), 4.50–4.53 (m, 2H), 4.37–4.39 (m, 2H), 2.68 (s, 6H), 2.16–2.18 (m, 2H), 0.99 (d, J = 6.9 Hz, 6H), 0.93 (d, J = 6.7 Hz, 6H); δC (150 MHz, DMSO-d6)164.0, 162.5, 155.1 (×2), 146.7 (x4), 110.1 (×2), 108.5 (×2), 69.6 (×2), 68.2 (×2), 30.0 (×2), 20.1 (×2), 18.9 (×2), 15.0 (×2); νmax(cm−1) 3420, 2962, 2928, 2874, 1726, 1660, 1612, 1564, 1388, 1325, 1214, 1150, 1084, 986, 953, 790, 750, 701, 597, 469. Found C: 37.52, H: 4.22, N: 7.28%; C25H32Cl6N4O4Zn2 (CHCl3 solvate) requires C: 37.72, H: 4.05, N: 7.04%.
Tetra-[3-(4S-isobutyl-4,5-dihydro-oxazol-2-yl)-6-methyl-2-ol] zinc(II) chloride, 14
Prepared using the procedure described above for compound 1, by refluxing a mixture of anhydrous ZnCl2 (1.500 g, 11.01 mmol), 2-hydro-6-methyl-nicotinonitrile (1.002 g, 7.47 mmol) and L-leucinol (4.022 g, 34.32 mmol) in 40 mL of dry chlorobenzene for 60 h. The product was obtained in 86% yield as colourless crystals after column chromatography (petroleum ether/CH2Cl2, 1/1). m.p. 120–124 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) =+30.0° (c = 0.08, THF). δH (600 MHz, CDCl3, 27 °C), 8.02 (d, J = 7.8 Hz, 2H), 7.98 (d, J = 7.8 Hz, 2H), 6.15 (d, J = 5.5 Hz, 2H), 6.14 (d, J = 5.4 Hz, 2H), 4.86 (t, J = 8.7 Hz, 2H), 4.48–4.56 (m, 6H), 4.29 (d, J = 7.8 Hz, 1H), 4.28 (d, J = 7.9 Hz, 1H), 3.94 (t, J = 8.6 Hz, 2H), 2.41(s, 6H), 2.44(s, 6H), 1.90–1.94 (m, 2H), 1.57–1.69 (m, 6H), 1.21–1.43 (m, 4H), 0.82 (t, J = 7.5 Hz, 12H), 0.74 (d, J = 6.6 Hz, 6H), 0.57 (d, J = 6.6 Hz, 6H). δC (150 MHz, CDCl3) 167.7, 167.5, 165.3, 164.8, 163.4, 163.2, 143.4, 143.3, 111.7, 111.5, 105.3, 105.1, 73.0, 72.8, 63.8, 63.4, 43.9, 43.1, 26.1 (×2), 25.3, 25.2, 22.7, 22.6, 22.5 (×2); νmax(cm−1) 2957, 2929, 2870, 1648, 1579, 1490, 1386, 1322, 1284, 1250, 1205, 1153, 1077, 1060, 953, 883, 787, 749, 707, 620, 595, 419. Found C: 46.98, H: 5.12, N: 7.99%; C52H68Cl4N8O8Zn4 requires C: 46.73, H: 5.13, N: 8.38%.
Tetra-{3-[4(R)-benzyl-4,5-dihydro-oxazol-2-yl]-6-methyl-2-ol}zinc complex, 15
Prepared using the procedure described above for compound 1, by refluxing a mixture of anhydrous ZnCl2 (1.562 g, 11.46 mmol), 2-hydro-6-methyl-nicotinonitrile (1.000 g, 7.46 mmol), and D-phenylalaninol (4.008 g, 26.51 mmol) in 40 mL of dry chlorobenzene for 60 h. The product was obtained in 82% yield as colourless crystals after column chromatography (petroleum ether/CH2Cl2, 1/2). m.p. 120–124 °C, \( \left[ {\alpha } \right]_{\text{D}}^{25} \) = −109.0° (c = 0.164, THF); δH (600 MHz, DMSO-d6, 27 °C) 12.36–12.41 (m, 3H), 9.78 (d, J = 8.0 Hz, 4H), 8.11 (d, J = 7.2 Hz, 2H), 7.13–7.22 (m, 17H), 6.23(d, J = 7.2 Hz, 2H), 4.90 (s, 3H), 4.08 (d, J = 5.2 Hz, 3H), 3.34–3.40 (m, 6H), 2.84–2.88 (m, 4H), 2.69–2.73 (m, 4H), 2.23 (s, 12H),δC (150 MHz, DMSO-d6) 162.7, 162.4, 150.3, 143.5, 138.5, 128.9 (×2), 127.8 (×2), 125.7, 116.7, 105.4, 61.6, 51.8, 36.6, 18.3. νmax(cm−1) 3435, 3061, 2922, 1644, 1581, 1488, 1454, 1385, 1323, 1245, 1206, 1152, 1085, 1059, 1031, 986, 968, 786, 784, 704, 619, 510. Found C: 52.03, H: 4.38, N: 7.25%; for C64H60N8O8Zn4Cl4 requires C: 52.20, H: 4.11, N: 7.61%.
Conclusions
One-pot synthesis of oxazolinyl-zinc(II) complexes from three-component reactions between ZnCl2, amino alcohols and a variety of nitrile precursors has been demonstrated. The reaction outcome is highly dependent upon the presence of additional donor atoms, reaction stoichiometry and nature of the δ-substituent at the stereogenic centre, giving rise to a variety of coordination modes, including mono- and bis-chelate complexes. Using excess of zinc salt led to the formation of multinuclear complexes.
Change history
20 September 2017
An erratum to this article has been published.
References
Desimoni G, Faita G, Jorgensen KA (2011) Update 1 of: C-2-symmetric chiral bis(oxazoline) ligands in asymmetric catalysis. Chem Rev 111:PR284–PR437
Hargaden GC, Guiry PJ (2009) Recent Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem Rev 109:2505–2550
O’Reilly S, Guiry PJ (2014) Recent Applications of C-1-Symmetric Bis(oxazoline)-Containing Ligands in Asymmetric Catalysis. Synthesis-Stuttgart 46:722–739
Gotoh R, Yamanaka M (2012) Chiral Zn(II)-bisamidine complex as a Lewis-Bronsted combined acid catalyst: application to asymmetric mukaiyama aldol reactions of alpha-ketoesters. Molecules 17:9010–9022
Makino K, Ogawa I, Hamada Y (2005) Synthesis of new chiral bis-oxazoline ligand with zinc triflate-selective chelating ability and its applications. Heterocycles 66:433–440
Abbina S, Du G (2014) Zinc-catalyzed highly isoselective ring opening polymerization of rac-lactide. ACS Macro Lett 3:689–692
Abbina S, Du G (2012) Chiral amido-oxazolinate zinc complexes for asymmetric alternating copolymerization of CO2 and cyclohexene oxide. Organometallics 31:7394–7403
Bleith T, Deng Q-H, Wadepohl H, Gade LH (2016) Radical changes in lewis acid catalysis: matching metal and substrate. Angew Chem Int Ed 55:7852–7856
Le Roux E, Merle N, Tornroos K W (2011) Synthesis and characterisation of trigonal C-2-chiral di- and tetra-substituted bis(oxazoline) alkyl zinc complexes and their reactivity towards protic reagents. Dalton T 40:1768–1777
Witte H, Seeliger W (1974) Formation of cyclic imidic esters by reaction of nitriles with amino alcohols. Liebigs Ann Chem. 1:996–1009
Bolm C, Weickhardt K, Zehnder M, Ranff T (1991) Synthesis of optically active bis(2-oxazolines): crystal structure of a 1,2-bis(2-oxazolinyl)benzene ZnCl2 complex. Chem Ber 124:1173–1180
Kögel JF, Kusaka S, Sakamoto R, Iwashima T, Tsuchiya M, Toyoda R, Matsuoka R, Tsukamoto T, Yuasa J, Kitagawa Y, Kawai T, Nishihara H (2016) Heteroleptic [Bis(oxazoline)](dipyrrinato)zinc(II) complexes: bright and circularly polarized luminescence from an originally achiral dipyrrinato Ligand. Angew Chem Int Ed 55:1377–1381
Telfer SG, McLean TM, Waterland MR (2011) Exciton coupling in coordination compounds. Dalton T 40:3097–3108
Authors’ contributions
Luo Mei: design the research, performed the research, and analyzed the data. Zhang jingcheng and pang wenmin help with NMR testing, King Kuok (Mimi) Hii wrote the paper and carried out some relevant instructions for analyzing the data. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by hefei university of technology.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Characterization spectra for compounds 1-15, this material can be seen in Additional file 2, and table, figures can also be seen in additional file 1 which are both available free of charge via the Internet at: https://ccj.springeropen.com/. Crystallographic information for all compounds 1-15 has been deposited with the Cambridge Crystallographic Data Center (CCDC) as supplementary publications CCDC 853709-853710, 931745-931746, 931745-931748, 931751-931753, 931756, 1014806-1014807 and 1540756; deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk.
Consent for publication
All authors consent to the publication.
Ethics approval and consent to participate
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional information
The original version of this article was revised. After the publication it was noticed that the legend for scheme 5: ‘(left 11, right 12)’ was accidentally included and should be removed. The drawing for scheme 6 is incorrect and was replaced with the correct version.
An erratum to this article is available at https://doi.org/10.1186/s13065-017-0315-z.
Additional files
13065_2017_305_MOESM1_ESM.pdf
Additional file 1. Table, figures, crystal data and structure determination, general remarks, and procedure for the synthesis of the complexes 1-15.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Luo, M., Zhang, J.C., Pang, W.M. et al. One-step multicomponent synthesis of chiral oxazolinyl-zinc complexes. Chemistry Central Journal 11, 81 (2017). https://doi.org/10.1186/s13065-017-0305-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-017-0305-1