
Abstract
Background
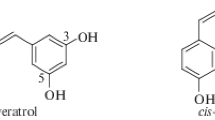
Compounds based on trans-1,2-diphenylethene are the subject of intense interest both for their optical properties and as potential leads for drug discovery, as a consequence of their anticancer, anti-inflammatory and antioxidant properties. Perhaps the best known of these is trans-3,5,4′-trihydroxystilbene (resveratrol), that has been identified as a promising lead in the search for anti-ageing therapeutics.
Results
We report here a new, convenient, one-pot stereo-selective synthesis of resveratrol and other trans-stilbene derivatives. A wide range of known and novel “Resveralogues” were synthesised by using this simple protocol, including examples with electron donating and electron withdrawing substituents, in uniformly high yield. The structures of all compounds were confirmed by standard methods including 1H and 13C NMR, IR and High Resolution Mass spectroscopy.
Conclusions
We have established a simple and convenient protocol for resveralogue synthesis. It is readily scalable, and sufficiently robust and simple for ready use in automated synthesis or for library development of resveralogues. This supersedes previously reported synthetic methods that required inert conditions, extensive purification and/or costly reagents.

One-pot preparation of diverse Resveralogues - high yields of product with minimal purification.
Similar content being viewed by others


Background
trans-1,2-Diphenylethene is the basic structural unit in a wide variety of naturally-occurring molecules. These, and synthetic analogues, have been deployed in photochemical dyes and fluorescent whitening agents [1], polymeric materials [2, 3], and are the subject of intense interest as potential leads for drug discovery as a consequence of their anticancer [4, 5] anti-inflammatory [6] and antioxidant properties [7]. Perhaps the best known of these is trans-3,5,4′-trihydroxystilbene (resveratrol), which has shown potential clinical value as a dietary restriction mimetic. Such mimetics are thought to slow the rate of deleterious processes associated with ageing and thus have the potential to prevent, or even remediate, multiple age-associated degenerative pathologies, including cognitive impairment, arthritis, cardiovascular disease and immune dysfunction [8, 9]. trans-Stilbenes thus represent attractive scaffolds for future compound development. However, resveratrol itself has been shown to have a range of activities, some of which may actually be detrimental to health. Equally, resveratrol is very limited in its bioavailability and, taken together, these issues leave uncertainties about its likely in vivo modes of action and consequent clinical utility. There is therefore a need for simple and versatile syntheses of a wide variety of structural analogues of resveratrol, or resveralogues, in order to facilitate detailed investigation of stilbenoid structure-activity relationships, and to allow development of potential therapeutic compounds with improved bioavailability.
trans-Stilbene derivatives are generally synthesised utilising Wittig or Horner- Wadsworth- Emmons (HWE) reactions and through catalytic methods such as Heck, Suzuki and Negishi coupling reactions or through the use of organozinc reagents [10]. Many of the examples in the literature suffer from incomplete conversion and low yields, or poor stereoselectivity. Of the existing methods, three have been previously modified to one-pot syntheses. These utilised the oxidative Wittig-Heck reaction [11, 12] two sequential Heck-type reactions of aryl bromide [13] and organozinc reagents [10]. Although some of these protocols provide good yields, they tend to require costly organometallic catalysts, inert reaction conditions, large excesses of reagents [13], long reaction times (>40 h in some instances) and/or complex solvent mixtures. We present here the first example of a robust, one-pot, readily scalable synthesis of diverse trans-stilbene derivatives and resveralogues.
Results and discussion
We have developed a simple and convenient one-pot method using sequential Michaelis-Arbuzov rearrangement and HWE chemistry. The overall reaction scheme is shown below (Scheme 1).

One-pot synthesis of resveralogues
Our synthetic protocol reduces both reaction time and reagent usage, by removing the need for isolation and purification both at the intermediate step and in the final product. A wide variety of analogues can conveniently be prepared from existing readily available benzyl bromides and benzaldehyde derivatives. We here describe the preparations of a range (1–16 below) of examples of resveralogues including several novel compounds (2, 4, 6, 8, 15 and 16), in good yield and purity, with minimal waste and manipulative steps.
Our method is facile, versatile and cost effective. We have applied this protocol to the synthesis of a range of differently substituted derivatives and in each case the required trans-stilbene was produced in good yield either without purification or through simple recrystallisation. The compounds were characterised by 1H NMR, 13C NMR, IR, high resolution mass spectroscopy and melting point, and where possible confirmed by comparison to literature values. The characteristic pattern of trans-coupling constant (J) value (more than 15 MHz) was observed in the 1H NMR spectra of all compounds synthesised, and indicated >95 % trans selectivity, in that signals from the alternate isomer were not observed.
Experimental
We selected the Michaelis-Arbuzov rearrangement as the most versatile method available for preparation of phosphonate esters [14, 15]. In this step, we reacted the starting substituted benzyl bromides with a single molar equivalent of triethylphosphite in the absence of solvent with heating to 150 °C. This abrogates the need for the large excesses of triethylphosphite utilised in earlier reports and also avoids potential by-product formed as a consequence of the reaction of ethyl bromide with the excess triethylphosphite. It is usual, at this stage, to isolate and then purify the diethylphosphonate intermediate via column chromatography or distillation [16]. In our protocol, however, we continue directly to the HWE reaction, giving the desired stilbenes in high yield and purity. In most cases the conversion to trans-stilbene is complete after 12 h of heating to reflux, and the product can be crystallised directly from the reaction mixture by simple addition of ice and a small quantity of methanol. Where necessary, further purification can be achieved by recrystallisation from ethanol. The choice of base for the HWE reaction is important to the success of this one pot synthesis. Our initial efforts focused on the reaction of 2- and 4- substituted phosphonates to give the relevant stilbenes and these proceed readily to completion with the use of 1.2 equivalents of potassium t-butoxide as base. However, when deactivating meta and/or electron donating substituents are present potassium t-butoxide is unsuccessful and a stronger base must be used. We found that 1.1 equivalents of sodium methoxide was sufficient to ensure the reaction proceeded successfully when such groups were present, although in some cases the yield was a little reduced.
Conclusions
Our one-pot protocol is very efficient and stereoselective for the synthesis of a wide range of resveralogues. The utility and applicability of this method is enhanced by its simple work up procedure, rendering it also suitable for use in automated synthesis.
Methods
General
The starting materials and solvent, reagents were obtained commercially and used directly without purification. The NMR spectra of compounds were recorded on Brüker FT-NMR 400Hz spectrometer in CDCl3 using tetramethylsilane (TMS) as an internal standard. The δ values represent chemical shifts reported in parts per million (ppm) and coupling constant (J) values are in Hz. Assignments correspond to R and R’ as per Table 1, numbering each ring from the carbon closest to the central double bond. 13C NMR spectra were definitively assigned with reference to HSQC correlation spectra (not presented). ESI-MS and ESI-HRMS were recorded on a Brüker MicroTOF instrument. Melting points were recorded on an Electrothermal melting point apparatus and are uncorrected. Flash chromatography was conducted by using Silica size (100–200 mesh). Thin layer chromatography was performed on TLC Silica Gel 60 F254 (Merck).
General procedure for synthesis of substituted trans-stilbenes
To a two-necked oven dried round-bottomed flask, the required benzyl bromide (500 mg, 1 equivalent) and triethylphosphite (1 equivalent) were added with N2 purging. This reaction mixture was heated at 150 °C for 4-5 h. The reaction was monitored by thin layer chromatography (eluent 10:90::ethyl acetate: petroleum ether). After completion, the reaction mixture was cooled to room temperature and then diluted tenfold with N,N-dimethylformamide. Base (for procedure A, KOtBu (1.1 equivalents) and for procedure B, NaOMe (1.2 equivalents)) was added and stirred at room temperature for 10 min. Aldehyde (0.8 equivalents) was then added to the reaction mixture which was then stirred at room temperature for a further hour. After this time it was heated to reflux for 12 h. The reaction was cooled to room temperature and quenched by adding ice and a small amount of methanol. The resulting precipitate was then filtered and dried to give the crude stilbene as title product. The crude product was recrystallised from ethanol or ethyl acetate to give fine and pure crystals.
(E)-2-(3,5-dimethoxystyryl)benzonitrile (1)
Procedure A gave 478 mg white solid (71 %), m.p. = 72–73 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.27, IR ν (cm-1) = 3068, 2948, 2838, 2219, 1588, 1425, 1352, 1152, 1056, 958, 755, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.84 (6H, s, -OMe), 6.45 (1H, t, J2Hz, H4), 6.72 (2H, d, J2Hz, H2, H6), 7.19 (1H, d, J16Hz, -C = C-H), 7.33 (1H, td, J7.6,1.1Hz, H4′), 7.41 (1H, d, J16Hz, -C = C-H), 7.57 (1H, td, J7.6, 1Hz, H5′), 7.65 (1H, dd, J8.0, 1Hz, H6′), 7.78 (1H, d, J8.0Hz, H3′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 101.2 (C4), 105.2 (C2, C6), 111.3 (C2′), 117.4 (CN), 124.6 (C = C), 125.4 (C3′), 127.6 (C4′), 132.7 (C5′), 133.2 (C6′), 133.5 (C = C), 138.2 (C1′), 140.4 (C1), 161.1 (C3, C5). HRMS m/z calculated 288.09950 [(M + Na)]+ and 533.20978 [2 M + Na]+, found 288.09945 and 553.20737.
(E)-4-(3-chlorostyryl)-N,N-dimethylaniline (2)
Procedure B gave 495 mg white solid (79 %). m.p. = 138–139 °C. Rf (10:90::ethyl acetate:petroleum ether) = 0.56, IR ν (cm-1) = 3031, 2893, 1608, 1582, 1065, 965, 808, 730, 1H NMR (400 MHz, CDCl3) δ (ppm) = 2.98 (6H, s, -N(CH3)2), 6.71 (distorted d, J8.8 Hz, H3, H5), 6.83 (1H, d, J16.4Hz, -C = C-H), 7.02 (1H, d, J16.4Hz, -C = C-H), 7.15 (1H, ddd, J8.0, 2.0,1.2, H6′), 7.24 (1H, t, J8Hz, H5′), 7.32 (1H, distorted d, J7.6Hz, H4′), 7.40 (2H, distorted d, J8.8Hz, H2, H6), 7.45 (1H, t, J3.5Hz, H2′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 40.4 (N(CH3)2), 112.4 (C3,C5), 122.8 (C = C), 124.2 (C4′), 125.4 (C1), 125.8 (C2′), 126.5 (C6′), 127.8 (C2, C6), 129.7 (C5′), 130.3 (C = C), 134.5 (C1′), 140.2 (C3′), 150.4 (C4). HRMS (ESI) calculated m/z 258.10440 [(M + H)]+, found 258.09288.
(E)-1,3-dimethoxy-5-(2-nitrostyryl)benzene (3)
Procedure A gave 528 mg yellow solid (80 %), m.p. = 72–73 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.27, IR ν (cm-1) = 3004, 2940, 2838, 1599, 1516, 1342, 1205, 1154, 1054, 951, 819, 744, 670, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (6H, s, -OMe), 6.44 (1H, t, J2.4Hz, H4), 6.68 (2H, d, J2.4Hz, H2, H6), 7.00 (1H, d, J16.4Hz, H-C = C-), 7.40 (1H, td, J8.4,1.3Hz, H4′), 7.59 (1H, d, J16Hz, H-C = C-), 7.59 (1H, t, J7.4Hz, H5′), 7.74 (d, J7.6Hz, H6′), 7.96 (1H, dd, J8.1,1.1Hz, H3′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 100.9 (C4), 105.2 (C2, C6), 124.0 (C = C), 124.8 (C3′), 128.0 (C4′), 128.2 (C6′), 132.9 (C5′), 133.1 (C = C), 133.9 (C1′), 138.5 (C1), 148.1 (C2′), 161.1 (C3, C5). HRMS calculated m/z 308.08933 [(M + Na)]+, found 308.10436.
(E)-1-(2-fluorostyryl)-3,5-dimethoxybenzene (4)
Procedure B gave 505 mg shiny white crystals (74 %), m.p. = 62 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.64, IR ν (cm-1) = 3001, 2997, 2954, 1590, 1488, 1357, 1152, 1053, 966, 825, 677, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (6H, s, -OMe), 6.41 (1H, t, J2.2 Hz, H4), 6.69 (d, J2.2Hz, H2, H6), 7.15-7.08 (3H, m (includes d, J16.4Hz), H3′, H6′, -C = C-H), 7.26-7.22 (2H, m (includes d, J16.4Hz), H5′, -C = C-H), 7.59 (1H, td, J = 7.7,1.6Hz, H4′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (-OMe), 100.4 (C4), 104.8 (C2, C6), 115.8 (C3′, J C-F 21.92Hz), 121.5 (C = C, J C-F 3.1Hz), 124.2 (C6′, J C-F 3.1Hz), 125.1 (C1′, J C-F 11.7Hz), 127.2 (C4′, J C-F 3.5Hz), 128.9 (C5′, J C-F 8.5Hz), 131.0 (C = C, J C-F 4.68Hz), 139.3 (C1), 161.0 (C3, C5), 160.5 (C2′, J C-F 247.9Hz). HRMS calculated m/z 281.09483 [(M + Na)]+, found 281.07161.
(E)-1-(4-methoxystyryl)-2-nitrobenzene (5)
Procedure A gave 466 mg yellow solid (79 %), m.p. = 66–69 °C (literature m.p. = 69.9 °C, [17]), Rf (10:90::ethyl acetate:petroleum ether) = 0.64, IR ν (cm-1) = 3081, 2977, 2938, 1600, 1505, 1422, 1337, 1250, 1054, 968, 822, 768, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.82 (3H, s, OMe), 6.90 (2H, dd, J8.8, 2Hz, H2, H6), 7.04 (1H, d, J16Hz, -C = C-H), 7.34 (td, J8.3,1.3Hz, H4′), 7.34–7.48 (3H, m (includes d, J16Hz), H3, H5, -C = C-H), 7.55 (1H, td, J7.4, 0.9Hz, H5′), 7.72 (1H, dd, J7.8, 0.9Hz, H6′), 7.91 (1H, dd, J8.2, 1.2Hz, H3′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 114.3 (C2, C6), 121.1 (C = C), 124.8 (C3′), 127.5 (C4′) 127.9 (C6′), 128.5 (C3, C5), 129.3 (C1′), 133.0 (C1), 133.3 (C5′), 133.5 (C = C), 147.9 (C2′), 160.1 (C4). HRMS calculated m/z 278.07876 [(M + Na)]+, found 278.07100.
(E)-1-(3-chlorostyryl)-3,5-dimethoxybenzene (6)
Procedure B gave, after purification, 454 mg colourless liquid (68 %), b.p. >300 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.47, IR ν (cm-1) = 3030, 2995, 2898, 1603, 1542, 1357, 1205, 1053, 968, 825, 677, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (6H, s, OMe), 6.41 (1H, t, J2.2Hz, H4), 6.66 (2H, d, J2.2Hz, H2, H6), 6.98 (1H, distorted d, J16.2Hz, -C = C-H), 7.03 (1H, distorted d, J16.2Hz, -C = C-H), 7.22 (1H, dt, J8.4,1.2Hz H6′), 7.26 (1H, dd, J8.8, 7.6Hz, H5′), 7.35 (dt, J7.5, 1.2Hz, H4′), 7.49 (1H, t, J1.6Hz, H2′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 100.4 (C4), 104.8 (C2, C6), 124.8 (C4′), 126.4 (C2′), 127.6 (C6′), 127.7 (C = C), 129.9 (C5′), 130.2 (C = C), 134.7 (C1′), 138.84 (C3′), 139.08 (C1), 161.06 (C3, C5). HRMS calculated m/z 297.06528 [(M + Na)]+ and 571.14134 [(2 M + Na)]+, found 297.06374 and 571.13963.
(E)-N,N-dimethyl-4-(3-(trifluoromethyl)styryl)aniline (7)
Procedure B gave 450 mg light green crystals (74 %), m.p. = 146 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.56, IR ν (cm-1) = 3026, 2810, 1601, 1521, 1327, 1115, 1071, 965, 1H NMR (400 MHz, CDCl3) δ (ppm) = 2.99 (6H, s, -N(CH3)2), 6.72 (2H, dd, J7.2, 2.0Hz, H2, H6), 6.91 (1H, d, J16.0Hz, -C = C-H), 7.10 (1H, d, J16.0Hz, -C = C-H), 7.40–7.44 (4H, m, H4′, H6′, H3, H5), 7.62 (1H, distorted t, H5′), 7.70 (1H, s, H2′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 40.4 (N(CH3)2), 112.4 (C2, C6), 122.5 (C2′, J C-F 3Hz), 122.7 (C = C), 123.0 (C6′, J C-F 3Hz), 125.0 (C3′), 125.6 (C1′), 127.84 (C3, C5), 128.94 (C5′), 128.99 (C4′), 130.7 (C = C), 131.0 (CF3, J C-F 32Hz), 139.1 (C1), 150.5 (C4). HRMS calculated m/z 292.13076 [(M + H)]+, found 292.13062.
(E)-1-(3,5-dimethoxystyryl)-3,5-dimethylbenzene (8)
Procedure B gave, after purification, 545 mg white solid (81 %), m.p. = 66–68 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.47, IR ν (cm-1) = 3256, 3009, 2923, 1598, 1463, 1326, 1164, 1071, 962, 839, 690, 1H NMR (400 MHz, d6-DMSO) δ (ppm) = 2.29 (6H, s, CH3), 3.78 (6H, s, OMe), 6.40 (1H, t, J2.4Hz, H4), 6.75 (2H, d, J2.4Hz, H2, H6), 6.91 (broad s, H4′), 7.11 (1H, d, J16.4Hz, C = C-H), 7.18 (1H, d, J16.4Hz, C = C-H), 7.25 (broad s, H2′, H6′), 13C NMR (100 MHz, d6-DMSO) δ (ppm) = 20.9 (Me), 55.1 (OMe), 99.8 (C4), 104.3 (C2, C6), 124.3 (C2′, C6′), 128.0 (C = C), 129.0 (C = C), 129.2 (C4′), 136.7 (C1′), 137.5 (C3′, C5′), 139.1 (C1), 160.6 (C3, C5). HRMS calculated m/z 291.13555 [(M + Na)]+, found 291.13523.
(E)-1-(3,5-dimethoxystyryl)-2,4-difluorobenzene (9)
Procedure B gave 494 mg white crystals (74 %), m.p. = 72–74 °C (literature m.p. = 78 °C, [18]), Rf (10:90::ethyl acetate:petroleum ether) = 0.64, IR ν (cm-1) = 3065, 2948, 2841, 1589, 1460, 1281, 1152, 1054, 965, 813, 676, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (6H, s, OMe), 6.42 (1H, t, J2.4Hz, H4), 6.67 (2H, d, J2.4Hz, H2, H6), 6.80–6.91 (2H, m, H5′ and H6′), 7.02 (1H, d, J16.4Hz, -C = C-H), 7.16 (1H, d, J16.4Hz, -C = C-H), 7.55 (1H, distorted dd, J HF15.8, 8.4Hz, H3′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 100.4 (C4), 104.2 (C6′, J C-F 25.6Hz), 104.7 (C2, C6), 111.7 (C5′, J C-F 21.31 and 3.63Hz), 120.5 (C = C), 121.5 (C1′, J C-F 8.2 Hz), 128.0 (C3′, J C-F 5.17Hz), 130.7 (C = C, J C-F 2.3Hz), 139.1 (C1), 160.4 (C4′, J C-F 250.6, 11.6 Hz), 161.0 (C3, C5), 162.2 (C2′, J C-F 248.3, 11.8Hz). HRMS calculated m/z 299.08541 [(M + Na)]+ and 575.18159 [(2 M + Na)]+, found 299.10167 and 575.20262.
(E)-1,3-dimethoxy-5-(4-methylstyryl)benzene (10)
Procedure A gave 577 mg white crystals (84 %), m.p. = 56–57 °C (literature m.p. = 50–52 °C, [19]), Rf (10:90::ethyl acetate:petroleum ether) = 0.54, IR ν (cm-1) = 3029, 2961, 2937, 1585, 1450, 1290, 1148, 955, 831, 754, 1H NMR (400 MHz, CDCl3) δ (ppm) = 2.36 (3H, s, Me), 3.83 (6H, s, OMe), 6.39 (1H, t, J2.2Hz, H4), 6.66 (2H, d, J2.2Hz, H2, H6), 7.02 (1H, d, J16.2Hz, C = C-H), 7.06 (1H, d, J16.2Hz, C = C-H), 7.16 (2H, d, J7.9Hz, H2′, H6′), 7.40 (2H, d, J8.0Hz, H3′, H5′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 21.2 (Me), 55.4 (OMe), 99.9 (C4), 104.5 (C2, C6), 126.5 (C3′, C5′), 127.7 (C = C), 129.9 (C = C), 129.4 (C2′, C6′), 134.4 (C1′), 137.7 (C4′), 139.6 (C1), 161.01 (C3, C5). HRMS calculated m/z 277.11990 [(M + Na)]+, found 277.12461.
(E)-4-(3,5-dimethoxystyryl)benzonitrile (11)
Procedure A gave 541 mg white solid (80 %), m.p. = 105–107 °C (literature m.p. = 106–107 °C, [20]), Rf (10:90::ethyl acetate:petroleum ether) = 0.54, IR ν (cm-1) = 2959, 2937, 2841, 2220, 1587, 1340, 1208, 1169, 1066, 948, 833, 679, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.84 (6H, s, OMe), 6.45 (1H, t, J2.2Hz, H4), 6.68 (2H, d, J2.2Hz, H2, H6), 7.05 (1H, d, J16.2Hz, -C = C-H), 7.13 (1H, d, J16.2Hz, -C = C-H), 7.59 (2H, d, J8.3Hz, H2′, H6′), 7.64 (2H, d, J8.3Hz, H3′, H5′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 100.9 (C4), 105.1 (C2, C6), 110.7 (C4′), 119.0 (CN), 126.9 (C2′, C6′), 127.2 (C = C), 132.4 (C = C), 132.6 (C3′, C5′), 138.3 (C1′), 141.7 (C1), 161.1 (C3, C5). HRMS calculated m/z 288.09950 [(M + Na)]+, found 288.12135.
(E)-1,3-dimethoxy-5-(4-methoxystyryl)benzene (12)
Procedure B gave, after purification, 403 mg white solid (60 %), m.p. = 59–61 °C (literature m.p. = 55–58 °C, [21]), Rf (10:90::ethyl acetate:petroleum ether) = 0.48, IR ν (cm-1) = 2960, 2912, 2837, 1605, 1517, 1442, 1239, 965, 824, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (9H, s, OMe), 6.38 (1H, t, J2.0Hz, H4), 6.65 (2H, d, J2.0Hz, H2, H6), 6.89 (2H, d, J8.8Hz, H2′, H6′), 6.90 (1H, d, J16.2Hz, -C = C-H), 7.04 (1H, d, J16.2Hz, -C = C-H), 7.44 (d, J8.8Hz, H3′, H5′), 13C NMR (100 MHz, CDCl3) δ(ppm) = 55.3 (OMe), 55.4 (2xOMe), 99.7 (C4), 104.4 (C2, C6), 114.2 (C2′, C6′), 126.6 (C = C), 127.8 (C3′,C5′), 128.8 (C = C), 130.0 (C1′), 139.7 (C1), 159.4 (C4′), 161.0 (C3, C5). HRMS calculated m/z 293.11482 [(M + Na)]+, found 293.09290.
Methyl (E)- 4-(3,5-dimethoxystyryl)benzoate (13)
Procedure A gave 384 mg white solid (59 %), m.p. = 121–122 °C (literature m.p. = 117–121 °C, [22]), Rf (10:90::ethyl acetate:petroleum ether) = 0.51, IR ν (cm-1) = 3080, 2999, 2945, 1708, 1587, 1433, 1273, 1152, 966, 697, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (6H, s, OMe), 3.92 (3H, s, COOMe), 6.43 (1H, t, J2.2Hz, H4), 6.69 (2H, d, J2.2Hz, H2, H6), 7.08 (1H, d, J16.4Hz, -C = C-H), 7.14 (1H, d, J16.4Hz, -C = C-H), 7.55 (2H, d, J8.3Hz, H2′, H6′), 8.02 (d, J8.2Hz, H3′, H5′), 13C NMR (100 MHz, CDCl3) δ (ppm) = 52.1 (COOMe), 55.4 (OMe), 100.6 (C4), 104.9 (C2, C6), 126.4 (C2′, C6′), 128.1 (C = C), 129.0 (C1′), 130.03 (C3′,C5′), 131.2 (C = C), 138.8 (C1), 141.65 (C4′), 161.1 (C3, C5), 166.8 (COOMe). HRMS calculated m/z 321.10973 [(M + Na)]+ and 619.23024 [(2 M + Na)]+, found 321.10598 and 619.22537.
(E)-1,3-dimethoxy-5-(4-nitrostyryl)benzene (14)
Procedure A gave 594 mg yellow solid (90 %), m.p. = 137–138 °C (literature m.p. = 135–137 °C, [23]), Rf (10:90::ethyl acetate:petroleum ether) = 0.48, IR ν (cm-1) = 3051, 2839, 1636, 1592, 1503, 1329, 1295,1205, 1106, 948, 824, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.86 (6H, s, OMe), 6.47 (1H, t, J2.4Hz, H4), 6.71 (2H, d, J2.4Hz, H2, H6), 7.11 (1H, d, J18.0Hz, -C = C-H), 7.20 (1H, d, J18.0Hz, -C = C-H), 7.63 (2H, d, J9.7Hz, H2′, H6′), 8.22 (2H, d, J9.7Hz, H3′, H5′), 13C NMR(100 MHz, CDCl3) δ(ppm) = 55.4 (OMe), 101.0 (C4), 105.12 (C2, C6), 124.1 (C3′, C5′), 126.8 (C = C), 126.9 (C2′,C6′), 133.3 (C = C), 138.1 (C1′), 143.7 (C1), 146.8 (C4′), 161.1 (C3, C5). HRMS calculated m/z 308.08933 [(M + Na)]+, found 308.08817.
(E)-2-(3,5-dimethoxystyryl)-1,3-difluorobenzene (15)
Procedure A gave 520 mg shiny white crystals (78 %), m.p. 85–86 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.64, IR ν (cm-1) = 3009, 2973, 2844, 1589, 1463, 1306, 1200, 1152, 979, 788, 682, 1H NMR (400 MHz, CDCl3) δ (ppm) = 3.83 (6H, s, OMe), 6.43 (1H, t, J2.2Hz, H4), 6.69 (2H, d, J2.2Hz, H2, H6), 6.92 (2H, t, J8.4Hz, H3′, H5′), 7.10 (d, J16.8Hz, -C = C-H) 7.15 (1H, tt, J8.4, 8.4Hz, H4′), 7.37 (1H, d, J16.4Hz, -C = C-H), 13C NMR (100 MHz, CDCl3) δ (ppm) = 55.4 (OMe), 100.6 (C4), 104.7 (C2, C6), 111.7 (C3′, C5′, J C-F 6.47), 114.7 (C1′, J C-F 15.1Hz), 115.7 (C = C), 128.0 (C4′, J C-F 10.7Hz), 135.1 (C = C, J C-F 8.35 Hz), 139.6 (C1), 160.9 (C2′, C6′, J C-F 250, 7.7 Hz), 161.0 (C3, C5). HRMS calculated m/z 299.08541 [(M + Na)]+, found 299.10582.
(E)-1,3-dichloro-2-(3,5-dimethoxystyryl)benzene (16)
Procedure B gave 508 mg, white solid (79 %), m.p. 66–67 °C, Rf (10:90::ethyl acetate:petroleum ether) = 0.64, IR ν (cm-1) = 2959, 2897, 2833, 1598, 1424, 1348, 1206, 1152, 1054, 965, 819, 763, 676, 1H NMR (400 MHz, d6-DMSO) δ (ppm) = 3.78 (6H, s, OMe), 6.48 (1H, t, J2.0Hz, H4), 6.78 (2H, d, J2.0Hz, H2, H6), 6.99 (1H, d, J16.4Hz, -C = C-H), 7.15 (1H, d, J16.4Hz, -C = C-H), 7.32 (1H, t, J8.0Hz, H4′), 7.52 (2H, d, J8.0Hz, H3′, H5′), 13C NMR (100 MHz, d6-DMSO) δ (ppm) = 55.3 (OMe), 100.9 (C4), 104.7 (C2, C6), 122.7 (C = C), 128.8 (C3′, C5′), 129.3 (C4′), 133.6 (C2′, C6′), 134.1 (C1’), 136.8 (C = C), 138.1 (C1), 160.7 (C3, C5). HRMS calculated m/z 331.02631 [(M + Na)]+ and 641.06044 [(2 M + Na)]+, found m/z 331.04468 and 641.09081.
References
Leaver IH, Milligan B. Fluorescent whitening agents—a survey (1974-82). Dyes Pigments. 1984;5(2):109–44.
Flores-Rojas GG, Lijanova IV, Morales-Saavedra OG, Sanchez-Montes K, Martínez-García M. Synthesis and NLO behavior of Oligo(phenylenevinylene)-Porphyrin Dendrimers. Dyes Pigments. 2013;96(1):125–9.
Lijanova IV, Moggio I, Arias E, Klimova T, Martínez-García M. Resorcinarene-dendrimers with stilbene moieties for optoelectronics. Tetrahedron. 2008;64(44):10258–66.
Carbo N, Costelli P, Baccino FM, Lopez-Soriano FJ, Argiles JM. Resveratrol, a natural product present in wine, decreases tumour growth in a rat tumour model. Biochem Biophys Res Commun. 1999;254(3):739–43.
Park JW, Choi YJ, Jang MA, Lee YS, Jun DY, Suh SI, et al. Chemopreventive agent resveratrol, a natural product derived from grapes, reversibly inhibits progression through S and G2 phases of the cell cycle in U937 cells. Cancer Lett. 2001;163(1):43–9.
Udenigwe CC, Ramprasath VR, Aluko RE, Jones PJ. Potential of resveratrol in anticancer and anti-inflammatory therapy. Nut Rev. 2008;66(8):445–54.
de la Lastra CA, Villegas I. Resveratrol as an antioxidant and pro-oxidant agent: mechanisms and clinical implications. Biochem Soc Trans. 2007;35(Pt 5):1156–60.
Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5(6):493–506.
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6.
Wang J-X, Fu Y, Hu Y. Carbon–carbon double-bond formation from the reaction of organozinc reagents with aldehydes catalyzed by a nickel(II) complex. Angew Chem Int Ed. 2002;41(22):4170–0.
Saiyed AS, Bedekar AV. One-pot synthesis of stilbenes by dehydrohalogenation–Heck olefination and multicomponent Wittig–Heck reaction. Tetrahedron Lett. 2010;51(48):6227–31.
Saiyed AS, Patel KN, Kamath BV, Bedekar AV. Synthesis of stilbene analogues by one-pot oxidation-Wittig and oxidation-Wittig–Heck reaction. Tetrahedron Lett. 2012;53(35):4692–6.
Jeffery T, Ferber B. One-pot palladium-catalyzed highly chemo-, regio-, and stereoselective synthesis of trans-stilbene derivatives. A concise and convenient synthesis of resveratrol. Tetrahedron Lett. 2003;44(1):193–7.
Michaelis A, Kaehne R. Ueber das Verhalten der Jodalkyle gegen die sogen. Phosphorigsäureester oder O-Phosphine. Berichte der deutschen chemischen Gesellschaft. 1898;31(1):1048–55.
Bhattacharya AK, Thyagarajan G. Michaelis-Arbuzov rearrangement. Chem Rev. 1981;81(4):415–30.
Callahan JF, Lin G, Wan Z, Yan H. Dual pharmacophores—PDE4-muscarinic antagonistics. Patent EP2249647 (A1). 2011.
Nandi D, Ghosh D, Chen S-J, Kuo B-C, Wang NM, Lee HM. One-Step Synthesis of Isocoumarins and 3-Benzylidenephthalides via Ligandless Pd-Catalyzed Oxidative Coupling of Benzoic Acids and Vinylarenes. J Organic Chem. 2013;78(7):3445–51.
Thomas E. COX-II INHIBITOR COMPOUNDS. Patent 2006, WO2006029436 (A1). 2006.
Letcher RM. The anisotropic effect of 4-substituents on the 1H NMR chemical shift of H-5 in phenanthrenes. Org Magnetic Res. 1981;16(3):220–3.
Sun B, Hoshino J, Jermihov K, Marler L, Pezzuto JM, Mesecar AD, et al. Design, synthesis, and biological evaluation of resveratrol analogues as aromatase and quinone reductase 2 inhibitors for chemoprevention of cancer. Bioorganic Med Chem. 2010;18(14):5352–66.
Schmidt B, Elizarov N, Berger R, Holter F. Scope and limitations of the Heck-Matsuda-coupling of phenol diazonium salts and styrenes: a protecting-group economic synthesis of phenolic stilbenes. Org Biomol Chem. 2013;11(22):3674–91.
Paul S, Mizuno CS, Lee HJ, Zheng X, Chajkowisk S, Rimoldi JM, et al. In vitro and in vivo studies on stilbene analogs as potential treatment agents for colon cancer. Eur J Med Chem. 2010;45(9):3702–8.
Liu H, Dong A, Gao C, Tan C, Liu H, Zu X, et al. The design, synthesis, and anti-tumor mechanism study of N-phosphoryl amino acid modified resveratrol analogues. Bioorg Med Chem. 2008;16(23):10013–21.
Acknowledgements
The authors would like to acknowledge the University of Brighton and the Glenn Foundation for providing financial support and Dr. Guy Standen and Dr. Howard Dodd for analytical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VB – developed the synthesis and co-wrote the manuscript, ANS – co-directed the study and co-wrote the manuscript, JM – undertook synthesis, purification and analytical studies, RGAF – co-directed the study and co-wrote the manuscript, ELO – conceived of and co-directed the study and co-wrote the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Birar, V.C., Sheerin, A.N., Milkovicova, J. et al. A facile, stereoselective, one-pot synthesis of resveratrol derivatives. Chemistry Central Journal 9, 26 (2015). https://doi.org/10.1186/s13065-015-0102-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-015-0102-7