
Abstract
Background
To date, targeted temperature management (TTM) is the only neuroprotective intervention after resuscitation from cardiac arrest that is recommended by guidelines. The evidence on the effects of TTM is unclear.
Methods/design
The Targeted Hypothermia Versus Targeted Normothermia After Out-of-hospital Cardiac Arrest (TTM2) trial is an international, multicentre, parallel group, investigator-initiated, randomised, superiority trial in which TTM with a target temperature of 33 °C after cardiac arrest will be compared with a strategy to maintain normothermia and active treatment of fever (≥ 37.8 °C). Prognosticators, outcome assessors, the steering group, the trial coordinating team, and trial statisticians will be blinded to treatment allocation. The primary outcome will be all-cause mortality at 180 days after randomisation. We estimate a 55% mortality in the targeted normothermia group. To detect an absolute risk reduction of 7.5% with an alpha of 0.05 and 90% power, 1900 participants will be enrolled. The secondary neurological outcome will be poor functional outcome (modified Rankin scale 4–6) at 180 days after cardiac arrest. In this paper, a detailed statistical analysis plan is presented, including a comprehensive description of the statistical analyses, handling of missing data, and assessments of underlying statistical assumptions. Final analyses will be conducted independently by two qualified statisticians following the present plan.
Discussion
This SAP, which was prepared before completion of enrolment, should increase the validity of the TTM trial by mitigation of analysis-bias.
Similar content being viewed by others

Background
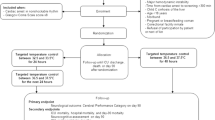
The Targeted Hypothermia Versus Targeted Normothermia After Out-of-hospital Cardiac Arrest (TTM2 trial) is a continuation of the collaboration that resulted in the Target Temperature Management after out-of-hospital cardiac arrest trial (TTM trial) [1].
The TTM trial (NCT01020916) [1] was a multicentre, multinational, outcome assessor-blinded, parallel group, randomised clinical trial comparing two target temperature regimens of 33 °C and 36 °C in unconscious patients who had sustained return of spontaneous circulation after out-of-hospital cardiac arrest [1]. The trial did not demonstrate any significant difference in mortality rates or intact neurological survival between the two groups. Recently, the Therapeutic Hypothermia after Cardiac Arrest in Nonshockable Rhythm (HYPERION) trial was published [2]. This trial showed that among patients with coma who had been resuscitated from cardiac arrest with nonshockable rhythm, moderate therapeutic hypothermia at 33 °C for 24 h compared with targeted normothermia led to a higher percentage of patients who survived with a favourable neurologic outcome at day 90 (P = 0.04) [2].
The TTM2 trial is an international, multicentre, parallel group, investigator-initiated, randomised, superiority trial in which TTM with a target temperature of 33 °C after out-of-hospital cardiac arrest of a presumed cardiac or unknown cause will be compared with early treatment of fever (≥ 37.8 °C).
This publication will describe the statistical analyses of the primary and secondary outcomes in the TTM2 trial.
Methods
The design of the TTM2 trial has been described in detail previously [3]. In short, the trial population will be adults (18 years of age or older) who experience a non-traumatic out-of-hospital cardiac arrest of a presumed cardiac or unknown cause with return of spontaneous circulation (ROSC). Patients will be eligible for enrolment if they meet all of the following inclusion criteria and none of the exclusion criteria.
Inclusion criteria
-
1.
Out-of-hospital cardiac arrest of a presumed cardiac or unknown cause
-
2.
Sustained ROSC—defined as 20 min with signs of circulation without the need for chest compressions
-
3.
Unconsciousness after sustained ROSC defined as FOUR score motor response < 4 and not able to obey verbal commands
-
4.
Eligible for intensive care without restrictions or limitations
-
5.
Screening for inclusion in the trial must be commenced no later than 180 min after ROSC
Exclusion criteria
-
1.
Unwitnessed cardiac arrest with an initial rhythm of asystole
-
2.
Temperature on admission < 30 °C
-
3.
On extracorporeal membrane oxygenation (ECMO) prior to ROSC
-
4.
Suspected pregnancy
-
5.
Intracranial bleeding
-
6.
Severe chronic obstructive pulmonary disorder with long-term home oxygen therapy
Co-enrolment with the TAME trial
The Targeted Therapeutic Mild Hypercapnia After Resuscitated Cardiac Arrest: A Phase III Multi-Centre Randomised Controlled Trial (TAME trial) (ACTRN12617000036314p) aims to determine whether targeted therapeutic mild hypercapnia improves neurological outcome at six months compared with standard care and targeted normocapnia. At certain sites, all TTM2 participants will also be enrolled in the TAME trial. We consider co-enrolment in TTM2 and TAME as an effective use of research resources. Adequate randomisation and a sample size as large as ours should lead to similar proportions of participants treated with targeted therapeutic mild hypercapnia in each of the TTM2 intervention groups. If there are no interactions between the TTM2 trial interventions and the TAME trial interventions, any beneficial or harmful effects of the TAME trial interventions will balance out.
An interaction between the TTM trial interventions and the TAME trial interventions is not likely. Theoretically, the TTM2 trial interventions are believed to have neuroprotective effects including reductions in metabolic rate and pathologic cell signalling, while the TAME trial interventions are believed to affect cerebral blood flow. Furthermore, we have studied the interaction between PaCO2 and temperature in the TTM trial and there was no statistically significant interaction (Pinteraction = 0.95) [4]. If we show significant interactions, this will be handled as described under the ‘Assessments of underlying statistical assumptions’ section.
Randomisation and blinding
Randomisation will be performed by an investigator in the emergency department, in the angiography unit, or in the intensive care unit via web-based application using permuted blocks with varying block sizes, stratified by site and co-enrolment in the TAME trial (no co-enrolment, TAME intervention arm 1, TAME intervention arm 2). Due to the nature of the intervention, the treating providers will not be blinded to the intervention. However, the outcome assessors, the prognosticators, the statisticians, the data managers, and the authors of the first version of the manuscript will be blinded to treatment allocation.
Trial interventions
The intervention period for both intervention groups will be 40 h and commence at the time of randomisation. Rapid cooling in the hypothermia group will be achieved by means of cold fluids and state-of-the-art cooling devices, i.e. intravascular/body-surface/nasal/oesophageal cooling (physical cooling). A feedback-controlled system will be used to maintain the target temperature. In the normothermia arm, the aim will be early treatment of fever (≥ 37.8 °C) using pharmacological measures and physical cooling when needed (up to 72 h). For participants who develop a temperature of 37.8 °C (trigger), a device will be used and set at 37.5 °C. All participants will be sedated, mechanically ventilated, and haemodynamically supported throughout the intervention period. Participants who are managed at 33 °C will begin rewarming 28 h after randomisation.
Participants who remain unconscious will be assessed according to a conservative protocol based on the European Resuscitation Council (ERC)’s recommendations for neurological prognostication after cardiac arrest [3].
The main results of the trial will be published following the 6-month follow-up, results from the long-term follow-up and the outcome assessment of neurocognitive function will be presented separately [5].
Outcomes
The outcomes were defined as primary and secondary [3]. The sample size was based on the primary outcome and our primary conclusions will be based on the results of the primary outcome. We ranked the outcomes in our outcome hierarchy according to clinical relevance and estimated the power of each outcome to ensure that we had sufficient power to confirm or reject the anticipated intervention effects [6].
Primary outcome
-
All-cause mortality (dichotomous outcome)
Secondary outcomes
-
Proportion of participants with a poor functional outcome (modified Rankin scale 4–6) (dichotomous outcome) [7], we will in a secondary analysis analyse the ordinal modified Rankin scale data (ordinal data)
-
Number of days alive after hospital discharge within 6 months after randomisation (count data)
-
Health-related quality of life using EQ5D-5 L (VAS) [8] (continuous outcome)
-
Time-to-death (survival data) for each participant from randomisation until 6 months after the last participant is randomised. If death has not occurred, participants will be censored at this point
Dichotomous and continuous outcomes will be assessed at 30 days, 6 months, and 24 months after randomisation. For primary and secondary analyses, only the 6 months time point will be used.
Sample size and power estimations
Based on the results of the previous TTM trial [1] and information in the International Cardiac Arrest Registry (INTCAR), we anticipate a mortality of 55% in the normothermia group [9]. Using an absolute risk reduction of 7.5% as anticipated intervention effect, an acceptable risk of type I error of 5%, and an acceptable risk of type II error of 10%, a total of 1862 (931 participants in each group) participants are required. This anticipated intervention effect corresponds to a relative risk reduction (RRR) of 13.6% and a number needed to treat (NNT) of 14 [10, 11]. Only 4/939 patients withdrew consent in TTM trial, and there were no missing data on mortality [1]. To allow for a possible loss to follow-up, we will recruit a total of 1900 participants.
We also estimated the statistical power of all secondary outcomes [6]. With an estimated sample size of 931 participants per group, the functional outcome measure (dichotomised mRS) has a power of 90% to detect a relative risk of 0.86 for a poor outcome (mRS 4–6) in 55% of cases in the control group. For the secondary outcome time-to-death, we estimate a power of > 90% based on the survival estimates mentioned above. We estimate a power of approximately 90% to detect a difference in 5 points on the EQ5D-5 L VAS-scale, based on a mean value of 70 in the control group and a standard deviation of 25 points [1, 3]. For the secondary outcome ‘days alive outside hospital’, we estimate a power of approximately 83%, based on simulations [3].
General analysis principles
All analyses will be conducted according to the intention-to-treat principle (ITT), i.e. all randomised participants will be included in the analysis. A per protocol analysis will be performed if the number of participants in whom temperature management is withheld due to palliative care, early death or other reasons during the first six hours after randomisation exceeds 5% of the total trial population.
We will both assess if the thresholds for statistical significance and clinical significance are crossed (Bayes factor calculations will be reported in supplementary material) [12]. Assessment of clinical significance will be based on the anticipated intervention effects used in the sample size/power estimations [12]. Our primary conclusion will be based on the primary outcome, so all tests of statistical significance (including subgroup analyses) will be two-sided with a type I error risk of 5% [12].
It is generally acknowledged that regression analyses ought to be adjusted for the stratification variables used in the randomisation [13,14,15]. The TTM2 trial uses two stratification variables in the randomisation, i.e. ‘site’ and ‘co-enrolment in the TAME trial’ (no co-enrolment, TAME intervention arm 1, TAME intervention arm 2). We will primarily adjust all regression analyses for ‘site’ and ‘co-enrolment in the TAME trial’ to balance prognostic baseline characteristics across TTM2 trial intervention groups. We will also assess whether there are significant interactions between TTM2 trial interventions and the stratification variables (see the ‘Assessments of underlying statistical assumptions’ sections).
We will also perform the following subgroup analyses: sex (male compared to female), first presenting cardiac rhythm (shockable compared to non-shockable), presence of shock on admission (no shock on admission compared to shock on admission), age (at or above the median compared to below the median), and duration of cardiac arrest (at or above the median compared to below the median). We will present the results in forest plots.
Statistical analyses
Analysis of dichotomous data
Dichotomised outcomes will be presented as proportions of participants in each group with the event, as well as risk ratios with 95% confidence intervals. Dichotomous outcomes will be analysed using mixed effects generalised linear models using a log link function with ‘site’ as a random intercept using an exchangeable covariance matrix, and co-enrolment will be included as a fixed effect.
Analysis of continuous data
Continuous outcomes will be presented as means and standard deviations for each group along with 95% confidence interval for the means of the groups and the mean differences between the groups. Continuous outcomes will be analysed using mixed effects linear regression with ‘site’ as a random intercept using an exchangeable covariance matrix, and co-enrolment will be included as fixed effect. We expect that a large proportion of the participants will die before assessment of quality of life. When assessing health-related quality of life, we will therefore in the primary analysis impute a ‘0’ for all participants who died or who are incapacitated and did not participate in the quality of life assessment. In a secondary analysis of quality of life, we will only include survivors at 6 months.
Analysis of count data
Count data will be presented as means, mean differences, and 95% confidence intervals or medians, interquartile ranges, and 95% confidence intervals (bootstrapping) depending on the observed distribution. Count data will be analysed by the van Elteren test stratified by ‘site’ [16].
Analysis of survival data
Survival data will be presented as median survival time, frequencies, and percentages per group as well as hazard ratios with 95% CIs. Survival data will be analysed using Cox regression adjusted for site and co-enrolment. We plan to present Kaplan-Maier curves.
Handling of missing data
All randomised participants will be included in the primary analysis of all outcomes except in the primary analysis of health-related quality of life (please see the ‘Analysis of continuous data’ section). We anticipate that the proportion of missing values on primary and secondary outcomes will be less than 5%. However, we will in a secondary analysis consider using multiple imputation and present best-worst and worst best case scenarios if it is not valid to ignore missing data [17]. Best-worst and worst-best case scenarios assess the potential range of impact of the missing data for the trial results [17]. In the ‘best-worst’ case scenario, it is assumed that all patients lost to follow-up in the hypothermia group have had a beneficial outcome (have survived, had no poor functional outcome, and so forth), and all those with missing outcomes in the control group have had a harmful outcome (have not survived, have had poor functional outcome, and so forth) [17]. Conversely, in the ‘worst- best’ case scenario, it is assumed that all patients who were lost to follow-up in the experimental group have had a harmful outcome and that all those lost to follow- up in the control group have had a beneficial outcome [17]. When continuous outcomes are used, a ‘beneficial outcome’ will be defined as the group mean plus two SDs of the group mean (fixed imputation), and a ‘harmful outcome’ will be defined as the group mean minus two SDs of the group mean (fixed imputation) [17].
Assessments of underlying statistical assumptions
We will systematically assess underlying statistical assumptions for all statistical analyses [18, 19]. For all regression analyses, both primary and secondary, we will test for major interactions between each covariate and the intervention variable. When assessing for major interactions, we will, in turn, include each possible first order interaction between included covariates and the intervention variable. For each combination, we will test if the interaction term is significant and assess the effect size. We will only consider that there is evidence of an interaction if the interaction is statistically significant after Bonferroni adjusted thresholds (0.05 divided by number of possible interactions (treatment variable interaction with ‘site’ and treatment variable interaction with ‘co-enrolment in the TAME trial’ = 0.025) and if the interaction shows a clinically important effect. If it is concluded that the interaction is significant, we will consider both presenting an analysis separately for each (e.g. for each site if there is significant interaction between the trial intervention and ‘site’) and an overall analysis including the interaction term in the model [18, 19].
Assessments of underlying statistical assumptions for dichotomous outcomes
We will assess if the deviance divided by the degrees of freedom is significantly larger than 1 to assess for relevant overdispersion. Overdispersion is the presence of greater variability (statistical dispersion) in a data set than would be expected based on a given statistical model, and this case considered using a maximum likelihood estimate of the dispersion parameter. To avoid analytical problems with either zero events or problems with all participants dying at a given site, we have only included sites planning to randomise a sufficient number of participants. However, we cannot exclude the risk that some sites might have problems with recruitment. We will, by checking if the number of participants is larger than 10 (rule of thumb) per site, consider pooling the data from small sites if the number of participants is too low [19].
Assessments of underlying statistical assumptions for linear regression
We will visually inspect quantile-quantile plots of the residuals [20, 21] to assess if the residuals are normally distributed and use residuals plotted against covariates and fitted values [20, 21] to assess for homogeneity of variances. If the plots show deviations from the model assumptions, we will consider transforming the outcome, e.g. using log transformation or square root and/or use robust standard errors [19,20,21].
Assessments of underlying statistical assumptions for Cox regression
We will visually inspect log-log plots stratified by treatment and adjusted for the effects of all covariates (continuous and categorical) [20, 22] to asses if the assumption of proportional hazards between the compared intervention groups is fulfilled. If the assumption of proportional hazards seems violated, we will consider using a non-parametric test (e.g. log rank test) or split the observation period into two (or more) separate observation periods [19].
Statistical reports
Blinded data on all outcomes will be analysed by two independent statisticians [19]. Two independent statistical reports will be sent to the chief principal investigator and will be shared with the steering group and author group, and if there are discrepancies between the two primary statistical reports, then possible reasons for that will be identified and the steering group will decide which is the most correct result. A final statistical report will be prepared, and all three statistical reports will be published as supplementary material [19].
Mock tables are presented in Mock Tables TTM2.
Discussion
The primary aim of this present publication is to minimise the risks of outcome reporting bias and erroneous data-driven results. We therefore present a pre-defined description of the statistical analysis plan for the TTM2 trial.
Strengths
Our methodology has several strengths as it is pre-defined and we have limited problems with multiplicity because we only assess one primary outcome and our conclusions will primarily be based on the results of the primary outcome [12]. Our chosen outcomes are all patient-centred. Our primary outcome, all-cause mortality, remains perhaps the most reliable and patient-centred outcome and we assess all-cause mortality as a dichotomous outcome at one time point, which simplifies both the statistical methodology and the clinical interpretability, i.e. it is intuitively easy to assess whether a shown difference (effect size) is clinically important when comparing two proportions at one time point. We will analyse data in accordance to the intention-to-treat principle and, if necessary, use multiple imputation and best-worst/worst-best case scenarios to assess the potential impact of the missing data on the results [17]. Furthermore, we plan to systematically assess whether underlying statistical assumptions are fulfilled for all statistical analyses.
Limitations
A potential limitation of the TTM2 trial are the potential heterogeneous intervention effects depending on the mode of cooling at different clinical sites, and the potential biased impact on the trial results if a large proportion of the randomised participants withdraw consent after regaining capacity. Another potential limitation is the planned co-enrolment with the TAME Trial; our results will be difficult to interpret if there are significant interactions between the TTM2 and TAME trial interventions. As mentioned (see the ‘Co-enrolment with the TAME trial’ section), we have studied the interaction between PaCO2 and temperature in the TTM trial and found no statistically significant interaction (Pinteraction = 0.95) [4], and if we show significant interactions, this will be handled (see the ‘Assessments of underlying statistical assumptions’ section). Co-enrolment with the TAME trial also made it possible to increase the planned sample size from 1200 to 1900 participants.
We only assess one primary outcome and our primary conclusions will be based on the result of the primary outcome, but we assess several secondary outcomes, exploratory outcomes, and subgroup analyses which increase the risks of type I errors. It is a limitation that we do not adjust our thresholds for significance according to the number of outcome comparisons. Furthermore, our anticipated intervention effects used in the sample size estimation and the power estimations for the secondary outcomes are not based on previous valid studies because we have not identified such studies. We have pragmatically chosen these anticipated intervention effects based on clinical judgement and previous trial results [1, 2]. This increased risk of type I errors and the uncertainty regarding the anticipated intervention effects need to be considered when interpreting our trial results.
Conclusion
We present a pre-defined description of the statistical analysis for the TTM2 trial. The risks of outcome reporting bias and erroneous data-driven results will be minimised if this statistical analysis plan is followed.
Availability of data and materials
All relevant data are available.
References
Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, Horn J, Hovdenes J, Kjaergaard J, Kuiper M, et al. Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med. 2013;369(23):2197–206.
Lascarrou JB, Merdji H, Le Gouge A, Colin G, Grillet G, Girardie P, Coupez E, Dequin PF, Cariou A, Boulain T, et al. Targeted temperature management for cardiac arrest with nonshockable rhythm. N Engl J Med. 2019;381(24):2327–37.
Dankiewicz J, Cronberg T, Gisela Lilja G, Jakobsen J, Bělohlávek J, Callaway C, Cariou A, Eastwood G, Erlinge D, Hovdenes J, et al. Targeted hypothermia versus targeted normothermia after out-of-hospital cardiac arrest (TTM2). A randomised clinical trial – rationale and design. Am Heart J. 2019;217:23–31.
Ebner F, Harmon MBA, Aneman A, Cronberg T, Friberg H, Hassager C, Juffermans N, Kjaergaard J, Kuiper M, Mattsson N, et al. Carbon dioxide dynamics in relation to neurological outcome in resuscitated out-of-hospital cardiac arrest patients: an exploratory Target Temperature Management Trial substudy. Crit Care. 2018;22(1):196.
Lilja G, Nielsen N, Ullen S, Blennow Nordstrom E, Dankiewicz J, Friberg H, Heimburg K, Jakobsen JC, Levin H, Callaway C, et al. Protocol for outcome reporting and follow-up in the Targeted Hypothermia versus Targeted Normothermia after Out-of-Hospital Cardiac Arrest trial (TTM2). Resuscitation. 2020;150:104–12.
Jakobsen JC, Ovesen C, Winkel P, Hilden J, Gluud C, Wetterslev J. Power estimations for non-primary outcomes in randomised clinical trials. BMJ Open. 2019;9:e027092. https://doi.org/10.1136/bmjopen-2018-027092.
Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke. 2007;38(3):1091–6.
Janssen MF, Pickard AS, Golicki D, Gudex C, Niewada M, Scalone L, Swinburn P, Busschbach J. Measurement properties of the EQ-5D-5L compared to the EQ-5D-3L across eight patient groups: a multi-country study. Qual Life Res. 2013;22(7):1717–27.
Nielsen N, Hovdenes J, Nilsson F, Rubertsson S, Stammet P, Sunde K, Valsson F, Wanscher M, Friberg H, Hypothermia N. Outcome, timing and adverse events in therapeutic hypothermia after out-of-hospital cardiac arrest. Acta Anaesthesiol Scand. 2009;53(7):926–34.
Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346(8):557–63.
Hypothermia after Cardiac Arrest Study G. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346(8):549–56.
Jakobsen JC, Gluud C, Winkel P, Lange T, Wetterslev J. The thresholds for statistical and clinical significance - a five-step procedure for evaluation of intervention effects in randomised clinical trials. BMC Med Res Methodol. 2014;14:34.
Kahan BC, Morris TP. Improper analysis of trials randomised using stratified blocks or minimisation. Stat Med. 2011;31:328–40.
Kahan BC, Morris TP. Reporting and analysis of trials using stratified randomisation in leading medical journals: review and reanalysis. BMJ. 2012;345:e5840.
Montgomery AA, Peters TJ, Little P. Design, analysis and presentation of factorial randomised controlled trials. BMC Med Res Methodol. 2003;3:26.
Jakobsen JC, Tamborrino M, Winkel P, Haase N, Perner A, Wetterslev J, Gluud C. Count data analysis in randomised clinical trials. J Biomet Biostat. 2015;6(1):227.
Jakobsen JC, Gluud C, Wetterslev J, Winkel P. When and how should multiple imputation be used for handling missing data in randomised clinical trials - a practical guide with flowcharts. BMC Med Res Methodol. 2017;17(1):162.
Nielsen EE, Norskov AK, Lange T, Thabane L, Wetterslev J, Beyersmann J, de Una-Alvarez J, Torri V, Billot L, Putter H, et al. Assessing assumptions for statistical analyses in randomised clinical trials. BMJ Evid Based Med. 2019;24(5):185–9.
Nørskov AK, Lange T, Nielsen EE, Gluud C, Winkel P, Beyersmann J, de Uña-Álvarez J, Torri V, Billot L, Putter H, et al. Assessment of assumptions of statistical analysis methods in randomised clinical trials: the what and how. BMJ Evid Based Med. 2020;bmjebm-2019-111268. https://doi.org/10.1136/bmjebm-2019-111268.
Frydenberg SJM. An introduction to Stata for health researchers. 4th ed. College Station: Stata Press; 2014.
Oliveira AG. Biostatistics decoded: Wiley; 2013.
Duval S, Tweedie R. Trim and fill: a simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics. 2000;56(2):455–63.
Acknowledgements
None.
Funding
The trial is supported by grants from: The Swedish Research Council (Vetenskapsrådet), The Swedish Heart- Lung Foundation, Stig and Ragna Gorthon Foundation and the Knutsson Foundation, the Skåne University Hospital Foundations, the Gyllenstierna-Krapperup Foundation, and governmental funding of clinical research within the Swedish National Health System. We have also received funding from Plus Laerdal Foundation. The authors are solely responsible for the design and conduct of this study, all study analyses, the drafting and editing of the manuscript, and its final contents.
Author information
Authors and Affiliations
Contributions
JCJ drafted the protocol. All authors amended the protocol. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval has been confirmed from the Regional Ethics Committee at Lund University (2015/228), and the study has been approved by all relevant ethics boards in all participating countries. Written informed consent is waived, delayed, or obtained from a legal surrogate, depending on the circumstances, and is obtained from participants who regain mental capacity. The trial is conducted in accordance with The Declaration of Helsinki. Registered within ClinicalTrials.gov (NCT02908308).
Consent for publication
Not applicable.
Competing interests
Dr. Cariou reports personal fees from Bard, outside the submitted work; Dr. Saxena has received an unrestricted grant from Bard BD and have spoken at educational events for BD Bard and provided advice on educational events and product development; Dr. Storm reports personal fees from BD Bard, personal fees from ZOLL GmbH, personal fees from Braincool Inc., personal fees from Xenios, and personal fees from Philips, outside the submitted work; Pr. Taccone reports lecture fees from BD Bard and ZOLL; Dr. Morgan reports personal fees from BD Bard for teaching use of surface cooling devices, outside the submitted work; Dr. Friberg reports receiving lecture fees from BD Bard and consulting fees from QuickCool, outside the submitted work; and Dr. Young reports personal fees from Bard Medical, outside the submitted work.
All remaining authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jakobsen, J.C., Dankiewicz, J., Lange, T. et al. Targeted hypothermia versus targeted normothermia after out-of-hospital cardiac arrest: a statistical analysis plan. Trials 21, 831 (2020). https://doi.org/10.1186/s13063-020-04654-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-020-04654-y