
Abstract
Background
Medical complications during pregnancy, including anaemia, gestational diabetes mellitus and hypertensive disorders of pregnancy place women are at higher risk of long-term complications. Scalable and low-cost strategies to integrate non-communicable disease screening into pregnancy care are needed. We aim to determine the effectiveness and implementation components of a community-based, digitally enabled approach, “SMARThealth Pregnancy,” to improve health during pregnancy and the first year after birth.
Methods
A pragmatic, parallel-group, cluster randomised, type 2 hybrid effectiveness-implementation trial of a community-based, complex intervention in rural India to decrease anaemia (primary outcome, defined as haemoglobin < 12g/dL) and increase testing for haemoglobin, glucose and blood pressure (secondary outcomes) in the first year after birth. Primary Health Centres (PHCs) are the unit of randomisation. PHCs are eligible with (1) > 1 medical officer and > 2 community health workers; and (2) capability to administer intravenous iron sucrose. Thirty PHCs in Telangana and Haryana will be randomised 1:1 using a matched-pair design accounting for cluster size and distance from the regional centre. The intervention comprises (i) an education programme for community health workers and PHC doctors; (ii) the SMARThealth Pregnancy app for health workers to support community-based screening, referral and follow-up of high-risk cases; (iii) a dashboard for PHC doctors to monitor high-risk women in the community; (iv) supply chain monitoring for consumables and medications and (v) stakeholder engagement to co-develop implementation and sustainability pathways. The comparator is usual care with additional health worker education. Secondary outcomes include implementation outcomes assessed by the RE-AIM framework (reach, effectiveness, adoption, implementation, maintenance), clinical endpoints (anaemia, diabetes, hypertension), clinical service delivery indicators (quality of care score), mental health and lactation practice (PHQ9, GAD7, EuroQoL-5D, WHO IYCF questionnaire).
Discussion
Engaging women with screening after a high-risk pregnancy is a challenge and has been highlighted as a missed opportunity for the prevention of non-communicable diseases. The SMARThealth Pregnancy trial is powered for the primary outcome and will address gaps in the evidence around how pregnancy can be used as an opportunity to improve women’s lifelong health. If successful, this approach could improve the health of women living in resource-limited settings around the world.
Trial registration
ClinicalTrials.gov NCT05752955. Date of registration 3 March 2023.
Similar content being viewed by others


Background and rationale {6a}
Over the past 20 years, significant progress has been made in India in reducing maternal and newborn deaths [1]; however, improvements have not been made evenly across the country and ensuring consistent quality care delivery across a vast population remains challenging. Globally, and in India, non-communicable diseases (NCDs) are women’s leading causes of death, and practical strategies are urgently needed to prevent premature mortality and morbidity [2, 3]. Complications that develop during pregnancy can identify women at increased risk for NCDs in the months and years following birth [4]. For example, hypertensive disorders of pregnancy (HDP), including gestational hypertension and preeclampsia, identify women at double the background risk for premature cardiovascular diseases [5], with around one in three women identified with an HDP having hypertension within the years immediately after birth, the leading risk factor for premature stroke [6]. Gestational diabetes mellitus (GDM), a form of glucose intolerance first detected in pregnancy and resolving with the birth of the baby and placenta, affects up to one in six pregnant people globally [7]. Those who develop GDM are at significantly increased risk of type 2 diabetes in the immediate postnatal years, with up to 50% having dysglycaemia within 5 years of the pregnancy [8]. Longer term, these women are also at increased risk of premature cardiovascular disease, a risk independent of type 2 diabetes [9].
Pregnancy as an opportunity to improve women’s lifelong health
The potential for pregnancy care to be part of an integrated approach to NCD prevention has been recommended by the International Federation of Gynaecology and Obstetrics (FIGO) [10] and other leaders in women’s health [11,12,13]. Anaemia, HDP and GDM have been identified as priority areas to improve maternal and newborn outcomes in rural India [14]. Intensive diet and exercise programmes, lactation and medications can delay or prevent the onset of type 2 diabetes after GDM [15, 16]. Postnatal and annual screening for diabetes following a pregnancy affected by GDM is therefore recommended. Screening and management of hypertension and other cardiovascular risk factors, such as controlling weight, stopping smoking and measuring lipids, is also recommended after a pregnancy affected by HDP [17]. Little evidence exists for how this can be effectively translated into practice in low-resource settings.
Anaemia is another endemic problem in India, and eradication is a Government health priority [18]. During pregnancy, anaemia is associated with poor pregnancy outcomes, including maternal mortality, caesarean section, low birth weight, small for gestational age, preterm birth and perinatal and neonatal mortality [19]. Anaemia Mukt Bharat (AMB), launched in 2018, is the comprehensive national programme that aims to reduce anaemia across the country [18]. Despite investment and coordinated efforts, the programme targets for anaemia prevalence by 2022 are < 32% in pregnant women and < 40% in breastfeeding women are yet to be met.
The SMARThealth programme
The George Institute for Global Health developed the Systematic Medical Appraisal, Referral and Treatment programme for cardiovascular disease prevention and management (SMARThealth) [20]. This global programme draws on innovative mobile-health technologies to promote evidence-based, clinical decision support systems. SMARThealth takes a health care ‘eco-system’ approach: (i) strengthening the capacity of primary health care workers and local communities; (ii) delivering fit-for-purpose electronic decision support systems and supportive technologies for self-management; and (iii) integrating with existing health system infrastructure. SMARThealth strengthens the skills of community health workers, known as ASHA (Accredited Social Health Activists) in India, to perform risk assessments for various conditions using an app deployed on a low-cost Android tablet. Data are stored securely on a cloud-based server with offline capabilities in areas of limited network connectivity. People at elevated risk of the screened conditions are electronically referred to the Primary Health Centre (PHC) for medical review. The PHC doctor has a complementary tablet app which communicates with the secure server and is provided with more complex decision support, including suggested medication management, based on national and international guidelines [21,22,23,24,25].
We have adapted the SMARThealth approach to improve the community-level identification, diagnosis, referral and management of women with anaemia, diabetes and hypertension during pregnancy and in the year after birth (SMARThealth Pregnancy) [26]. We demonstrated the acceptability and feasibility of SMARThealth Pregnancy in a pilot cluster randomised trial in four sites in rural India during 2019–2020 involving 200 pregnant women and 50 frontline health workers (unpublished work).
The SMARThealth Pregnancy programme evaluates both clinical and implementation effectiveness. A separate protocol will be published on the process evaluation, with detailed implementation outcomes and strategies.
Objectives {7}
To determine if the SMARThealth Pregnancy complex intervention can improve women’s health in the year after pregnancy, specifically by decreasing the prevalence of anaemia by 9% and improving screening, referral and follow-up following a pregnancy affected by either anaemia, diabetes or hypertensive disorders of pregnancy. We also aim to evaluate our within-trial implementation strategy and identify the key determinants needed to support implementation beyond the trial phase.
Our hypotheses is that community-based screening, referral and follow-up in women with high-risk pregnancy conditions will improve health status at 12 months after birth and will be a low-cost and sustainable service delivery model.
Trial design {8}
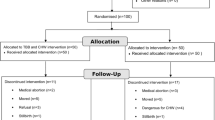
A pragmatic, type 2 hybrid effectiveness/implementation, parallel-group cluster randomised superiority trial in two states in India (Haryana and Telangana). Thirty PHCs in Telangana and Haryana will be randomised 1:1 using a matched-pair design accounting for cluster size and distance from the regional centre. As the intervention is aimed at health workers working as teams (community health workers and PHC doctors), effectiveness is best evaluated through a cluster randomised trial with clustering at the PHC level (Fig. 1).

Consort diagram
Methods: participants, interventions and outcomes
Study setting {9}
The study will be community-based and conducted in rural and semi-rural districts in India: Siddipet district, Telangana State, and Jhajjar and Rohtak districts, Haryana State. These have been chosen to represent two geographically, linguistically and culturally diverse populations.
Each PHC is responsible for primary care services for a population of around 30,000 people across several villages. We will select two villages per PHC as the study sites for the trial. All community health workers and PHC staff will be eligible to participate at each study site.
Eligibility criteria {10}
Eligibility for primary health centres (PHCs)
PHCs will need to be located in one of the study districts, with the chief doctor (or equivalent) consenting to participate and support the programme for the duration of the study. In addition, at the time of enrolment, to be eligible, PHCs must have:
-
≥ 1 Medical officer
-
≥ 1 Laboratory technician
-
≥ 1 ANM
-
≥ 2 ASHAs
-
The ability to administer iron sucrose infusions. Iron sucrose is the Government of India’s recommended treatment for moderate or severe anaemia in pregnancy that can be administered at the PHC level [18].
Exclusion criteria PHCs
PHCs with populations < 10,000 people, unwillingness to engage with the technology, or participation in a competing research study will be excluded.
Eligibility for women
Up to 5000 pregnant women will be screened until the sample size has been reached. Potentially eligible women will be identified by ANMs and ASHAs with the support of laboratory technicians. Women will be eligible if they are ≥ 18 years old. Women can be enrolled at any time during the pregnancy at or after 12 weeks of gestation, as determined by ultrasound scan, date of last menstrual period or best clinical estimate.
Exclusion criteria women
Women who decline participation; plan to move away and not return to the same village in the next 12 months; and do not speak or understand the local language (Telugu or Hindi). Women in India frequently return to their own mother’s village for the first few weeks after the birth. This will not be an exclusion criterion, provided they intend to return to the original village after this time.
Who will take written informed consent? {26a}
We have based our approach to consent on a professional-cluster design approach proposed by Hemming and Eldridge [27, 28]. Consent will be collected at three levels:
-
(1)
At the cluster level, the study team will seek consent for each PHC from the designated clinical lead:
-
a
To be randomised
-
b
For all health workers affiliated with the PHC to participate in the study.
-
a
-
(2)
From the health workers, we will seek consent:
-
a
To participate in the study,
-
b
For data collection during and after the educational programme.
-
a
-
(3)
From individual women, a member of the study team will seek consent:
-
a
To participate in the additional study assessments at the start and end of the study.
-
b
For the study team to access and use their data for the purposes outlined in the informed consent form (supplementary file).
-
a
Additional consent provisions for collection and use of participant data and biological specimens {26b}
This study does not involve the collection of biological specimens. As part of the consent form, participants will be asked if they agree for their data until that point to be included if they withdraw from the study. Participants will be asked permission for the research team to share relevant data with the international study team.
Interventions
The explanation for the choice of comparators {6b}
The study has been designed as a pragmatic trial to assess the impact of the intervention in a rural Indian context. In addition to current usual care, training will be held in all participating PHCs on screening for GDM using the oral glucose tolerance test, according to the Government of India recommendations [29] (enhanced usual care).
Intervention description {11a}
SMARThealth Pregnancy is a complex intervention comprising five main components (Fig. 2):
-
(1)
A health worker education programme
A 3-day training package for community healthcare workers (ASHAs) has been developed to improve knowledge of high-risk pregnancy conditions and raise awareness about the long-term implications of certain pregnancy-related conditions on women’s lifelong health. In addition, ASHAs are trained to perform point-of-care assessments, and how to use the SMARThealth Pregnancy app. PHC physicians will receive individual training on the SMARThealth Pregnancy programme, and on how to use the app.
-
(2)
The SMARThealth Pregnancy app
The system was co-developed with end users with clinical decision support based on local guidelines.
The app functions include:
-
Secure storage of individual participant records
-
Diary and schedule assistant to prioritise workload
-
Facility to record blood pressure and haemoglobin results and track trends
-
Facility to record iron folic acid compliance and supply
-
Electronic decision support for blood pressure, haemoglobin, GDM screening, tetanus toxoid vaccination and deworming
-
Electronic referral if indicated for PHC physician review
-
Compliance monitoring for medication adherence (where relevant)
-
Short health promotional videos aimed at women to be recorded in local languages about hypertension and diabetes, healthy diet and exercise, breastfeeding and future pregnancies
The ASHAs will be reminded by the app to assess women periodically during their pregnancy and in the first year after birth, timed where possible with other health encounters such as scheduled antenatal visits and infant immunisations. ASHAs will be trained and equipped to perform point-of-care testing for common pregnancy complications. Anaemia will be screened using a point-of-care fingerstick device for haemoglobin measurement (TrueHb™). Blood pressure will be measured using an automated device validated for pregnancy (Omron™). ASHAs will ensure the woman is seated comfortably, with both feet flat on the floor and the cuff placed on the arm at the level of the heart. ANMs will be trained to administer the oral glucose tolerance test. The test is performed in the non-fasting state. ANMs will mix 75 g of glucose powder in one cup of water for the woman to drink. After 2 h, a fingerstick capillary glucose test will be performed. GDM will be diagnosed if the value is > 140 mg/dL.
At the PHC, physicians will be issued a tablet computer with a version of the SMARThealth Pregnancy app designed to support guideline-based clinical decision-making in primary care setting. The main functions of the physician app are to generate recommendations for managing anaemia, hypertension and diabetes based on current Indian guidelines for pregnant and non-pregnant adults, as relevant to each woman.
-
-
(3)
The SMARThealth Pregnancy dashboard
The SMARThealth Pregnancy study dashboard has been designed to provide real-time analytics and report generation that allows service oversight, including tracking of specified participant cohorts.
-
(4)
Supply chain and logistics support
Through the SMARThealth Pregnancy app, we will monitor instances of stock-outs of iron folic acid tablets, iron sucrose, disposables for point-of-care testing, glucose for GDM screening and antihypertensive tablets. This information will be fed back to PHCs and district officials.
-
(5)
Stakeholder engagement
Community stakeholder engagement events will be held throughout the study to increase awareness of the programme and the importance of high-risk pregnancy conditions, and co-develop pathways to implementation and sustainability after completion of the trial. We will also regularly update and engage with district officials and PHC physicians to ensure the programme addresses local priority needs and is seen as being owned by the communities.

SHP intervention
Community stakeholder engagement events will be held throughout the study to increase awareness of the programme and the importance of high-risk pregnancy conditions, and co-develop pathways to implementation and sustainability after completion of the trial. We will also regularly update and engage with district officials and PHC physicians to ensure the programme addresses local priority needs and is seen as being owned by the communities.
Criteria for discontinuing or modifying allocated interventions {11b}
There will be no predefined criteria for stopping the allocated intervention. If new national clinical guidelines are published during the trial, this may justify modification of the content of the intervention. The decision for modification will be made by the Steering Committee. The overall form of the intervention will remain unchanged.
Participants may withdraw from the trial at any time. Withdrawal will not negatively affect their ability to access usual care during pregnancy and after birth. As this study focuses on improving the woman’s health, stillbirth, neonatal or infant death will not exclude her from participating after delivery.
Strategies to improve adherence to interventions {11c}
Study staff will support ASHAs by attending visits (with participant permission) and providing support for using the technology until the ASHA feels confident of performing assessments independently. Periodic quality control visits will be made to observe the intervention delivered by ASHAs and assessed by the study team. Every 6 months, the study team will conduct a 1-day refresher training session for the ASHAs.
Compliance with the intervention visits will be monitored centrally by tracking the visits recorded in the SMARThealth Pregnancy app. If trends of non-engagement across multiple participants from the same ASHA worker or PHC are noted, the study team will arrange a site visit.
For some women in India, it is traditional to move back to their mother’s village for the baby’s birth and stay there for up to 6 months. If the mother’s village is another intervention site, then visits will be transferred to the ASHA in the mother’s village, if it is part of the SMARThealth Pregnancy intervention. If the village is not an intervention site or is a EUC site, then visits will be replaced with phone calls, resuming face-to-face visits when the woman returns to her original village. Phone call follow-up was demonstrated to be acceptable and feasible for ASHA workers and women in the pilot study following COVID-19 disruptions.
Relevant concomitant care permitted or prohibited during the trial {11d}
This is a pragmatic trial; hence, women will be free to attend any other health providers (including private health providers, traditional medicine practitioners and care providers from other government facilities) to seek additional care. These health utilisation patterns will be captured as part of the process evaluation.
Provisions for post-trial care {30}
After the trial, any women with diabetes or hypertension will be referred to the local NCD services for ongoing management.
Theoretical underpinning for implementation
The Medical Research Council’s (MRC) framework for evaluating complex interventions [30] was used to inform the design of the overall study, intervention development, implementation and evaluation processes. Drawing on the findings of the pilot study and the experience of the collaborators, the study interventions were tailored and adapted for effective implementation and study delivery. The Reach Effectiveness Adoption Implementation Maintenance (RE-AIM) Framework, which has been widely applied to address challenges of combined effectiveness-implementation trials [31], has been used to guide the selection of implementation measures, alongside Proctor et al.’s [32] implementation outcomes typology.
The SHP2 trial applies a complex set of implementation strategies. The development of the implementation strategies was informed by the Expert Recommendations for Implementing Change (ERIC) project [33]. The ERIC compendium will also be used to identify implementation strategies that can help overcome arising issues that are affecting implementation during the trial. Due to the set-up of the trial, it was not feasible to randomise the implementation side of the trial (i.e. to allocate participants to different implementation strategies), and all participants in the intervention arm were allocated to the same strategies. We will examine the effectiveness of the implementation strategies used in the SHP2 trial in a separate process evaluation.
Outcomes {12}
Primary outcome
The primary outcome will be the difference in the proportion of participants with anaemia, defined as haemoglobin measured using a point-of-care test < 12 g/dL at 12 months after delivery between the intervention and EUC clusters. Anaemia has been chosen as the primary outcome as it is the most common condition affecting women during pregnancy and breastfeeding across India. Measurement using a point-of-care device was chosen as this has been recommended in national guidelines and is a common method for diagnosing anaemia in rural settings, thereby reflecting real-world practice. The measurement will be performed by a member of the study team.
Secondary outcomes
Secondary outcomes will be collected across four areas:
-
(1)
Implementation outcomes
-
(2)
Clinical endpoints
-
(3)
Clinical service delivery indicators
-
(4)
Women’s wellbeing and mental health
(1) Implementation outcomes
Using the RE-AIM framework [31], we will report the following implementation outcomes (Table 1):
-
Reach: The absolute number, proportion and representativeness of individuals that participated in the trial (both arms), and the intervention compared to the total population of pregnant women in the setting.
-
Effectiveness: The impact of the intervention on clinical outcomes (described above); quality of life, measured using the EQ5D tool [34]; and economic outcomes (health expenses incurred during pregnancy, at the time of birth and in the postnatal first year, and incremental economic costs).
-
Adoption: The absolute number, proportion and representativeness of health care workers who participated in the study compared to all health workers in the study locations. Qualitative work will explore reasons for non-participation or drop-out.
-
Implementation: Fidelity to the intervention protocol for the trial and for the intervention itself and any adaptations during the trial. This will be measured assessing data completeness, and through qualitative methods with health workers.
-
Maintenance: We will explore the system-level factors needed for institutionalisation of the intervention and level of intent to continue to programme in the study settings and more widely using key stakeholder interviews and focus group discussions, as well as policy discourse analysis around maternal health and digital health policy.
-
(2)
Clinical endpoints
-
Anaemia: Haemoglobin will be compared between intervention and EUC clusters as a continuous variable (measured in grams per decilitre on point-of-care test) and as categories of anaemia using the WHO thresholds used for non-pregnant adults: no anaemia > 12.0 g/dL; mild anaemia 11.0–11.9 g/dL; moderate anaemia 8.0–10.9 g/dL; severe anaemia < 8 g/dL
-
Diabetes: Between the intervention and EUC, we will compare venous blood glucose following a 2-h, 75-g oral glucose challenge at 1 year after birth in participants diagnosed with GDM during pregnancy. Blood glucose will be assessed as a continuous measure (in milligrams per decilitre) and as categories of glucose impairment: Normal: < 140 mg/dl, impaired glucose tolerance (IGT): 140–199mg/dl, Diabetes: ≥ 200 mg/dl
-
Hypertension: Blood pressure (BP) will be compared between women who developed HDP. BP will be assessed as a continuous measure (systolic, diastolic and mean arterial pressure) in millimetres of mercury (mmHg) using a validated sphygmomanometer and as a binary category of hypertension (yes/no) defined as BP > 140/90 mmHg
-
-
(3)
Clinical service delivery indicators
-
Quality-of-care score: We will measure differences between the intervention and EUC clusters using a quality-of-care score we have developed specifically for this study. The score comprises an equally weighted composite of clinical outcomes (anaemia, diabetes, hypertension) with measures of access and health professional visit frequency during pregnancy and in the first 12 months after delivery.
-
Follow-up after anaemia, GDM or HDP condition: We will compare how many women with anaemia undergo a haemogloblin test; how many women after HDP or GDM undergo BP measurement, and how many women after GDM undergo a glucose assessment (fasting or random capillary glucose, OGTT or HbA1c) within 6 weeks and 6 months and 12 months after birth.
-
-
(4)
Women’s wellbeing, mental health, lactation practice
-
Wellbeing: The impact of the intervention on women’s general wellbeing, mental health. We will use locally validated mental health and wellbeing tools (EQ-5D-3L, PHQ9 and GAD7 [35]) to compare changes between baseline and at 12 months.
-
Lactation: is one of the few interventions for which evidence supports a positive impact on maternal and newborn long-term health following a high-risk pregnancy, specifically one affected by GDM [36]. We will assess whether the additional support provided through the intervention can improve lactation practice using WHO Infant and Young Child Feeding indicators [37].
-
Participant timeline {13}
PHCs will be recruited and randomised before individual participant recruitment commences. After randomisation, ASHAs in the intervention clusters will attend the education programme to receive training on the clinical conditions, use of the point-of-care devices and SMARThealth Pregnancy app. Individual participants will be approached by their ASHA worker to participate after 12 weeks’ gestation. If they consent to participate in the study, a study team member will complete a baseline assessment, which includes questionnaires, blood pressure and haemoglobin measurement. A study team member will perform a mid-point assessment between 9 and 12 weeks after birth to capture events and healthcare utilisation including out-of-pocket costs around the time of birth. A study team member will conduct the final study visit 9–12 months after delivery, which will involve completing questionnaires, and measuring haemoglobin and blood pressure (Table 2).
Sample size {14}
The sample size was calculated using anaemia as the primary outcome. Three aspects were considered in determining the sample size:
-
(1)
Intracluster correlation
-
(2)
Cluster size and number
-
(3)
Variability in the primary outcome and estimated reduction that could be achieved by the intervention
These risks were balanced with operational considerations
Intracluster correlation
The nature of the intervention as a health system intervention precludes individual randomisation. Estimating intracluster correlation coefficients (ICCs) was based on the SMARThealth Pregnancy pilot study extrapolations [38]. Across the four sites in the pilot study, the ICC for anaemia was 0.019; thus, an ICC of 0.02 has been assumed for the study.
Cluster size and number
Clusters (i.e. PHCs) of various population sizes and birth numbers will be enrolled in the study. For operational reasons, a maximum cluster number of 30 is proposed, with two villages per cluster selected. Each village has one affiliated ASHA; thus, a minimum of 120 ASHAs will be needed.
Variability in the primary outcome and estimated effect size
The pilot study’s prevalence of anaemia during pregnancy was higher than the reported national levels, likely due to selection differences and differences in testing techniques (venous sample vs POC device used in the pilot study).
The prevalence of anaemia is slightly higher in non-pregnant than pregnant women (national estimate for India 53 and 50% respectively, NFHS 5 [39]). The national target for anaemia reduction is three percentage points per year as part of the Anaemia Mukt Bharat programme [18].
We estimate that our intervention will improve anaemia rates in these populations by 9% over 3 years (i.e. 53 to 44%). Determining a minimally clinically important threshold for anaemia is challenging. We selected the target decrease in anaemia as it was in keeping with the AMB programme [18], and to be an achievable and programmatically meaningful target with optimal support for anaemia prevention. Whilst anaemia is anticipated to decrease in the population because of the ongoing government programmes (including AMB), evidence from our pilot study is that compliance and implementation issues exist on the ground. Our intervention was designed to enhance delivery of the government programme and reaching of national anaemia reduction targets.
Given 30 clusters and an ICC of 0.02, 86 women per cluster are needed to detect a 9% absolute reduction (53 to 44%) with 80% power (a total sample size of 2580). Allowing for losses to follow-up of 20%, a sample size of 108 women per cluster is needed (a total sample size of 3240).
Recruitment {15}
Each district will produce a list of all PHCs, with population size, number of ASHAs and number of births in the previous 12 months. After obtaining relevant permissions from the District Medical and Health Officer, the head of each PHC deemed eligible will be approached. Once they have consented to participate, the PHC will be randomised to either intervention or enhanced usual care.
Individual women will be identified by ASHAs affiliated with each PHC and by review of PHC records for women registered in the area for antenatal care. Women will be approached by the ASHA and given information about the study, then asked whether they agree to participate.
Assignment of interventions: allocation
Sequence generation {16a}
Before randomisation, each PHC will be paired with another similar PHC from the same state and with similar characteristics. The pairing will be based on:
-
State (Telangana or Haryana)
-
Distance in km to the district headquarters
-
PHC total population size
-
Local advice on population similarity (socio-economic, demographic, etc.).
Within each PHC pair, one PHC will be randomly allocated to intervention using a computer-generated randomisation programme. The other PHC will then be designated as the control. To ensure a 1:1 balance (the same number of PHCs allocated to intervention and to control) within each state, we will select an even number of PHCs in each state. Sixteen PHCs will be enrolled from Telangana and 14 from Haryana.
Concealment mechanism {16b}
PHCs will be identified, and permission given to participate before allocation. After the central allocation of all clusters has been performed, each PHC will be notified of their allocation by the project manager. Given the pragmatic nature of this care-delivery trial, individual participants can only be recruited after allocation, and there will be no concealment before recruitment.
Implementation {16c}
The allocation sequence will be generated by the study statistician (LB). Clusters will be enrolled before revealing the allocation. PHC physicians will be informed of their allocation by the Project Manager. Given the cluster design, participants will automatically be allocated to the group according to their PHC.
Assignment of interventions: blinding
Who will be blinded {17a}
Owing to the pragmatic nature of the trial, participants, care providers and outcome assessors will not be blinded to the allocation group. Data analysts, the Principal Investigator and the Steering Committee will remain blinded until the end of the study and database lock when the primary analysis will be performed.
Procedure for unblinding if needed {17b}
All members of the clinical care team will remain unblinded throughout the study. As the statistician, Principal Investigator and Steering Committee are not directly involved in patient care in this study; there are no foreseeable reasons why the study would need to be unblinded.
Data collection and management
Plans for assessment and collection of outcomes {18a}
Independent, GCP-certified, trained, study team members will be involved in data collection at each stage. Data collection will occur on three occasions at both the intervention and control sites: baseline (after 12 weeks’ gestation), 9–12 weeks and then 9–12 months post-birth. All data will be captured on tablet computers using the SMARTHhealth app adapted for the clinical research forms (CRFs). Data will be de-identified, saved and stored on a secure server at the George Institute of Global Health, India. Strategies to ensure data quality include using published, validated questionnaires and the CRFs developed and user tested in the SMART Health Pregnancy pilot study. The field team supervisor will check approximately 10% of all CRFs for accuracy against source data or by direct interview with participants.
Plans to promote participant retention and complete follow-up {18b}
Community education events will be held at intervention and control sites to promote the study and awareness of women’s health, anaemia, cardiovascular disease and diabetes. Refreshments and transportation costs will be provided to women at the baseline and annual study assessments.
ASHAs and PCPs will receive a small payment per participant to compensate for the time spent on the study. For women who move away from the study sites during their follow-up period, information on pregnancy complications and outcomes will be collected by phone where possible.
Data management {19}
Data management will be performed in line with the study Data Management Plan. All baseline and outcome data will be entered digitally into the SMARThealth Pregnancy researcher platform. Access to data will be restricted to delegated study staff only and will require multifactor identification with a digital log kept of all logins. The trial manager and data managers will be primarily responsible for data quality. The trial manager will work with the project managers at both sites to ensure data quality, including completeness of data, data validation, data query management and training of database users. Monthly audits will be conducted to ensure the completion and quality of data captured.
Confidentiality {27}
Throughout the study, all efforts will be made to maintain participant confidentiality. Each subject enrolled in the project will have a unique identifier (ID) of six digits. The first digits denote the study state (00 = Telangana, 01 = Haryana). The final four digits will denote the participant’s unique number, e.g. 00–0001 will be in Telangana and will be the first participant screened for both sites; 01–0002 will be in Haryana and will be the second participant screened for both sites. Participants who are screened but do not give consent or withdraw from the study will also receive a study ID to calculate intervention reach. Tablet computers used for data collection as part of the intervention and for the study assessments will be password protected with multifactor authentication. They will be used only by authorised users and stored in a secure location when not in use. Local data stored on mobile devices will be deleted daily following online database synchronisation. Participant-identifiable data will be collected in a separate database to the research data set and linked using a unique identifier (see above for details). This information will be held only for the study duration to enable follow-up by the study team.
Plans for collection, laboratory evaluation and storage of biological specimens for genetic or molecular analysis in this trial/future use {33}
No biological samples will be collected or stored. Haemoglobin will be estimated using validated point-of-care devices. For the small number of women who develop GDM and require endline HbA1c, private local laboratories will process the samples with no sample storage.
Statistical methods
Statistical methods for primary and secondary outcomes {20a}
The primary analysis will be by intention to treat conducted at the participant level, using either random-effects models or generalised estimating equations adjusted for PHC clustering. We used a matched-pair design to balance cluster characteristics prior to randomisation, as recommended for community-based trials with a limited number of clusters [40]. Given the relatively small number of clusters, the analysis will not adjust for matching since this has been shown to provide unbiased estimates of the randomised intervention effect [41] and provide similar power to a matched analysis [40]. An unmatched analysis has the advantage that it can easily be conducted using participant-level data whilst allowing estimation of the intra-class correlation coefficient.
The final analysis is anticipated to occur 36 months after recruitment of the first participant. For the groups to be declared significantly different, the P value for the primary outcome must be ≤ 0.05. We will report the point estimate and confidence interval and present an assessment of the clinical relevance of our findings.
Baseline data will be presented for both groups. Continuous variables will be assessed for distribution, with appropriate statistical transformation (e.g. log or cubic splines) to correct for non-normality. Subgroup analyses will be performed by study location (Telangana or Haryana) to explore the effects of contextual factors on outcomes of interest. A detailed statistical analysis plan will be developed and publicly available before unblinding.
Interim analyses {21b}
No interim analysis is planned as (i) part of the objective of the study is to determine if the intervention is sustainable over 12 months, and (ii) we are interested in the longer-term effects of the intervention on anaemia, blood pressure and blood glucose and early changes detectable between the groups may not be sustained.
Methods for additional analyses (e.g. subgroup analyses) {20b}
Subgroup analyses will be performed for women with GDM and HDP separately and with both conditions. The analysis of secondary outcomes will be performed on all women reaching the study endpoint. A structured observation of health worker performance will be performed on a subsample of 200 women from 10 intervention and control PHC facilities to assess fidelity to the study intervention.
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing data {20c}
The primary analysis population will include all participants with endline clinical assessment and haemoglobin measurement recorded. An intention to treat population will be defined as those participants who received at least one assessment during pregnancy and one after birth who also have a complete endline assessment. In case of substantial missing primary outcome data, sensitivity analyses based on multiple imputations will be performed.
Plans to give access to the full protocol, participant-level data and statistical code {31c}
The full protocol, participant-level data and statistical code will be made available upon publication of the primary analysis as supplementary material. These will also be deposited in a data repository for future use.
Oversight and monitoring
Composition of the coordinating centre and trial steering committee {5d}
The coordinating centre will be in Hyderabad at the George Institute for Global Health. The Steering Committee comprises Indian and international experts in clinical trials, maternal health, health systems, implementation science and digital health. The trial Steering Committee will meet virtually or in person every 6 months to review trial progress, and be informed of severe adverse events, quality monitoring updates, modifications to the trial protocol, requests for data from other groups and plans for publications and dissemination.
The trial operations group will meet fortnightly (or more often if required) throughout the trial to ensure smooth day-to-day running and recruitment progress and provide organizational support to the field teams. The operations group comprises the project manager, the research staff directly involved and the Principal Investigators from the UK and India. The project manager will meet virtually or in person with each site manager weekly. The site managers have oversight responsibility for the field supervisors in their state.
Composition of the data monitoring committee, its role and reporting structure {21a}
Given the pragmatic nature of this study and that there will not be an interim analysis, there will not be a data monitoring committee.
Adverse event reporting and harms {22}
As the intervention aims to support the delivery of guideline-based antenatal and postnatal care already recommended in this setting, adverse events related to the intervention are not anticipated. That said, there may be unintended adverse effects from improved diagnosis of high-risk conditions (e.g. stigma), which could have physical or emotional consequences. All efforts will be taken by the study team to minimise potential harm. The project manager and field supervisors will identify and report any adverse events during the trial and promptly escalate these to the Principal Investigator (JEH). JEH will have the final say on whether such events are categorised as an adverse event (AE) or severe adverse event (SAE). She will be responsible for reporting any SAEs to the study sponsor and ensuring any actions are taken in the mandated timeframe. At the Steering Committee meetings, all adverse events will be reviewed. AEs and SAEs will be transparently reported in the primary publication, the final report to the REC and the final report to the study sponsor (The George Institute for Global Health, India).
Frequency and plans for auditing trial conduct {23}
Trial conduct will be audited by the Data Management team at the George Institute at the start of recruitment and every 6 months, independently of the operational team. We will report annually to the study sponsor.
Plans for communicating important protocol amendments to relevant parties (e.g. trial participants, ethical committees) {25}
Any protocol modifications will be reported to the George Institute India and Oxtrec ethics committees for minor or major amendment approval. Once approved, changes will be notified by the project manager to the site managers and field teams, who will inform the relevant health workers and participants. The trial register will be updated with any protocol amendments.
Dissemination plans {31a}
The trial results will be communicated to the scientific and clinical communities through journal publications, and presentations at international and national conferences. Results will be communicated to local collaborators through written information and education sessions at the study end. Results will be shared with communities and women through local media and events.
Discussion
This study will test the effectiveness and implementation of a community-based high-risk pregnancy screening and referral programme up to 1 year after birth. In rural India, as in many settings, women must attend antenatal appointments in a clinic or hospital to detect high-risk conditions. We hope to demonstrate that screening by community health workers in the woman’s village or home can be a practical approach to improving the detection of women with high-risk conditions, timely referral and evidence-based management. Uniquely, our intervention considers the impact of conditions detected during pregnancy on women’s longer-term health, with repeated assessments during the first year after birth. We hope to demonstrate that this is a practical approach to implement effective interventions for reducing the endemic problem of anaemia amongst women in India and engaging women who develop GDM or HDP with postnatal screening for diabetes and hypertension. In particular, the mixed quantitative and qualitative data collection in this hybrid effectiveness implementation study will help us analyse implementation outcomes and identify implementation strategies which are context-sensitive and can address implementation barriers of the programme.
The SMARThealth Pregnancy intervention aims to support government efforts to reduce anaemia rates through improved understanding by ASHAs and capacity to perform point-of-care haemoglobin testing. This is in keeping with AMB recommendations.
Our study has several strengths. The pragmatic nature of the trial will generate evidence of the effectiveness of strategies to improve the care of women at high risk of postnatal anaemia, type 2 diabetes or hypertension. Community follow-up after a pregnancy affected by these conditions is not routinely performed in rural India, which misses critical opportunities for prevention.
Our study will be conducted in two states with very different socio-cultural environments, which will improve the generalisability of our findings to other states in India. We are conducting this study as a hybrid effectiveness-implementation design. A separate protocol will be published on the planned implementation outcomes that we will capture through a detailed process evaluation. The analysis of implementation outcomes and strategies will inform the iterative development and steps needed for the eventual scale-up and adoption of the intervention more widely; alternatively, it will explain the factors that contributed to a null trial. As part of the process analysis, we will continue refining our logic model and assumptions regarding how the intervention works and the barriers to implementation, as well as the economic benefits and implications.
We acknowledge our study design has limitations. Owing to funding constraints, we can only involve community health workers from two villages affiliated with each PHC. Thus, the intervention will not be at the population level which might affect primary care physician engagement. We will explore this in detail in our process analysis. The nature of the trial design means there could be benefits above usual care in the control clusters. Participants in both study arms will have their haemoglobin tested and their blood pressure measured at the study visits. Ideally, after a high-risk pregnancy condition, follow-up would extend for life, which is clearly impractical and prohibitively expensive in the context of this study. We believe demonstrating engagement beyond 6 weeks after birth will be a strength. The nature of the complex intervention means that blinding of participants is not practical or feasible, increasing the likelihood of information bias. To minimise this, we will ensure that the trial statistician and investigator group remain blinded to allocation until the primary analysis has been performed.
Given the longitudinal nature of recruitment, not all potential participants will be pregnant before the study starts; therefore, randomisation must occur before participants are recruited. This could bias recruitment, as community health workers will know their allocation group when approaching pregnant women. However, in our pilot study, this was not the case: we achieved equal recruitment and retention rates across both groups.
In summary, we present a study protocol for a pragmatic, cluster randomised hybrid effectiveness-implementation trial to test the SMARThealth Pregnancy complex intervention in rural India. It is hoped that the trial will demonstrate the effectiveness of the intervention and implementation in reducing anaemia and improving, during and after pregnancy, the detection and follow-up of other high-risk pregnancy conditions.
Trial status
The current protocol number is version 2.0, November 2022. The SMARThealth Pregnancy trial is currently recruiting. Recruitment commenced on 1 July 2022, and the trial is anticipated to run until 30 June 2024.
Availability of data and materials {29}
The Principal Investigator, project manager, database manager and study statistician will have access to the final study dataset. The study team maintain the right to exclusive use of the data until the primary analysis is published or for 24 months after the end of the trial (whichever comes sooner). After this time, the data will be made publicly accessible as per the University of Oxford Open Access policy.
Abbreviations
- AE:
-
Adverse event
- AMB:
-
Anemia Mukt Bharat: the Government of India programme for anaemia
- ANM:
-
Auxiliary nurse midwife
- ASHA:
-
Accredited Social Health Activist
- CRF:
-
Case record form
- EQ5D: :
-
Euro-Quol Quality of Life measure - five domains
- ERIC:
-
Expert recommendations for implementing change
- EUC:
-
Enhanced usual care
- FGD:
-
Focus group discussion
- FIGO:
-
International Federation of Gynaecology and Obstetrics
- GAD7:
-
Generalised Anxiety and depression scale
- GDM:
-
Gestational diabetes mellitus
- GCP:
-
Good Clinical Practice for research
- HbA1c:
-
Glycated haemoglobin
- HDP:
-
Hypertensive disorder of pregnancy as per ISSHP 2018 guidelines
- ICC:
-
Intracluster correlation coefficient
- ISSHP:
-
International Society for the Study of Hypertension in Pregnancy
- LMUP:
-
London Measure of Unplanned Pregnancy
- MRC:
-
Medical Research Council, UK
- NCD:
-
Non-communicable diseases
- NFHS:
-
National Family Health Survey of India
- OGTT:
-
Oral glucose tolerance test
- PHC:
-
Primary Health Centre
- PHQ9:
-
Patient Health Questionnaire
- SAE:
-
Severe adverse event
- SHP:
-
SMARThealth Pregnancy
- UKRI:
-
United Kingdom Research and Innovation
References
Trends in maternal mortality 2000 to 2020: estimates by WHO, UNICEF, UNFPA, World Bank Group and UNDESA/Population Division. Geneva: World Health Organization; 2023. Licence: CC BY-NC-SA 3.0 IGO.
Global, regional, and national under-5 mortality, adult mortality, age-specific mortality, and life expectancy, 1970–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1084–150.
Mehran R, Vogel B, Ortega R, Cooney R, Horton R. The lancet commission on women and cardiovascular disease: time for a shift in women’s health. Lancet. 2019;393(10175):967–8.
Hirst JE, Nagraj S, Henry A, Mackillop L, Norton R, Kennedy S. The obstetrician’s role in preventing cardiometabolic disease. Obstet Gynaecol. 2019;21(4):247–54.
Wu P, Haththotuwa R, Kwok CS, Babu A, Kotronias RA, Rushton C, et al. Preeclampsia and future cardiovascular health: a systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes. 2017;10(2):e003497.
Behrens I, Basit S, Melbye M, Lykke JA, Wohlfahrt J, Bundgaard H, et al. Risk of post-pregnancy hypertension in women with a history of hypertensive disorders of pregnancy: nationwide cohort study. BMJ. 2017;358:j3078.
International Diabetes Federation. IDF Diabetes Atlas. https://diabetesatlas.org. (Accessed 15th May 2023).
Bhavadharini B, Anjana RM, Mahalakshmi MM, Maheswari K, Kayal A, Unnikrishnan R, et al. Glucose tolerance status of Asian Indian women with gestational diabetes at 6weeks to 1year postpartum (WINGS-7). Diabetes Res Clin Pract. 2016;117:22–7.
Kramer CK, Campbell S, Retnakaran R. Gestational diabetes and the risk of cardiovascular disease in women: a systematic review and meta-analysis. Diabetologia. 2019;62(6):905–14.
McAuliffe FM. Impact of pregnancy on long-term health: Advances in postpregnancy care-an opportunity to improve long-term maternal health. Int J Gynaecol Obstet. 2023;160(Suppl 1):4–6.
Brown HL, Warner JJ, Gianos E, Gulati M, Hill AJ, Hollier LM, et al. Promoting risk identification and reduction of cardiovascular disease in women through collaboration with obstetricians and gynecologists: a presidential advisory from the American heart association and the American College of obstetricians and gynecologists. Circulation. 2018;137(24):e843–52.
National Institute of Clinical Excellence (NICE). Hypertension in pregnancy: diagnosis and management. 2019
National Institute of Clinical Excellence (NICE). Diabetes in pregnancy: Management of diabetes and its complications from preconception to the postnatal period. 2015.
Nagraj S, Kennedy SH, Norton R, Jha V, Praveen D, Hinton L, et al. Cardiometabolic risk factors in pregnancy and implications for long-term health: identifying the research priorities for low-resource settings. Front Cardiovasc Med. 2020;7:40.
Aroda VR, Christophi CA, Edelstein SL, Zhang P, Herman WH, Barrett-Connor E, et al. The effect of lifestyle intervention and metformin on preventing or delaying diabetes among women with and without gestational diabetes: the diabetes prevention program outcomes study 10-year follow-up. J Clin Endocrinol Metab. 2015;100(4):1646–53.
Pathirana MM, Ali A, Lassi ZS, Arstall MA, Roberts CT, Andraweera PH. Protective influence of breastfeeding on cardiovascular risk factors in women with previous gestational diabetes mellitus and their children: a systematic review and meta-analysis. J Hum Lact. 2022;38(3):501–12.
Poon LC, Nguyen-Hoang L, Smith GN, Bergman L, O’Brien P, Hod M, et al. Hypertensive disorders of pregnancy and long-term cardiovascular health: FIGO best practice advice. Int J Gynaecol Obstet. 2023;160(Suppl 1):22–34.
Government of India. Anemia Mukt Bharat. https://anemiamuktbharat.info. (Accessed 15th May 2023).
Rahman MM, Abe SK, Rahman MS, Kanda M, Narita S, Bilano V, et al. Maternal anemia and risk of adverse birth and health outcomes in low- and middle-income countries: systematic review and meta-analysis. Am J Clin Nutr. 2016;103(2):495–504.
Praveen D, Patel A, McMahon S, Prabhakaran D, Clifford GD, Maulik PK, et al. A multifaceted strategy using mobile technology to assist rural primary healthcare doctors and frontline health workers in cardiovascular disease risk management: protocol for the SMARTHealth India cluster randomised controlled trial. Implement Sci. 2013;8:137.
Peiris D, Praveen D, Mogulluru K, Ameer MA, Raghu A, Li Q, et al. SMARThealth India: A stepped-wedge, cluster randomised controlled trial of a community health worker managed mobile health intervention for people assessed at high cardiovascular disease risk in rural India. PLoS ONE. 2019;14(3):e0213708.
Praveen D, Patel A, Raghu A, Clifford GD, Maulik PK, Mohammad Abdul A, et al. SMARTHealth India: development and field evaluation of a mobile clinical decision support system for cardiovascular diseases in Rural India. JMIR Mhealth Uhealth. 2014;2(4):e54.
Patel A, Praveen D, Maharani A, Oceandy D, Pilard Q, Kohli MPS, et al. Association of multifaceted mobile technology-enabled primary care intervention with cardiovascular disease risk management in Rural Indonesia. JAMA Cardiol. 2019;4(10):978–86.
Mukherjee A, Daniel M, Kallakuri S, Kaur A, Devarapalli S, Raman U, et al. Protocol for process evaluation of SMART Mental Health cluster randomised control trial: an intervention for management of common mental disorders in India. BMJ Open. 2022;12(6):e058669.
Daniel M, Maulik PK, Kallakuri S, Kaur A, Devarapalli S, Mukherjee A, et al. An integrated community and primary healthcare worker intervention to reduce stigma and improve management of common mental disorders in rural India: protocol for the SMART mental health programme. Trials. 2021;22(1):179.
Nagraj S, Kennedy SH, Jha V, Norton R, Hinton L, Billot L, et al. SMARThealth pregnancy: feasibility and acceptability of a complex intervention for high-risk pregnant women in Rural India: protocol for a pilot cluster randomised controlled trial. Front Glob Womens Health. 2021;2:620759.
Hemming K, Eldridge S, Forbes G, Weijer C, Taljaard M. How to design efficient cluster randomised trials. BMJ. 2017;358:j3064.
Eldridge S, Kerry S. A Practical guide to Cluster Randomised Trials in Health Services Research. London: Wiley; 2012.
WMaternal Health Division Ministry of Health and Family Welfare, Government of India. National Guidelines for Diagnosis & Managment of Gestational Diabetes Mellitus 2014. https://nhm.gov.in/images/pdf/programmes/maternal-health/guidelines/National_Guidelines_for_Diagnosis_&_Management_of_Gestational_Diabetes_Mellitus.pdf. (Accessed 15th may 2023).
Skivington K, Matthews L, Simpson SA, Craig P, Baird J, Blazeby JM, et al. A new framework for developing and evaluating complex interventions: update of medical research council guidance. BMJ. 2021;374:n2061.
Glasgow RE, Harden SM, Gaglio B, Rabin B, Smith ML, Porter GC, et al. RE-AIM Planning and Evaluation Framework: adapting to new science and practice with a 20-year review. Front Public Health. 2019;7:64.
Proctor E, Silmere H, Raghavan R, Hovmand P, Aarons G, Bunger A, et al. Outcomes for implementation research: conceptual distinctions, measurement challenges, and research agenda. Adm Policy Ment Health. 2011;38(2):65–76.
Powell BJ, Waltz TJ, Chinman MJ, Damschroder LJ, Smith JL, Matthieu MM, et al. A refined compilation of implementation strategies: results from the Expert Recommendations for Implementing Change (ERIC) project. Implement Sci. 2015;10:21.
Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727–36.
De Man J, Absetz P, Sathish T, Desloge A, Haregu T, Oldenburg B, et al. Are the PHQ-9 and GAD-7 suitable for use in India? Psychomet Anal Front Psychol. 2021;12:676398.
Tanase-Nakao K, Arata N, Kawasaki M, Yasuhi I, Sone H, Mori R, et al. Potential protective effect of lactation against incidence of type 2 diabetes mellitus in women with previous gestational diabetes mellitus: a systematic review and meta-analysis. Diabetes Metab Res Rev. 2017;33(4):e2875.
World Health Organization. Indicators for assessing infant and young child feeding practices: definitions and measurement methods. 2021. https://apps.who.int/iris/handle/10665/340706. (Accessed 15th May 2023).
Nagraj S, Kennedy S, Jha V, Norton R, Hinton L, Billot L, et al. SMARThealth Pregnancy: Pilot cluster randomized controlled trial of a mobile clinical decision support system for high-risk pregnant women in rural India. JMIR Format Res. 2023. https://doi.org/10.2196/44362.
IIPS. National Family Health Survey (NFHS-5), India, 2019–21. Mizoram, Mumbai: International Institute for Population Sciences; 2021.
Diehr P, Martin DC, Koepsell T, Cheadle A. Breaking the matches in a paired t-test for community interventions when the number of pairs is small. Stat Med. 1995;14(13):1491–504.
Chondros P, Ukoumunne OC, Gunn JM, Carlin JB. When should matching be used in the design of cluster randomized trials? Stat Med. 2021;40(26):5765–78.
Acknowledgements
We acknowledge the support of the health workers, women and district medical officers in the study districts and Dr Shobhana Nagraj, who led the pilot study and initial intervention development.
Funding
This trial is funded by URKI as part of a personal fellowship awarded to Prof Jane Hirst (UKRI Future Leaders Fellowship, Grant Ref: MR/T040750/1). The funder has no role in the design of the study, collection, analysis, interpretation or writing of this manuscript.
Author information
Authors and Affiliations
Contributions
JEH is the Chief Investigator; she conceived the study, led the proposal and protocol development. DP is the co-principal investigator in India who jointly led protocol development. LB is the trial statistician. NV is the lead for the implementation science outcomes. VA, AP, DP, VJ, EM, RN, ER, ST, SK, and AS contributed to study design and development of the proposal. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate {24}
This trial first received ethical approval from OXTREC (reference 27–21) on 24 November 2021, and the George Institute for Global Health ethics committee (reference 22/2021) on 1 December 2021, with version 2.0 approved in November 2022. Version 2.0 included details on the process analysis and the addition of a mid-line study visit in both study arms to facilitate accurate collection of birth outcomes. Written, informed consent to participate will be obtained from all participants: Primary Health Centres, health workers and individual women.
Consent for publication {32}
Not applicable—no identifying images or other personal or clinical details of participants are presented here or will be presented in reports of the trial results. Informed consent materials are attached as supplementary materials.
Competing interests {28}
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. PARTICIPANT CONSENT FORM.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hirst, J.E., Votruba, N., Billot, L. et al. A community-based intervention to improve screening, referral and follow-up of non-communicable diseases and anaemia amongst pregnant and postpartum women in rural India: study protocol for a cluster randomised trial. Trials 24, 510 (2023). https://doi.org/10.1186/s13063-023-07510-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-023-07510-x