
Abstract
Background
Many thiourea derivatives have exhibited biological activities including anticancer activity through several mechanisms. On the other hand, benzenesulfonamide derivatives have proven to be good anticancer agents. Hybrids of both moieties could be further developed to explore their biological activity as anticancer.
Results
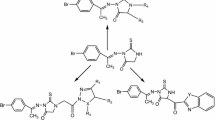
Novel series of thioureidobenzenesulfonamides incorporating miscellaneous biologically active moieties 3–17 were designed and synthesized utilizing 4-isothiocyanatobenzenesulfonamide 2 as strategic starting material. The structures of the newly synthesized compounds were established on the basis of elemental analyses, IR, 1H-NMR, 13C-NMR and mass spectral data. All the newly synthesized compounds were evaluated for their in vitro anticancer activity against various cancer cell lines. Most of the synthesized compounds showed good activity, especially compounds 3, 6, 8, 9, 10, 15 and 16 which exhibited good activity higher than or comparable to the reference drugs, DCF and Doxorubicin, except breast cancer line. As a trial to suggest the mechanism of action of the active compounds, molecular docking on the active site of mitogen kinase enzyme (MK-2) was performed and good results were obtained especially for compound 3.
Conclusion
Compounds 3, 6, 8, 9, 10, 15 and 16 may represent good candidates for further biological investigations as anticancer agents. Their cytotoxic activity could be due to their action as MK-2 enzyme inhibitors.

Compound 3 on the active site of MK-2 enzyme
Similar content being viewed by others

Background
Various types of cancer are now considered as the second cause of death after cardiovascular disorders [1]. The inability of the present anticancer chemotherapeutic agents to discriminate between normal cells and cancer cells comprises the biggest challenge for successful cancer treatment [2]. Serious side effects of anticancer chemotherapeutic agents limit their usage and in many cases surgery or radiotherapy replace them [3]. The continuous seek for safer and more effective anticancer agents is still a major goal for medicinal chemists.
Thiourea is a versatile synthetic block for the synthesis of a wide variety of new organic compounds with biological activity including antimicrobial, antifungal, antidiabetic, antimalarial, anti HIV and CNS active drugs [4–11]. Many of aryl thiourea derivatives have applications in medicine, industry and agriculture [12–15]. Thiourea was incorporated in many tyrosine kinase inhibitors because of its ability to form powerful hydrogen bonds in the ATP binding pocket of the enzymes [16]. The thiourea derivative YH345 A has shown strong protein farnesyl transferase inhibition activity [17]. Also several heterocyclic thiourea derivatives have shown strong DNA topoisomerase inhibitory activity [18].
On the other hand, sulfonamide derivatives posses a wide range of biological activity including antibacterial, anticonvulsant, anti-inflammatory and anticancer activity [19–22]. The mechanism of anticancer activity may involve a wide range of different mechanisms, such as cell cycle arrest in the G1 phase [23] and inhibition of carbonic anhydrase [24], histone deacetylases (HDACs) [25], methionine amino peptidases (MetAPs) [26], matrix metalloproteinase (MMPs) [27], nicotinamide adenine dinucleotide (NADH) oxidase [28], cyclin-dependentkinase (CDK) [29], binding to β-Tubulin, and disruption of microtubule assembly [30]. Indisulam (E7070) B is an example of an anticancer agent that contains sulfonamide moiety [31].
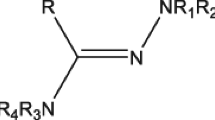
Based on the previous facts and as a continuation of our previous work in the seek of novel anticancer agents [32–38], we herein report the synthesis and biological evaluation of new sulfonamide thiourea derivatives 3–17 presented by general structure C as hybrid molecules of benzensulfonamide and thiourea moieties as anticancer agents. Molecular docking of the active newly synthesized compounds was performed on the active site of mitogen activated kinase enzyme (MK-2) in a trial to suggest a mechanism of action for their cytotoxic activity.
Results and discussion
Chemistry
The aim of this work was to design and synthesize a new series of thioureidobenzenesulfonamide derivatives having miscellaneous biologically active moieties to evaluate their anticancer activity. Thus, interaction of 4-isothiocyanatobenzenesulfonamide 2 with several amines in dry N,N-dimethylformamide containing triethlyamine as catalyst afforded the corresponding thioureidobenzenesulfonamude derivatives 3–17 (Schemes 1 and 2). The structures of the obtained compounds were established on the basis of elemental analyses and spectral data.IR spectra of compounds 3–17 showed the absence of N=C=S group and presence absorption bands for (NH), (CH arom.), (CH aliph.), (C=S) and (SO2). 1H-NMR spectra of compounds 3–17 exhibited a singlets at 7.8–13.8 ppm assigned to 2NH groups of thiourea which were exchanged upon duetration.IR spectrum of compound 3 showed the characteristic bands at 3312, 3214 cm−1 (NH), 3099 cm−1 (CH arom.), 2202 cm−1 (C≡N), 1655 cm−1 (C=O), 1387, 1157 cm−1 (SO2), 1250 cm−1 (C=S). 1H-NMR spectrum of compound 3 exhibited a triplet signal at 0.8 ppm due to CH3, a multiplet at 1.2–1.4 ppm due to 5CH2, a mutiplet at 3.3 ppm due to NHCH2 and singlets at 9.3 and 10.4 ppm assigned to 2NH groups which were exchangeable with D2O. Mass spectrum of compound 3 revealed a molecular ion peak m/z at of 329 (M+) (14.41) with a base peak appeared at 155 (100). 13C-NMR spectrum of compound 3 exhibited signals at 177.4 ppm assigned to (C=S). IR spectrum of compound 4 showed the characteristic bands at 3363 cm−1 (OH), 3280, 3143 cm−1 (NH, NH2), 3090 cm−1 (CH arom.), 1393, 1182 cm−1 (SO2), 1274 cm−1 (C=S). 1H-NMR spectrum of compound 4 exhibited signals at 10.2, 11.4 attributed to 2NH groups and 13.1 ppm assigned to OH group which exchangeable with D2O. Mass spectrum of compound 4 revealed a molecular ion peak m/z at of 323 (M+) (9.03) with a base peak appeared at 91 (100). 13C-NMR spectrum of compound 4 showed signals at 180.1 ppm assigned to (C=S).IR spectrum of compound 5 revealed the characteristic bands at 3317, 3254, 3173 cm−1 (NH, NH2), 3100 cm−1 (CH arom.), 2963, 2938, 2829 cm−1 (CH aliph.), 1363, 1156 cm−1 (SO2), 1259 cm−1 (C=S). 1H-NMR spectrum of compound 5 exhibited singlet at 3.9 ppm attributed to 3OCH3 groups and singlet at 9.8 ppm assigned to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 5 revealed a molecular ion peak m/z at of 367 (M+) (17.8) with a base peak appeared at 76 (100). 13C-NMR spectrum of compound 5 exhibited signals at 179.3 ppm assigned to (C=S).IR spectrum of compound 6 showed the characteristic bands at 3353, 3243, 3171 cm−1 (NH, NH2), 3009 cm−1 (CH arom.), 1340, 1161 cm−1 (SO2), 1290 cm−1 (C=S). 1H-NMR spectrum of compound 6 revealed singlet at 10.3 ppm assigned to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 6 exhibited a molecular ion peak m/z at of 366 (M+) (15.8) with a base peak appeared at 133 (100). 13C-NMR spectrum of compound 6 exhibited singlet at 180.1 ppm assigned to (C=S). IR spectrum of compound 7 exhibited the characteristic bands at 3325, 3241 cm−1 (NH, NH2), 3100 cm−1 (CH arom.), 1331, 1156 cm−1 (SO2), 1241 cm−1 (C=S). 1H-NMR spectrum of compound 7 showed singlet at 9.5 ppm assigned to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 7 revealed a molecular ion peak m/z at of 351 (M+) (34.64) with a base peak appeared at 93 (100). 13C-NMR spectrum of compound 7 showed signal at 180.6 ppm assigned to (C=S). IR spectrum of compound 8 exhibited the characteristic bands at 3384, 3348, 3206 cm−1 (NH, NH2), 3003 cm−1 (CH arom.), 1377, 1185 cm−1 (SO2), 1294 cm−1 (C=S). 1H-NMR spectrum of compound 8 showed singlet at 7.8 ppm attributed to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 8 exhibited a molecular ion peak m/z at of 365 (M+) (18.42) with a base peak appeared at 135 (100). 13C-NMR spectrum of compound 8 showed signal at 161.1 ppm attributed to (C=S). IR spectrum of compound 9 revealed the characteristic bands at 3434, 3354 cm−1 (NH, NH2), 3100 cm−1 (CH arom.), 2997, 2906, 2851 (CH aliph.), 1396, 1186 (SO2), 1282 (C=S). 1H-NMR spectrum of compound 9 exhibited multiplet at 1.9 ppm due to 6CH2, a multiplet at 2.2–2.4 due to 3CH and singlet at 11.4 ppm due to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 9 exhibited a molecular ion peak m/z at of 366 (M+) (9.32) with a base peak appeared at 154 (100). 13C-NMR spectrum of compound 9 showed singlet at 179.9 ppm due to (C=S). IR spectrum of compound 10 showed the characteristic bands at 3359, 3257, 3143 cm−1 (NH, NH2), 3031 cm−1 (CH arom.), 2954, 2851 cm−1 (CH aliph.), 1594 cm−1 (C=N), 1381, 1186 cm−1 (SO2), 1296 cm−1 (C=S). 1H-NMR spectrum of compound 10 revealed singlets at 10.2 and 13.0 ppm assigned to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 10 showed a molecular ion peak m/z at of 393 (M+) (16.9) with a base peak appeared at 162 (100). 13C-NMR spectrum of compound 10 showed singlet at 180.0 ppm attributed to (C=S). IR spectrum of compound 11 revealed the characteristic bands at 3410, 3334, 3195 (NH, NH2), 3069 (CH arom.), 2974, 2925, 2843 (CH aliph.), 1595 (C=N), 1393, 1123 (SO2), 1256 (C=S). 1H-NMR spectrum of compound 11 revealed a triplet at 1.3 due to CH3, a quartet at 4.0 ppm due to CH2 and a singlet at 10.3 and 11.2 ppm due to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 11 exhibited a molecular ion peak m/z at of 409 (M+) (1.85) with a base peak appeared at 156 (100). 13C-NMR spectrum of compound 11 revealed singlet at 180.1 ppm assigned to (C=S). IR spectrum of compound 12 revealed the characteristic bands at 3384, 3261, 3165 (NH, NH2), 3097 (CH arom.), 1595 (C=N), 1331, 1185 (SO2), 1252 (C=S). 1H-NMR spectrum of compound 12 exhibited singlet at 10.5 and 12.0 ppm due to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 12 exhibited a molecular ion peak m/z at of 409 (M+) (13.43) with a base peak appeared at 178 (100). 13C-NMR spectrum of compound 12 showed singlet at 179.9 ppm assigned to (C=S). IR spectrum of compound 13 revealed the characteristic bands at 3326, 3175 (NH, NH2), 3088 (CH arom.), 1572 (C=N), 1356, 1192 (SO2), 1211 (C=S). 1H-NMR spectrum of compound 13 exhibited singlet at 12.4 ppm. due to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 13 exhibited a molecular ion peak m/z at of 388 (M+) (11.81) with a base peak appeared at 157 (100). 13C-NMR spectrum of compound 13 showed singlet at 178.6 ppm attributed to (C=S). IR spectrum of compound 14 showed the characteristic bands at 3378, 3240, 3155 (NH, NH2), 3100 (CH arom.), 1601 (C=N), 1346, 1199 (SO2), 1270 (C=S). 1H-NMR spectrum of compound 14 revealed singlets at 11.3, 13.0 ppm attributed to 2NH groups. Mass spectrum of compound 14 showed a molecular ion peak m/z at of 309 (M+) (12.83) with a base peak appeared at 79 (100). 13C-NMR spectrum of compound 14 showed singlet at 179.0 ppm attributed to (C=S). IR spectrum of compound 15 revealed the characteristic bands at 3413, 3354, 3152 (NH, NH2), 3083 (CH arom.), 2982, 2935, 2831 (CH aliph.), 1351, 1159 (SO2), 1264 (C=S). 1H-NMR spectrum of compound 15 exhibited multiplet at 1.8–2.8 ppm due to 4CH2 and singlet at 9.0 ppm due to 2NH groups. Mass spectrum of compound 15 exhibited a molecular ion peak m/z at of 361 (M+) (26.34) with a base peak appeared at 177 (100). 13C-NMR spectrum of compound 15 showed singlet at 181.5 ppm assigned to (C=S). IR spectrum of compound 16 revealed the characteristic bands at 3373, 3246, 3164 (NH, NH2), 3077 (CH arom.), 1595 (C=N), 1365, 1150 (SO2), 1293 (C=S). 1H-NMR spectrum of compound 16 exhibited singlet at 10.8 ppm. attributed to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 16 exhibited a molecular ion peak m/z at of 358 (M+) (17.53) with a base peak appeared at 156 (100). 13C-NMR spectrum of compound 16 showed singlet at 178.6 ppm attributed to (C=S). IR spectrum of compound 17 revealed the characteristic bands at 3363, 3218, 3154 (NH, NH2), 3034 (CH arom.), 2943, 2836 (CH aliph.), 1590 (C=N), 1324, 1154 (SO2), 1241 (C=S). 1H-NMR spectrum of compound 17 exhibited singlets 10.1 and 13.8 ppm. attributed to 2NH groups, which exchangeable with D2O. Mass spectrum of compound 17 exhibited a molecular ion peak m/z at of 372 (M+) (21.22) with a base peak appeared at 141 (100). 13C-NMR spectrum of compound 17 showed singlet at 179.3 ppm attributed to (C=S).

Synthesis of compounds 1–9

Synthesis of compounds 10–17
In-vitro anticancer evaluation
The synthesized compounds were evaluated for their in vitro anticancer activity against human lung cancer cell line (A549-Raw), cervical (Hela) cancer cell line, colorectal cell line (Lovo) and breast cancer cell line (MDA-MB231) using 2′7′dichlorofluorescein (DCF) and Doxorubicin as reference drugs in this study. The relationship between surviving fraction and drug concentration was plotted to obtain the survival curve of cancer cell lines. The response parameter calculated was the IC50 value, which corresponds to the concentration required for 50 % inhibition of cell viability. The results are presented in Table 1.
Regarding the cytotoxic activity on lung cancer cell line (A549), compounds 2, 3, 6, 8, 9, 15 and 16 were active with IC50 ranging between 29.12 and 114.28 µg ml−1. The most active compound was the n-heptane thiourea derivative 3. In case of cervical cancer cell line (Hela), compounds 3, 6, 8, 9, 10 and 15 were active with IC50 ranging between 35.63 and 93.42 µg ml−1. The most active compounds was again the n-hepatne thiourea derivative 3.
For the colorectal cell line (Lovo), compounds 2, 3, 8, 9 and 10 were active with IC50 ranging between 39.83 and 148.33 µg ml−1 and once again the most active compound was n-hepatne thiourea derivative 3. Finally, the activity on breast cancer cell line (MDA-MB231) was exhibited by compounds 3, 6, 8, 9, 10, 15 and 16 with IC50 ranging between 26.28 and 69.04 µg ml−1 with less activity than Doxorubicin. The same compound (n-hepatne thiourea derivative 3) was the most active compound.
Structure activity relationship
In a closer look to the biological results we can see that: the thiourea derivatives 3, 6, 8, 9, 10, 15 and 16 were the active compounds on most of the cell lines while the rest of the compounds were inactive. It was obvious that incorporating an n-heptane aliphatic substitution as in compound 3 gave the most activity on all cell line. This activity was reduced upon replacing this substituent with another tricyclic aliphatic one as in compound 9. In case of aromatic substitution the activity was retained but markedly decreased as in the 2-methyl-6-nitrophenyl thiourea derivative 6, the 3-benzo[d][1,3]dioxol-5-ylmethyl thiourea derivative 8, the 3-(5,6-dimethylbenzo[d]thiazol-2-yl)thiourea derivative 10, the tetrahydronaphthalen derivative 15 and the quinoline derivative 16.
Comparing compound 3 which was the most active compound among the newly synthesized compounds with the reference drug Doxorubicin we can see that: compound 3 was more active that Doxorubicin as cytotoxic agents on lung cancer cell line, Hella cells and colorectal cancer cells with IC50 value of 29.12, 35.63 and 39.83 µg ml−1, respectively. However, in case of breast cancer cell line compound 3 was less active than Doxorubicin with IC50 value of 26.28 µg ml−1.
Molecular docking
Mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK-2 or MK-2) is an important enzyme in signal transduction pathway controlling several pathways in cell proliferation [39]. MK-2 inhibition is one of the strategies of discovering new anticancer agents [40]. Recently, several urea and thiourea derivatives have shown good inhibitory activity on MK-2 [41]. Based on the thiourea scaffold of our newly synthesized compounds and as a trial to suggest a mechanism of action for their cytotoxic activity, molecular docking was performed on the active site of MK-2 for the most active compound. The protein data bank file (PDB:3WI6). The file contains MK-2 enzyme co-crystalized with an inhibitor. All docking procedures were achieved by MOE (Molecular Operating Environment) software 10.2008 provided by chemical computing group, Canada. The inhibitor interacts with MK-2 active site with four hydrogen bonds involving Glu 190, Leu 141, Asn 191 ans Asp 207 (Fig. 1). The docking protocol was validated by redocking of the ligand on the active site of MK-2 enzyme with energy score (S) = −15.4978 kcal mol−1 and root mean square deviation (RMSD) = 1.1457.

a YH345, b Indisulam, c general structure for the designed compounds
The active compounds were docked on the active site of MK-2 using the same docking protocol. Energy scores and amino acid interactions were displayed in Table 2.
All the docked compounds were fit on the active site of MK-2 with energy scores ranging between −10.2371 and −20.1443 kcal mol−1. Best docking score was exhibited by compound 16 which interacted with Lys 188 with one hydrogen bond (Fig. 2) while the best amino acid interaction was exhibited by compound 3 which interacted with Leu 141 by two hydrogen bonds and Asp 207 with one hydrogen bond (Figs. 3, 4). The same previous two amino acids interacted also with the co-crystalized inhibitor in a comparable manner.

Co-crystalized lignd in the active site of mitogen activated kinase (MK-2)

Compound 16 in the active site of mitogen activated kinase (MK-2)

Compound 3 in the active site of mitogen activated kinase (MK-2)
Experimental
General chemistry
Melting points (uncorrected) were and determined in open capillary on a Gallen Kamp melting point apparatus (Sanyo Gallen Kamp, UK). Precoated silica gel plates (Kieselgel 0.25 mm, 60 F254, Merck, Germany) were used for thin layer chromatography. A developing solvent system of chloroform/methanol (8:2) was used and the spots were detected by ultraviolet light. IR spectra (KBr disc) were recorded using an FT-IR spectrophotometer (Perkin Elmer, USA). 1H-NMR spectra were scanned on an NMR spectrophotometer (Bruker AXS Inc., Switzerland), operating at 500 MHz for 1H- and 125.76 MHz for 13C. Chemical shifts are expressed in δ-values (ppm) relative to TMS as an internal standard, using DMSO-d 6 as a solvent. Elemental analyses were done on a model 2400 CHNSO analyser (Perkin Elmer, USA). All the values were within ±0.4 % of the theoretical values. All reagents used were of AR grads.
Synthesis of thioureidobenzenesulfonamide derivatives (3–17)
General procedure
A mixture of 4-isothiocyanatobenzenesulfonamide 2 (2.14 g, 0.01 mol) and amines (0.012 mol) in dry dimethylformamide (15 ml) containing three drops of triethylamine was refluxed for 24 h, then left to cool. The solid product formed upon pouring onto ice/water was collected by filtration and recrystallized from ethanol–dimethylformamide to give 3–17, respectively.
4-(3-Heptylthioureido)benzenesulfonamide (3)
Yield, 92 %; m.p. 124.7 °C. IR (KBr, cm−1): 3218, 3143 (NH, NH2), 3087 (CH arom.), 2926, 2853 (CH aliph.), 1376, 1150 (SO2), 1254 (C=S). 1H-NMR (DMSO-d2): 0.8 [t, 2H, CH3], 1.2–1.4 [m, 10H, 5CH2], 3.3 [m, 2H, NHCH2], 7.3–7.9 [m, 6H, Ar–H + SO2NH2], 9.3, 10.4 [2 s, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 14.2, 22.4, 26.2, 28.6, 29.0, 31.5, 43.9, 119.4 (2), 127.4 (2), 134.7, 143.0, 177.4.MS m/z (%): 329 (M+) (14.41), 155 (100). Anal.Calcd. For C14H23N3O2S2 (329): C, 51.03; H, 7.04; N, 12.75. Found: C, 51.29; H, 6.79; N, 12.45.
4-(3-(4-Hydroxyphenyl)thioureido)benzenesulfonamide (4)
Yield, 88 %; m.p.192.9 °C. IR (KBr, cm−1): 3363 (OH), 3280, 3143 (NH, NH2), 3090 (CH arom.), 1393, 1182 (SO2), 1274 (C=S).1H-NMR (DMSO-d2): 6.7–7.9 [m, 10H, Ar–H + SO2NH2], 10.2, 11.4, [2 s, 2H, 2NH, exchangeable with D2O], 13.1 [s, 1H, OH, exchangeable with D2O], 13C-NMR (DMSO-d6): 112.9 (2), 122.8 (2), 126.7 (2), 127.1 (2), 127.9, 139.8, 140.3, 157.6, 180.1. MS m/z (%): 323 (M+) (9.03), 91 (100). Anal.Calcd. For C13H13N3O3S2 (323): C, 48.28; H, 4.05; N, 12.99. Found: C, 48.55; H, 4.31; N, 13.29.
4-(3-(3,5-Dimethoxyphenyl)thioureido)benzenesulfonamide (5)
Yield, 77 %; m.p. 160.3 °C. IR (KBr, cm−1): 3317, 3254, 3173 (NH, NH2), 3100 (CH arom.), 2963, 2938, 2829 (CH aliph.), 1363, 1156 (SO2), 1259 (C=S). 1H-NMR (DMSO-d2): 3.9 [s, 6H, 2OCH3], 6.3–7.8 [m, 8H, Ar–H + SO2NH2], 9.8 [s, 2H, 2NH, exchangeable with D2O].13C-NMR (DMSO-d6): 56.1 (2), 96.8, 102.0 (2), 123.2 (2), 126.6 (2), 141.4 (2), 143.1, 160.6 (2), 179.3. MS m/z (%): 367 (M+) (17.8), 76 (100). Anal.Calcd. For C15H17N3O4S2 (367): C, 49.03; H, 4. 66; N, 11.44. Found: C, 48.74; H, 4.29; N, 11.17.
4-(3-(2-Methyl-6-nitrophenyl)thioureido)benzenesulfonamide (6)
Yield, 81 %; m.p. 226.0 °C. IR (KBr, cm−1): 3353, 3243, 3171 (NH, NH2), 3009 (CH arom.), 1340, 1161 (SO2), 1290 (C=S).1H-NMR (DMSO-d2): 2.2 [s, 3H, CH3], 6.5–7.8 [m, 9H, Ar–H + SO2NH2], 10.3 [s, 2H, 2NH exchangeable with D2O]. 13C-NMR (DMSO-d6): 18.3, 123.3, 123.9 (2), 126.7, 126.8, 127.8 (2), 131.3, 136.5 (2), 139.8, 142.8, 180.1. MS m/z (%): 366 (M+) (15.8), 133 (100). Anal.Calcd. For C14H14N4O4S2 (366): C, 45.89; H, 3.85; N, 15.29. Found: C, 45.57; H, 3.54; N, 15.61.
4-(3-Benzo[d][1,3]dioxol-5-ylthioureido)benzenesulfonamide (7)
Yield, 86 %; m.p. 136.6 °C. IR (KBr, cm−1): 3325, 3241 (NH, NH2), 3100 (CH arom.), 1331, 1156 (SO2), 1241 (C=S). 1 H-NMR (DMSO-d2): 6.0 [s, 2H, CH2], 6.7–7.9 [m, 9H, Ar–H + SO2NH2], 9.5 [s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 101.2, 107.1, 109.1, 117.8, 123.1 (2), 126.5 (2), 133.4, 134.6, 142.4, 143.2, 147.3, 180.6. MS m/z (%): 351 (M+) (34.64), 93 (100). Anal.Calcd. For C14H13N3O4S2 (351): C, 47.85; H, 3.73; N, 11.96. Found: C, 47.49; H, 3.43; N, 11.62.
4-(3-Benzo[d][1,3]dioxol-5-ylmethyl)thioureido)benzenesulfonamide (8)
Yield, 68 %; m.p. 140.8 °C. IR (KBr, cm−1): 3384, 3348, 3206 (NH, NH2), 3003 (CH arom.), 1377, 1185 (SO2), 1294 (C=S).1H-NMR (DMSO-d2): 4.3 [s, 2H, CH2], 6.0 [s, 2H, OCH2O], 6.7–7.7 [m, 9H, Ar–H + SO2NH2], 7.8 [s, 2H, +2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 63.8, 101.2, 106.4, 108.2, 120.8, 121.4 (2), 131.2 (2), 133.5, 134.0, 146.4, 147.6, 148.3, 161.1. MS m/z (%): 365 (M+) (18.42), 135 (100). Anal.Calcd. For C15H15N3O4S2 (365): C, 49.30; H, 4.14; N, 11.50. Found: C, 49.05; H, 4.46; N, 11.19.
4-(3-(1-Adamanylamine)thioureidobenzenesulfonamide (9)
Yield, 80 %; m.p. 174.5 °C. IR (KBr, cm−1): 3434, 3354 (NH, NH2), 3100 (CH arom.), 2997, 2906, 2851 (CH aliph.), 1396, 1186 (SO2), 1282 (C=S).1H-NMR (DMSO-d2): 1.6–1.9 [m, 12H, 6CH2], 2.2–2.4 [m, 3H, 3 CH], 6.9–7.9 [m, 6H, Ar–H + SO2NH2], 11.4 [s, 2H, 2NH, exchangeable with D2O].13C-NMR (DMSO-d6): 28.8 (3), 35.3 (3), 40.5 (3), 44.9, 126.4 (2), 129.1 (2), 131.8, 142.7, 179.9. MS m/z (%): 366 (M+) (9.32), 154 (100). Anal.Calcd. For C17H23N3O2S2 (366): C, 55.86; H, 6.34; N, 11.50. Found: C, 55.50; H, 6.68; N, 11.18.
4-(3-(5,6-Dimethylbenzo[d]thiazol-2-yl)thioureido)benzenesulfonamide (10)
Yield, 84 %; m.p. 252.1 °C. IR (KBr, cm−1): 3359, 3257, 3143 (NH, NH2), 3031 (CH arom.), 2954, 2851 (CH aliph.), 1594 (C=N), 1381, 1186 (SO2), 1296 (C=S).1H-NMR (DMSO-d2): 2.2 [s, 6H, 2CH3], 7.2–8.0 [m, 8H, Ar–H + SO2NH2], 10.2, 13.0 [2 s, 2H, 2NH, exchangeable with D2O].13C-NMR (DMSO-d6): 19.9, 20.4, 118.5, 121.5, 123.3 (2), 126.6, 127.1, 127.8 (2), 133.0, 136.2, 139.8, 143.1, 151.9, 180.0 (2).MS m/z (%): 393 (M+) (16.9), 162 (100). Anal.Calcd. For C16H16N4O2S3 (393): C, 48.96; H, 4.11; N, 14.27. Found: C, 48.66; H, 3.85; N, 14.54.
4-(3-(6-Ethoxybenzo[d]thiazol-2-yl)thioureido)benzenesulfonamide (11)
Yield, 78 %;m.p. 153.6 °C. IR (KBr, cm−1): 3410, 3334, 3195 (NH, NH2), 3069 (CH arom.), 2974, 2925, 2843 (CH aliph.), 1595 (C=N), 1393, 1123 (SO2), 1256 (C=S). 1H-NMR (DMSO-d2): 1.3 [t, 3H, CH3], 4.0 [q, 2H, CH2], 6.9–8.0 [m, 9H, Ar–H + SO2NH2], 10.3, 11.2 [2 s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 15.0, 66.8, 106.0, 115.3, 118.2, 120.4 (2), 127.7 (2), 132.7, 138.2, 140.9, 142.1, 157.7, 177.1, 180.1. MS m/z (%): 409 (M+) (1.85), 156 (100). Anal.Calcd. For C16H16N4O3S3 (409): C, 47.04; H, 3.95;N, 13.71. Found: C, 47.34; H, 3.67; N, 13.39.
4-(3-(6-Nitrobenzo[d]thiazol-2-yl))thioureido)benzenesulfonamide (12)
Yield, 65 %; m.p. 205.8 °C. IR (KBr, cm−1): 3384, 3261, 3165 (NH, NH2), 3097 (CH arom.), 1595 (C=N), 1331, 1185 (SO2), 1252 (C=S).1H-NMR (DMSO-d2): 7.1–8.9 [m, 9H, Ar–H + SO2NH2], 10.5, 12.0 [2 s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 119.6 (2), 123.1 (3), 126.6 (3), 139.8 (2), 142.8, 161.2179.9 (2). MS m/z (%): 409 (M+) (13.43), 178 (100). Anal.Calcd. For C14H11N5O4S3 (409): C, 41.07; H, 2.71; N, 17.10. Found: C, 41.31; H, 2.40; N, 17.43.
4-(3-(5-(Bromopyridin-2-yl)thioureido)benzenesulfonamide (13)
Yield, 72 %; m.p. 247.0 °C. IR (KBr, cm−1): 3326, 3175 (NH, NH2), 3088 (CH arom.), 1572 (C=N), 1356, 1192 (SO2), 1211 (C=S). 1H-NMR (DMSO-d2): 6.8–8.3 [m, 8H, Ar–H + SO2NH2], 12.4 [s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 105.5, 124.8 (2), 128.9 (2), 134.6, 140.8, 158.5, 159.3 (2), 178.6.MS m/z (%): 388 (M+) (11.81), 157 (100). Anal.Calcd. For C11H10BrN5O2S2 (388): C, 34.03; H, 2.60; N, 18.04. Found: C, 34.28; H, 2.27; N, 18.37.
4-(3-Pyrazin-2-ylthioureido)benzenesulfonamide (14)
Yield, 80 %; m.p. 185.3 °C. IR (KBr, cm−1): 3378, 3240, 3155 (NH, NH2), 3100 (CH arom.), 1601 (C=N), 1346, 1199 (SO2), 1270 (C=S).1H-NMR (DMSO-d2): 7.2–8.7 [m, 9H, Ar–H + SO2NH2], 11.3, 13.0 [2 s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 123.1 (2), 126.7 (2), 137.1, 138.4, 138.5, 139.9, 140.3, 149.7, 179.0. MS m/z (%): 309 (M+) (12.83), 79 (100). Anal.Calcd. For C11H11N5O2S2 (309): C, 42.71; H, 3.58; N, 22.64. Found: C, 42.38; H, 3.84; N, 22.29.
4-(3-(5,6,7,8-Tetrahydronaphthalen-1-yl)thioureido)benzenesulfonamide (15)
Yield, 76 %; m.p. 171.8 °C. IR (KBr, cm−1): 3413, 3354, 3152 (NH, NH2), 3083 (CH arom.), 2982, 2935, 2831 (CH aliph.), 1351, 1159 (SO2), 1264 (C=S).1H-NMR (DMSO-d2): 1.8–2.8 [m, 8H, 4CH2, cyclo], 7.0–8.0 [m, 9H, Ar–H + SO2NH2], 9.0[s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 22.7 (2), 24.8, 29.6, 117.4, 120.8 (2), 124.0, 125.7, 127.4 (2), 134.5, 137.1, 137.4, 137.6, 146.4, 181.5.MS m/z (%): 361 (M+) (26.34), 177 (100). Anal.Calcd. For C17H19N3O2S2 (361): C, 56.48; H, 5.30; N, 11.62. Found: C, 56.12; H, 5.03; N, 11.36.
4-(3-Quinolin-3-ylthioureido)benzenesulfonamide (16)
Yield, 66 %; m.p. 214.6 °C. IR (KBr, cm−1): 3373, 3246, 3164 (NH, NH2), 3077 (CH arom.), 1595 (C=N), 1365, 1150 (SO2), 1293 (C=S). 1H-NMR (DMSO-d2): 6.8–8.5 [m, 12H, Ar–H + SO2NH2], 10.8 [s, 2H, 2NH, exchangeable with D2O].13C-NMR (DMSO-d6): 127.3, 127.7, 128.6, 129.1 (2), 130.0 (2), 132.0, 134.3 (2), 137.9 (2), 142.8, 178.6. MS m/z (%): 358 (M+) (17.53), 156 (100). Anal.Calcd. For C16H14N4O2S2 (358): C, 53.61; H, 3.94; N, 15.63. Found: C, 53.36; H, 3.62; N, 15.36.
4-(3-(2-Methylquinolin-4-yl)thioureido)benzenesulfonamide (17)
Yield, 71 %; m.p. 192.3 °C. IR (KBr, cm−1): 3363, 3218, 3154 (NH, NH2), 3034 (CH arom.), 2943, 2836 (CH aliph.), 1590 (C=N), 1324, 1154 (SO2), 1241 (C=S). 1H-NMR (DMSO-d2): 2.6 [s, 3H, CH3], 6.6–8.8 [m, 11H, Ar–H + SO2NH2], 10.1, 13.8[2 s, 2H, 2NH, exchangeable with D2O]. 13C-NMR (DMSO-d6): 19.9, 102.0, 108.3, 121.1 (2), 122.9, 124.0, 126.1, 127.8, 128.0 (2), 137.3, 139.5, 143.1, 151.7, 158.0, 179.3. MS m/z (%): 372 (M+) (21.22), 141 (100). Anal.Calcd. For C17H16N4O2S2 (372): C, 54.82; H, 4.33; N, 15.04. Found: C, 54.51; H, 4.09; N, 15.31.
In vitro anticancer evaluation
Cell culture
Human cancer cell lines HeLa (cervical), A549 (lungs) and Lovo (colorectal) were grown in DMEM + GlutaMax (Invitrogen), and MDA MB321 (breast) were grown in DMEM-F12 + GlutaMax) medium (invitrogen), supplemented with 10 % heat-inactivated bovine serum (Gibco) and 1× penicillin–streptomycin (Gibco) at 37 °C in a humified chamber with 5 % CO2 supply.
Cytotoxicity assay
The in vitro anticancer screening was done at pharmacognosy Department, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia. Cells were seeded (105 cells/well) in 96-well flat-bottom plates (Becton–Dickinson Labware) a day before treatment and grown overnight. Compounds were dissolved in dimethyl sulfoxide (DMSO; Sigma) and finally prepared as 1.0 mg ml−1 stocks, respectively in the culture media. The final concentration of DMSO never exceeded 0.1 % in the treatment doses. Four different doses of compounds (50, 25, 12.5 and 6.25 µg ml−1) were further prepared by diluting the stocks in culture media, and cells were treated (in triplicate/dose). 2′7′ dichlorofluorescein (DCF) was included as standard reference drug (positive control) and untreated culture was considered as negative control. The treated cultures were further incubated for 48 h. At 48 h post-treatment, cell viability test was performed using TACS MTT Cell Proliferation and Viability Assay Kit (TACS) as per manufacturer’s instructions. The optical density (OD) was recorded at 570 nm in a microplate reader (BioTek, ELx800) and cell survival fraction was determined. The cell survival fraction was calculated as [(A − B)/A], where A and B are the OD of untreated and of treated cells, respectively [42]. The IC50 values of the tested compound were estimated using the best fit regression curve method in Excel.
Microscopy
A direct visual investigation was made under an inverted microscope (Optica, 40× and 100×) to observe any morphological changes in the cells cultured with different treatment doses at 24 and 48 h.
Molecular docking
All the molecular modeling studies were carried out on an Intel Pentium 1.6 GHz processor, 512 MB memory with Windows XP operating system using Molecular Operating Environment (MOE, 10.2008) software. All the minimizations were performed with MOE until a RMSD gradient of 0.05 kcal mol−1 Å with MMFF94X force field and the partial charges were automatically calculated. The protein data bank file (PDB: 3WI6) was selected for this purpose. The file contains MK-2 enzyme co-crystalized with a ligand obtained from protein data bank. The enzyme was prepared for docking studies where: (1) Ligand molecule was removed from the enzyme active site. (2) Hydrogen atoms were added to the structure with their standard geometry. (3) MOE Alpha Site Finder was used for the active sites search in the enzyme structure and dummy atoms were created from the obtained alpha spheres. (4). The obtained model was then used in predicting the ligand enzymes interactions at the active site.
Conclusions
In summary, we had synthesized a novel series of sulfonamide thiourea derivatives. Seven compounds 3, 6, 8, 9, 10, 15 and 16 showed good anticancer activity against lung (A594 Raw), Hela, and Colorectal (Lovo) cancer cell lines with better or comparable activity to DCF. Moreover, molecular docking for these active compounds showed proper fitting on the active site of MK-2 enzyme suggesting their action as inhibitors for this enzyme but more investigation should be carried out in the future to explore precisely the mechanism of the action of the synthesized derivatives.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Zwick E, Bange J, Ullrich A (2001) Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer 8:161–173
Heffeter P, Jakupec MA, Korner W, Wild S, von Keyserlingk NG, Elbling L, Zorbas H, Korynevska A, Knasmuller S, Sutterluty H, Micksche M, Keppler BK, Berger W (2006) Anticancer activity of the lanthanum compound [tris(1,10-phenanthroline)lanthanum(III)]trithiocyanate (KP772; FFC24). Biochem Pharmacol 71:426–440
Karipcin F, Atis M, Sariboga B, Celik H, Tas M (2013) Structural, spectral, optical and antimicrobial properties of synthesized 1-benzoyl-3-furan-2-ylmethyl-thiourea. J Mol Struct 1048:69–77
Vega-Perez JM, Perinan I, Argandona M, Vega-Holm M, Palo-Nieto C, Burgos-Morón E, López-Lázaro M, Vargas C, Nieto JJ, Iglesias-Guerra F (2012) Isoprenylthiourea and urea derivatives as new farnesyl diphosphate analogues: synthesis and in vitro antimicrobial and cytotoxic activities. Eur J Med Chem 58:591–612
Saeed A, Saeed N, Hummera R, Sadaf R, Hameed A (2009) Synthesis, characterization and antibacterial activity of some 1-aroyl-3-aryl thioureas. Chemistry 18:152–158
Saeed S, Rashid N, Jones PG, Ali M, Hussain R (2010) Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents. Eur J Med Chem 45:1323–1331
Ekoue-Kovi K, Yearick K, Iwaniuk DP, Natarajan JK, Alumasa J, de Dios AC, Roepe PD, Wolf C (2009) Synthesis and antimalarial activity of new 4-amino-7-chloroquinolyl amides, sulfonamides, ureas and thioureas. Bioorg Med Chem 17:270–283
Liav A, Angala SK, Brennan PJ, Jackson M (2008) N-d-Aldopentofuranosyl-N′-[p-(isoamyloxy)phenyl]-thiourea derivatives: potential anti-TB therapeutic agents. Bioorg Med Chem Lett 18:2649–2651
Manjula SN, Noolvi NM, Parihar KV, Manohara Reddy SA, Ramani V, Gadad AK, Singh G, Gopalan Kutty N, Mallikarjuna Rao C (2009) Synthesis and antitumor activity of optically active thiourea and their 2-aminobenzothiazole derivatives: a novel class of anticancer agents. E J Med Chem 44:2923–2929
Tsogoeva SB, Hateley MJ, Yalalov DA, Meindl K, Weckbecker C, Huthmacher K (2005) Thiourea-based non-nucleoside inhibitors of HIV reverse transcriptase as bifunctional organocatalysts in the asymmetric Strecker synthesis. Bioorg Med Chem 13:5680–5685
Holla BS, Veerenda B, Shivananda K, Boja P (2003) Synthesis characterization and anticancer activity studies on some Mannich bases derived from 1,2,4-triazoles. Eur J Med Chem 38:759–767
Abdel-Megeed AM, Abdel-Rahman HM, Alkaramany GE, El-Gendy MA (2009) Design, synthesis and molecular modeling study of acylated 1,2,4-triazole-3-acetates with potential anti-inflammatory activity. Eur J Med Chem 44:117–123
Kaplancikli ZA, Turan-Zitouni G, Ozdemir A, Revial G (2008) New triazole and triazolothiadiazine derivatives as possible antimicrobial agents. Eur J Med Chem 43:155–159
Koçyiğit-Kaymakçıoğlu B, Çalışır MM, Özbek B, Ötük G (2010) Synthesis and antimicrobial activities of schiff bases derived from 4-amino-5-(1-phenylethyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione. E J Chem 7:458–464
Choudhary A, Raines RT (2011) An evaluation of peptide-bond isosteres. Chembiochem 12:1801–1807
Lee J, La S, Ahn BR, Jeong TC, Kim DH (2004) Metabolism of 1-(3-[3-(4-cyanobenzyl)-3H-imidazol-4-yl]-propyl)-3-(6-methoxypyridin-3-yl)-1-(2-trifluoromethylbenzyl)thiourea (YH3945), a novel anti-cancer drug, in rats using the 14C-labeled compound. Rapid Commun Mass Spectrom 18:1901–1910
Esteves-Souza A, Pissinate K, Nascimento MG, Grynberg NF, Echevarria A (2006) Synthesis, cytotoxicity, and DNA-topoisomerase inhibitory activity of new asymmetric ureas and thioureas. Bioorg Med Chem 14:492–499
Hassan SM, El-Maghraby AA, Abdel Aal MM, Bashandy MS (2009) Heteroaromatization with sulfonamido phenyl ethanone, part I: synthesis of novel pyrrolo[2,3-d]pyrimidine and pyrrolo[3,2-E][1, 2, 4]Triazolo[1,5-C]pyrimidine derivatives containing dimethylsulfonamide moiety. Phosphorus Sulfur Silicon Relat Elem 184:291–308
Hilmy KM, Khalifa MM, Hawata MA, Keshk RM, el-Torgman AA (2010) Synthesis of new pyrrolo[2,3-d]pyrimidine derivatives as antibacterial and antifungal agents. Eur J Med Chem 45:5243–5250
Temperini C, Cecchi A, Scozzafava A, Supuran CT (2008) Carbonic anhydrase inhibitors. Sulfonamide diuretics revisited—old leads for new applications? Org Biomol Chem 6:2499–2506
Suthar SK, Bansal S, Lohan S, Modak V, Chaudhary A, Tiwari A (2013) Design and synthesis of novel 4-(4-oxo-2-arylthiazolidin-3-yl)benzenesulfonamides as selective inhibitors of carbonic anhydrase IX over I and II with potential anticancer activity. Eur J Med Chem 66:372–379
Fukuoka K, Usuda J, Iwamoto Y, Fukumoto H, Nakamura T, Yoneda T, Narita N, Saijo N, Nishio K (2001) Mechanisms of action of the novel sulfonamide anticancer agent E7070 on cell cycle progression in human non-small cell lung cancer cells. Invest New Drugs 19:219–227
Supuran CT, Briganti F, Tilli S, Chegwidden WR, Scozzafava A (2001) Carbonic anhydrase inhibitors: sulfonamidesas antitumoragents. Bioorg Med Chem 9:703–714
Payne JE, Bonnefous C, Hassig CA, Symons KT, Guo X, Nguyen PM, Annable T, Wash PL, Hoffman TZ, Rao TS, Shiau AK, Malecha JW, Noble SA, Hager JH, Smith ND (2008) Identification of KD5170: a novel mercaptoketone-based histone deacetylase inhibitor. Bioorg Med Chem Lett 18:6093–6096
Kawai M, BaMaung NY, Fidanze SD, Erickson SA, Tedrow JS, Sanders WJ, Vasudevan A, Park C, Hutchins C, Comess KM, Kalvin D, Wang J, Zhang Q, Lou P, Tucker-Garcia L, Bouska J, Bell RL, Lesniewski R, Henkin J, Sheppard GS (2006) Development of sulfonamide compounds as potent methionine aminopeptidase type II inhibitors with antiproliferative properties. Bioorg Med Chem Lett 16:3574–3577
Casini A, Scozzafava A, Supuran CT (2002) Sulfonamide derivatives with protease inhibitory action as anticancer, anti-inflammatory and antiviral agents. Expert Opin Ther Pat 12:1307–1327
Villar R, Encio I, Migliaccio M, Gil MJ, Martinez- Merino V (2004) Synthesis and cytotoxic activity of lipophilic sulphonamide derivatives of the benzo[b]thiophene 1,1-dioxide. Bioorg Med Chem 12:963–968
Huang S, Connolly PJ, Lin R, Emanuel S, Middleton SA (2006) Synthesis and evaluation of N-acyl sulfonamides aspotential prodrugs of cyclin-dependent kinase inhibitor JNJ-7706621. Bioorg Med Chem Lett 16:3639–3641
Kenneth R, Hande KR, Hagey A, Berlin J, Cai Y, Meek K, Kobayashi H, Lockhart AC, Medina D, Sosman J, Gordon GB, Rothenberg ML (2006) The pharmacokinetics and safety of ABT-751, a novel, orallybioavailable sulfonamide antimitotic agent: results of a phase 1 study. Clin Cancer Res 12:2834
Kesteren CV, Mathôt RAA, Raymond E, Armand JP, Dittrich C, Dumez H, Roché H, Droz JP, Punt C, Ravic M, Wanders J, Beijnen JH, Fumoleau P (2002) Schellens for the Early Clinical Studies Group of the European Organization for research and treatment of cancer. J Clin Oncol 20:4065–4073
Al-Dosari MS, Ghorab MM, Alsaid MS, Nissan YM (2013) Discovering some novel 7-chloroquinolines carrying a biologically active benzenesulfonamide moiety as a new class of anticancer agents. Chem Pharm Bull 61:50–58
Al-Dosari MS, Ghorab MM, Alsaid MS, Nissan YM, Ahmed AB (2013) Synthesis and anticancer activity of some novel trifluoromethylquinolines carrying a biologically active benzenesulfonamide moiety. Eur J Med Chem 69:373–383
Ghorab MM, Ragab FA, Heiba HI, Nissan YM, Ghorab WM (2012) Novel brominated quinoline and pyrimidoquinoline derivatives as potential cytotoxic agents with synergistic effects of γ-radiation. Arch Pharm Res 35:1335–1346
Ghorab MM, Alsaid MS, Nissan YM (2012) Dapson in heterocyclic chemistry, part V: synthesis, molecular docking and anticancer activity of some novel sulfonylbiscompounds carrying biologically active dihydropyridine, dihydroisoquinoline, 1,3-dithiolan, 1,3-dithian, acrylamide, pyrazole, pyrazolopyrimidine and benzochromenemoieties. Chem Pharm Bull 60:1019–1028
Ghorab MM, Ceruso M, AlSaid MS, Nissan YM, Arafa RK, Supuran CT (2014) Novel sulfonamides bearing pyrrole and pyrrolopyrimidine moieties as carbonic anhydrase inhibitors: synthesis, cytotoxic activity and molecular modeling. Eur J Med Chem 87:186–196
Ghorab MM, Ceruso M, AlSaid MS, Nissan YM, Supuran CT (2014) Carbonic anhydrase inhibitors: synthesis, molecular docking, cytotoxic and inhibition of the human carbonic anhydrase isoforms I, II, IX, XII with novel benzenesulfonamides incorporating pyrrole, pyrrolopyrimidine and fused pyrrolopyrimidine moieties. Bioorg Med Chem 22:3684–3695
Ghorab MM, Alsaid MS, Nissan YM (2013) Anti breast cancer of some novel pyrrolo and pyrrolopyrimidine derivatives bearing a biologically active sulfonamide moiety. Life Sci J 10:2170–2183
Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk H (1999) MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol 1:94–97
Haddad J (2001) VX-745. Vertex pharmaceuticals. Curr Opin Invest Drugs 2:1070–1076
Lin S, Lombardo M, Malkani S, Hale JJ, Mills SG, Chapman K, Thompson JE, Zhang WX, Wang R, Cubbon RM, O’Neill EA, Luell S, Carballo-Jane E, Yang L (2009) Novel 1-(2-aminopyrazin-3-yl)methyl-2-thioureas as potent inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). Bioorg Med Chem Lett 19:3238–3242
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112
Authors’ contributions
MMG, MSAl said designed and contributed in synthesis. MSAl-Dosari carried out biological screening. YMN carried out molecular docking study. SMA contributed in experimental interpretation. All authors read and approved the final manuscript.
Acknowledgements
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award number (13-MED 997-02).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ghorab, M.M., Alsaid, M.S., Al-Dosary, M.S. et al. Design, synthesis and anticancer activity of some novel thioureido-benzenesulfonamides incorporated biologically active moieties. Chemistry Central Journal 10, 19 (2016). https://doi.org/10.1186/s13065-016-0161-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-016-0161-4