
Abstract
The rapid rise in the availability and scale of scRNA-seq data needs scalable methods for integrative analysis. Though many methods for data integration have been developed, few focus on understanding the heterogeneous effects of biological conditions across different cell populations in integrative analysis. Our proposed scalable approach, scParser, models the heterogeneous effects from biological conditions, which unveils the key mechanisms by which gene expression contributes to phenotypes. Notably, the extended scParser pinpoints biological processes in cell subpopulations that contribute to disease pathogenesis. scParser achieves favorable performance in cell clustering compared to state-of-the-art methods and has a broad and diverse applicability.
Similar content being viewed by others


Background
The scRNA-seq technology has emerged as a popular and powerful tool for profiling the transcriptomic landscape of individual cells within complex and heterogeneous systems. It has been revolutionizing our ability to dissect and understand various aspects of biology at the single-cell level, including developmental biology [1] and gene regulation [2], and has opened new avenues for exploring developmental biology, gene regulation, tissue heterogeneity, disease mechanisms, and evolutionary dynamics.
The scRNA-seq study usually involves the integrative analysis of transcriptomic data of individual cells measured with different technologies and derived from multiple tissues [1] of individuals with different phenotypes [2,3,4] across statuses [5] and even species. Indeed, the integrative analysis of heterogeneous scRNA-seq data to identify cell types or states and to compare gene expression across biological conditions has tremendous potential to transform our understanding of complex and heterogeneous biological systems [6]. A successful example of this practice is that joint analysis of scRNA-seq data from multiple melanoma tumors identifies an immune resistance program in malignant cells, which predicts clinical responses to immunotherapy in melanoma patients [7]. However, the heterogeneous variation due to different sequencing experiments and different biological conditions, including donors, tissues, or phenotypes, makes the integrative analysis challenging.
Methods have been developed for the integrative analysis of scRNA-seq data [8,9,10,11,12,13,14]. Computational approaches, including BBKNN [8] and FastMNN[11], are proposed, which assume that the batch effect in scRNA-seq data is almost orthogonal to the biological variation [11]. Thus, their abilities to correct batch effects originating biologically are limited [15]. Seurat [12] matches cell states across biosamples by a shared correlation space defined by canonical correlation analysis. LIGER [6] employs nonnegative matrix factorization (NMF) to delineate shared and dataset-specific features of cells across biosamples. Harmony [9] integrates scRNA-seq data by projecting cells into a shared embedding. Scanorama [13] leverages the matches of cells with similar transcriptional profiles across biosamples to perform batch correction and integration. Most of the above methods focus on batch effect correction and cell identity annotation. They do not model the heterogeneous variation from different biological conditions and thus lack interpretability on how biological conditions affect the gene expression of cells. Moreover, it is also meaningful to model the effect of biological conditions on different cell subpopulations to elucidate the specific cell subpopulation and its related biological process that contributes to the disease pathogenesis.
To fill in the gap and address the above challenges, we develop a general and flexible statistical framework, scParser, which is based on an ensemble of matrix factorization and sparse representation learning. scParser directly models the variation from different biological conditions (e.g., donor, disease status, experimental time points) via gene modules, which bridge gene expression with the phenotype of interest and unveil the relevant biological processes together with their contributing genes. The rationale behind our modeling is that the biological conditions affect the activities of certain biological processes, which in turn affect gene expression; the gene modules in scParser are learnt adaptively from the data and encode the biological processes that are affected by the biological conditions. We also develop an extended version of scParser, which further models the interactive effects of biological conditions on different cell subpopulations and pinpoints the relevant biological processes within the specific cell subpopulation that may contribute to the disease pathogenesis. Empowered by gene modules, scParser can boost the signal of disease-associated genes. In addition, scParser can correct for batch effects and achieves favorable performance in cell clustering. To make scParser scalable to large-scale datasets, we incorporate a batch-fitting strategy. Importantly, the wide applicability of our proposed framework has been demonstrated via extensive applications to various biomedical studies.
Results
Overview of scParser
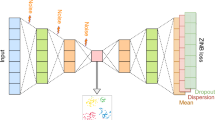
We propose a flexible computational method scParser (sparse representation learning for scalable single-cell RNA sequencing data analysis), presented in Fig. 1. Here we use donors and phenotypes (e.g., disease status) as an example of biological conditions. The RNA expression profiles of cells from different donors with different phenotypes are profiled (Fig. 1A). To facilitate interpretation, scParser models variation from multiple biological conditions (e.g., donor and phenotype) with matrix factorization and models cellular variation with sparse representation learning (Fig. 1B).

Overview of scParser. A The scRNA-seq data is obtained from biosamples from multiple biological conditions. Variations from biological conditions (e.g., donor and phenotypes) and technical artifacts may bring about batch effects in the scRNA-seq data. B scParser models the variation from donor and phenotype with matrix factorization and cellular variation with sparse representation learning. After cell population annotation, an extension of scParser enables revealing the effects of biological heterogeneous conditions (e.g., phenotypes) on different cell populations. C The output from scParser enables various interpretive data analyses, such as cell population annotation, connecting gene expression to biological conditions via biologically meaningful gene modules, revealing the heterogeneous effect of phenotypes on different cell populations via gene modules, and uncovering donors’ molecular characteristics
More specifically, the expression level of gene \(m\) for cell \(i\), \({z}_{im}\), which is obtained from donor \(j\) with phenotype \(t\), is modeled as
where \({d}_{j},{p}_{t},{v}_{m}\) are vectors of length \({K}_{1}\), \({s}_{i},{g}_{m}\) are vectors of length \({K}_{2}\). The donor and phenotype information of samples is known. Here we name the above formulation as the vanilla scParser. The equation above has the following matrix form
where \({V}^{{K}_{1}\times M}\) is the key component in scParser and represents \({K}_{1}\) metagenes/gene modules (here we use the two phrases metagenes and gene modules interchangeably), and \(M\) is the total number of genes. These gene modules in the matrix \(V\) summarize the expression patterns of thousands of genes to a few metagenes/gene modules (as \({K}_{1}\) is much smaller than \(M\)), which provides a high-level summary of the gene activities affected by the biological conditions; \({D}^{{N}_{1}\times {K}_{1}}\) and \({P}^{{N}_{2}\times {K}_{1}}\) are matrices for the latent representations of \({N}_{1}\) donors and \({N}_{2}\) phenotypes, respectively; \({D}^{{N}_{1}\times {K}_{1}}\) and \({P}^{{N}_{2}\times {K}_{1}}\) can also be interpreted as the expression level of the \({K}_{1}\) gene modules in the \({N}_{1}\) donors and \({N}_{2}\) phenotypes; \({S}^{N\times {K}_{2}},{G}^{{K}_{2}\times M}\) denote latent representations for \(N\) cells and \(M\) genes after modeling variation from donors and phenotypes; \({X}^{D},{X}^{P}\) are indicator matrices of the donor and phenotype labels for the cells, and they have \(N\) rows and \({N}_{1}\) and \({N}_{2}\) columns, respectively. The objective function for the equation with matrix representation is as follows:
where \({\lambda }_{1},\) \({\lambda }_{2},\alpha\) are tuning parameters, and \({\Vert \cdot \Vert }_{F}\) represents the Frobenius norm. \(c\) is a constant, restricting the scale of \({G}_{k}\), the \(k\) th row of \(G\). Alternating block coordinate descent (BCD) is utilized to optimize the objective.
The outputs from scParser provide informative inputs for various downstream analyses (Fig. 1C): cell populations/types can be identified with the cell latent representation matrix \(S\) from Eq. 1, the connection between gene expression and biological conditions can be uncovered via biologically meaningful gene modules using \(P\) and \(V\), the effect of phenotypes on gene expression can be revealed with \(P*V\), and the molecular characteristics of donors can be characterized with \(D\). In addition to the vanilla version of scParser, we also developed an extension of scParser that models the heterogeneous effects of biological conditions on cell populations (details in the “Methods” section) after cell identity annotation with \(S\). Notably, the extended scParser pinpoints the related biological processes in the specific cell subpopulations that may contribute to disease pathogenesis.
Datasets
In our applications, we applied scParser to the three scRNA-seq datasets from studies on the pancreas of type 2 diabetic and normal donors (T2D dataset) [2], human airway epithelium of donors with different smoking habits (Smoking dataset) [5], and peripheral blood cells of patients experiencing mild to severe COVID-19 infection (COVID-19 dataset) [3]. The number of cells in these datasets ranges from ~6000 to over 30,000. To demonstrate the scalability of scParser, we further applied scParser with the batch-fitting strategy to the immune dataset, offering scRNA-seq data of > 300,000 immune cells from 16 different tissues of 12 donors [1], and the GBM (glioblastoma) data covering > 200,000 human glioma, immune, and stromal cells from GBM patients [4]. In our applications, we first applied the vanilla scParser. With the cell populations annotated after fitting the vanilla scParser, we employed the extended scParser.
scParser connects gene expression to phenotypes through gene modules
The vanilla scParser captures variation from heterogeneous biological conditions in scRNA-seq data with biologically meaningful gene modules, which serve as a bridge to connect gene expression to phenotypes via path analysis. The path analysis empowered by gene modules gives us deeper insights into the underlying biological context/process where genes participate in disease pathology, which is usually not provided in standard differential expression (DE) analysis.
Let us use the T2D dataset from the study [2] for demonstration. The gene modules M1 and M2 connect the eight selected most variable genes with diabetes status (Fig. 2A). As revealed by the biological process (BP) enrichment analysis on top genes in gene modules M1 and M2, M1 encodes the BPs related to insulin and peptide secretion, and M2 encodes the BPs involved in the regulation of transcription in response to stress and response to unfolded protein.

Various downstream analysis with outputs from the vanilla scParser in our applications. A The path diagram links the expression of the eight selected most variable genes to diabetes status via two gene modules in the application to the T2D dataset. B Changes in the expression of the most variable (top 10) genes across control, mild, and severe COVID-19 from the analysis of the COVID-19 dataset are shown. C Changes in the expression of the most variable (top 10) genes across different types of GBM from the analysis of the GBM dataset are shown. The words “LGG”, “ndGBM,” and “rGBM” stand for low-grade gliomas, newly diagnosed GBM, and recurrent GBM, respectively. D The top 20 severe COVID-19-associated genes ranked by adjusted p-values are displayed. E The top 20 recurrent GBM-associated genes ranked by adjusted p-values are presented. F The up-regulated BPs enriched by genes in the upper 5% quantile of the differences in the adjusted expression profiles between mild COVID-19 and normal are shown. G The up-regulated BPs enriched by genes in the upper 5% quantile of the differences in the adjusted expression profiles between severe and mild COVID-19 are shown. H The up-regulated BPs enriched by genes in the upper 5% quantile of the difference in the adjusted expression profiles between the recurrent GBM and newly diagnosed GBM are shown
On the one hand, we observed that the expression of M1 is much higher in normal than in diabetes: 1.34 vs. 0.97 (Fig. 2A), suggesting that normal patients have a higher activity level of BPs in insulin and peptide secretion than diabetic patients. This observation is also consistent with the fact that type 2 diabetic patients can have impaired insulin secretion. Additionally, the genes that have large loading in M1 include INS (the loading is 1.14), CHGB (the loading is 0.93), and FXYD2 (the loading is 0.47). This suggests that these genes may affect type 2 diabetic patients through BPs in insulin and peptide secretion. In fact, the role of these genes in type 2 diabetes is supported by previous studies: INS encodes insulin [16], FXYD2 was suggested to be a novel target for diabetes [17], and loss of CHGB impairs glucose-stimulated insulin secretion [18].
On the other hand, we see that the expression of M2 is much higher in diabetes compared to normal: 1.10 vs. 0.62 (Fig. 2A), implying that the diabetic has a higher level of BPs in response to unfolded protein, compared with the normal. This is also supported by the finding that response to unfolded protein leads to endoplasmic reticulum (ER) stress [19], which contributes to insulin resistance [20]. Moreover, the loadings of FOS and JUN are high for M2 (0.58 and 0.56, respectively). This implies that the two genes (FOS and JUN) may contribute to type 2 diabetes via BPs in the regulation of transcription in response to stress and response to unfolded protein. The implication is also consistent with the finding that the AP-1 transcription factor, which is composed of c-Jun, encoded by gene JUN [21], and c-Fos, encoded by gene FOS [22], is necessary for the induction of the ER stress [23], which contributes to pancreatic β-cell loss and insulin resistance [24].
In brief, scParser summarizes the expression patterns of thousands of genes to a few metagenes/gene modules, which provides a high-level summary of the gene activities. These metagenes represent the genes that collectively carry out certain biological functions. Hence, the path analysis empowered by scParser via gene modules not only identifies the genes associated with the disease status but also unearths the relevant biological context/process through which the genes participate in the disease pathology. Notably, these gene modules are learned adaptively from the data.
scParser facilitates the identification of phenotype-associated genes and their enriched biological processes
The changes in gene expression across different phenotypes can be unveiled by \(P*V\), which is the product of the phenotype representation matrix \(P\) and the gene module matrix \(V\) in Eq. 1. We also name \(P*V\) as the adjusted expression profiles for phenotypes because the unwanted variation from donors is controlled in them. The product \(P*V\) together with the matrices \(P\) and \(V\) facilitates the identification of phenotype-associated genes.
We first demonstrate it with the application to the COVID-19 dataset. The top variable genes, which are identified by inspecting \(P*V\), show dynamic changes across severe COVID-19, mild COVID-19, and normal (Fig. 2B). Besides, we identified the genes associated with the disease status by examining the correlation between the rows of \(P\) and the columns of \(V\) [25]). The top identified genes, including HBB (Fig. 2B and D) and GRINA and FTL (Fig. 2D), are shown to be differentially expressed between severe COVID-19 cases and normal in independent studies [26,27,28]. Additionally, the high expression levels of HBB, HBA1, and HBA2 (Fig. 2B) in severe COVID-19 patients are supported by the evidence that the level of hemoglobin, whose subunits are encoded by HBB, HBA1, and HBA2, was found to be significantly higher in severe COVID-19 patients than in mild COVID-19 ones and normal controls [29].
To further verify the usefulness of the adjusted expression profiles for phenotypes \(P*V\), we conducted additional experiments with disease targets provided by Open Targets [30]. First, we extracted target genes for COVID-19 from Open Targets, where the associations between COVID-19 and target genes are indicated by scores (from 0 to 1). Then, we also computed the absolute expression difference of genes across phenotypes (mild COVID-19 vs. normal, and severe COVID-19 vs. mild COVID-19) using \(P*V\). Subsequently, we examined the correlation between the absolute expression difference of genes across phenotypes and the association scores provided by Open Targets.
We found that there is a positive correlation between them: p-values for a positive correlation between the absolute gene expression difference in mild COVID-19 vs. normal and the association score in Open Targets are 0.0005 for both Kendall and Spearman correlation, and p-values for a positive correlation between absolute gene expression difference in severe vs. mild COVID-19 and the association score are 6.47E−05 for Kendall correlation and 6.919E−05 for Spearman correlation, respectively. These findings suggest that the genes that have a bigger difference in expression levels across phenotypes tend to have a higher association score with the corresponding disease.
To complement path analysis by considering all gene modules, BP enrichment analysis can be performed on the list of genes that demonstrate the greatest difference across COVID-19 statuses with the adjusted expression profile \(P*V\), which controls for the variation of donors. The up-regulated BPs in cells from donors with mild COVID-19 compared to those from the normal include the viral life cycle, viral process, response to viral, and the BPs related to immune response, such as (myeloid) leukocyte and neutrophil chemotaxis (Fig. 2F), suggesting elevated viral activity and immune response in mild COVID-19 patients compared to the normal; in the comparison of cells from severe COVID-19 cases to those from mild COVID-19 ones, the BPs related to apoptotic signaling pathways are activated (Fig. 2G), consistent with the previous finding that T cell apoptosis characterizes severe COVID-19 [31].
We also implemented the analyses on the glioblastoma (GBM) dataset. There is a wider difference in gene expression between glioblastoma (GBM) and low-grade gliomas (LGG), compared to that between recurrent and newly diagnosed GBM (Fig. 2H). Also, some listed top genes (FTL [32], FTH1[33], GAPDH [34], and VIM [35]) (Fig. 2C and E) are shown to be associated with GBM. Moreover, recurrent GBM shows higher levels of BPs related to inflammation and immune response compared with newly diagnosed GBM (Fig. 2H). The result is also supported by Open Targets [30]: p-values for a positive correlation between absolute differences in gene expression between recurrent GBM and newly diagnosed GBM and the association score of genes for GBM are 0.0122 for Kendall correlation and 0.0123 for Spearman correlation, respectively.
scParser can also boost the signal of phenotype-associated genes by removing the variation of the less relevant gene modules (Additional file 1: section S1): after filtering less relevant gene modules, the absolute gene expression differences between disease statuses (computed with \(P*V\)) have a more significant and higher positive correlation with the association scores provided by Open Targets and that the BPs enrichment analysis also becomes statistically more significant. In addition, we implemented transcription factor (TF) enrichment analysis to identify potential drivers for the changes in gene expression across biological conditions in the T2D, Smoking, and GBM datasets, and most TFs identified in our analyses are supported by previous studies (Additional file 1: section 12).
scParser pinpoints the relevant biological processes in cell subpopulations that contribute to disease pathogenesis
Previously, we presented how biological conditions affect gene expression through gene modules in the vanilla scParser. However, it is important to note that the same biological condition may exert heterogeneous effects on different cell populations. Motivated by this, we develop the extended version of scParser, defined in Eq. 6 in the “Methods” section, which directly models the interactive effects of biological conditions on different cell subpopulations annotated with \(S\) in Eq. 4. The matrices \(W\) and \(V\) from Eq. 6 enable path analysis. We use the results from our application to the T2D dataset for demonstration. We first show that path analysis informs us of the difference in biological functions for three subpopulations of beta cells (B1, B2, and B3) in normal controls (Fig. 3A), and then demonstrate that path analysis can unveil the relevant biological processes that are affected in B1 beta cells in diabetic patients that may contribute to the disease pathogenesis (Fig. 3B).

Further exploratory analyses with cell population representations from scParser in the application to the T2D dataset. A The path diagram shows that scParser links the expression of 8 beta cell marker genes to the three beta cell populations (B1, B2, and B3) from the normal via two gene modules. B The path diagram shows that scParser links the expression of 8 beta cell marker genes to the beta cell population (B1) across diabetes status via two gene modules
More specifically, we focus on two gene modules M1 and M2. As revealed by enrichment analysis on top genes in M1 and M2, BPs encoded by M1 involve the secretion of hormones and peptides (not including insulin), but BPs encoded by M2 further include insulin secretion. We first looked at the three subpopulations of beta cells (B1, B2, and B3) in normal controls to study their difference. The subpopulation B3 has lower loadings in M1 and M2 (−2.31 and 0.23, respectively), compared to those for B1 (0.47 and 1.54, respectively) and B2 (1.31 and 0.37, respectively) (Fig. 3A), and the loading of M1 and M2 are also different between B1 and B2 (Fig. 3A). This evidence suggests that the beta cell populations from the normal show heterogeneity in biological functions: B1 and B2 (especially B1) may have much higher levels of insulin secretion (encoded in gene module M2) than B3.
To further investigate the role of the beta cell populations in diabetes pathogenesis, we next focus on comparing beta cells B1 in normal vs diabetes through path analysis (Fig. 3B) because B1 has a very high loading in M2 in the normal and it may play a key role in insulin secretion. We see that the expression of M2 for B1 is much higher in the normal than in the diabetic: 1.54 vs. 0.87 (Fig. 3B), suggesting that B1 from the diabetic has a lower level of BPs in insulin secretion, compared to that from the normal. This implies that B1 may be directly associated with diabetes pathogenesis. This implication is consistent with the fact that T2D is due to the dysfunction of beta cells [36].
Besides pinpointing the relevant biological context/process shown above, scParser quantifies the contribution of individual genes to the biological context/process (Fig. 3). As mentioned previously, the gene module M1 encodes BPs for the secretion of hormones and peptides (not including insulin), and M2 further encodes BPs in insulin secretion. The genes (RBP4, IAPP, PCSK1, and INS) with large loadings on M1 (Fig. 3) either encode peptide hormone or participate in peptide hormone secretion, as shown in previous studies [16, 37,38,39]. In addition, the loadings of INS and IAPP are high for M2, and the loading of RBP4 is negative for M2 (Fig. 3). The three genes (INS, IAPP, RBP4) were shown to associate with insulin secretion in previous studies [16, 39, 40], where RBP4 is negatively associated with insulin secretion [40], explaining its negative loading in M2. The findings imply that these genes may contribute to heterogeneity in the function of beta cells and participate in the contribution of the cell populations to the diabetes status through BPs involved in insulin, peptide, and hormone secretion.
In brief, the extended scParser reveals the key biological context/process in the cell subpopulation that contributes to the disease status. In addition, it sheds light on the involvement of genes in the key biological context/process. This unique advantage of scParser provides us with a much deeper understanding of the contribution of cell populations to disease pathology via gene modules.
scParser reveals the heterogeneous effects of biological conditions on different cell subpopulations or types
In the previous subsection, the heterogeneous effects of biological conditions on cell subpopulations have been demonstrated with two gene modules with the T2D dataset as an example. To complement the path analysis by considering all gene modules, we computed the adjusted expression profiles for cell subpopulations (types) under different biological conditions with \(W*V\), which is the product of the representation matrix for cell subpopulations (types) under different biological conditions \(W\) and the gene module matrix \(V\) in Eq. 6. The product of \(W*V\) reveals expression dynamics of genes in cell subpopulations (types) across biological conditions and identify the phenotype- or disease-critical cell populations by BP enrichment analysis.
Let us first demonstrate the results from our application to the T2D dataset. The top variable genes in the beta cell subpopulation B1, which are obtained by examining the rows of \(W*V\) corresponding to B1, show distinct differences across diabetes status (Fig. 2B). Some identified genes (MTRNR2L1 [41], CPE [42], RBP4 [43], FXYD2 [44, 45], and CHGB [18]) are shown to play an important role in beta cells and be involved in diabetes (Fig. 4A), suggesting the contributing role of beta cells in diabetes. Then, the BP enrichment analysis on the list of top variable genes for each cell subpopulation (type) across diabetes status was conducted to reveal the effects of biological conditions on the cell subpopulation (type). Here the list of top variable genes is obtained with the adjusted expression profile \(W*V\) corresponding to the cell subpopulation (type), which controls for the variation of donors. We observed that the levels of BPs in the secretion and regulation of insulin are lower in the two beta subpopulations (B1 and B2) from diabetic donors, compared to normal controls (Fig. 4C and D). However, this finding is not observed for the other three cell types (Additional file 1: Fig. S1A, S1B, and S1C) and the beta cell subpopulation B3 (Additional file 1: Fig. S1D). This implies that the two beta cell subpopulations are directly associated with diabetes.

scParser reveals the heterogeneous effects of biological conditions on different cell populations (types). A The figure shows the changes in expression of the most variable (top 10) genes for the beta cell subpopulation (B1) across diabetes status with the T2D dataset. B The figure shows the changes in expression of the most variable (top 10) genes for the T cell population across normal, mild COVID-19, and severe COVID-19 with the COVID-19 dataset. C The figure shows the down-regulated BPs enriched by genes in the lower 5% quantile of the difference in the adjusted expression profiles for B1 between the diabetic and normal conditions. D The figure shows the down-regulated BPs enriched by genes in the lower 5% quantile of the difference in the adjusted expression profiles for B2 between the diabetic and normal conditions. E The up-regulated BPs enriched by genes in the upper 5% quantile of the difference in the adjusted expression profiles for T cells between severe and mild COVID-19 are displayed. F The up-regulated BPs enriched by genes in the upper 5% quantile of the difference in the adjusted expression profiles for monocytes between severe and mild COVID-19 are displayed
Meanwhile, the average proportions of B1 and B2 are greater in normal controls (35.00% and 8.88%, respectively) than in diabetes (30.56% and 4.58%), and the average proportion of B3 is smaller in normal controls (5.29%) than in diabetes (8.99%). (Additional file 2: Table S1). This leads to the finding that diabetes is associated with a decrease in the subpopulations of beta cells (B1 and B2) that secrete insulin and an increase in the subpopulation of beta cells (B3) that have a low level of insulin secretion.
To verify the finding above, we first obtained a list of eight genes, which are shown to associate with insulin resistance, T2D, or insulin secretion, through literature search (RBP4 [46,47,48], NPY [49, 50], DLK1 [51,52,53,54], PCSK1 [55, 56], MAFA [57,58,59], SIX3 [60,61,62], PFKFB2 [63,64,65], TMEM37 [66]). Then, we conducted two-sided t-tests to compare the expression levels of these genes between normal controls and diabetes across all six cell populations, including the two beta cell populations (B1 and B2) (Additional file 2: Table S2). The p-values tend to be much smaller in B1 and B2 than in other cell populations (Additional file 2: Table S2). In brief, the additional experiment supports the conclusion above. This phenomenon is supported and systematically reviewed by the study [67], suggesting that beta cells are morphologically and functionally heterogeneous and that this is strongly associated with diabetes.
We also performed the above analysis to the COVID-19 datasets. The relationship between the three genes listed in Fig. 4B (HBB, HBA1, and HBA2) and severe COVID-19 has been discussed in our previous section, and two other identified genes (TNFAIP3 [68] and GNLY [69]) were reported to play an important role in COVID-19 (Fig. 4B). Further, we observed that the BPs related to the differentiation of T and lymphocyte cells are up-regulated in monocytes from severe COVID-19 donors, compared to monocytes from mild ones (Fig. 4F). More importantly, the BPs involved in response to viral are up-regulated in T cells in the comparison (Fig. 4E), and the levels of the BPs related to interleukin-2 production, which plays a key role in the human immune system, are also high in T cells, but not in monocytes (Fig. 4E and F). This may suggest that monocytes promote the differentiation of T cells and that T cells play a key role in the human immune response to severe COVID-19.
To further verify the association of monocytes and T cells in severe COVID-19 patients, we conducted the following analysis. Specifically, we first computed the absolute differences in gene expression between severe and mild COVID-19 (here \(W*V\) is used for the gene expression level for cell populations across COVID-19 status, which is adjusted for the variation of donors) for T cells and monocytes, and then computed the correlation between the absolute expression differences and the scores of genes associated with COVID-19 provided by Open Targets [30]. Our results show that there are statistically significant and positive relationships between the absolute expression difference for T cells (P Kendall = 0.0170 and P Spearman = 0.0192) as well as monocytes (P Kendall = 0.0091 and P Spearman = 0.0080) and the gene association scores for COVID-19, corroborating the role of monocytes and T cells in COVID-19. On the one hand, the important role of monocyte cells in T cell formation has been supported by the literature [70,71,72]: monocytes play an important role in the formation of memory T cells [72], and monocyte‐derived cell populations promote the differentiation of CD4+ T cells [71] and CD8+ T cells [70]. Additionally, the contributing role of monocytes in COVID-19 pathogenesis is supported by the literature [73, 74]. On the other hand, the relationship between severe COVID-19 and T cells is also supported by the literature [31, 75,76,77]: T cells play a crucial protective role in the human immune response to COVID-19 [75,76,77], and T cell apoptosis characterizes severe COVID-19 infection [31]. These findings support the conclusion above that monocyte cells may promote the differentiation of T cells and that T cells may play a key role in the immune response to severe COVID-19.
scParser reveals the molecular characteristics of donors
Here we demonstrate how scParser can help reveal the molecular characteristics of donors with the application to the GBM dataset. First, we directly plotted the donor representations of 11 newly diagnosed GBM donors (Additional file 1: Fig. S2). Donors 01, 06, and 10 show obvious differences in molecular characteristics compared with the other eight donors (Additional file 1: Fig. S2). For demonstration, we focus on discussing the molecular differences of these donors with metagenes 12, 13, and 15, and the biological meaning of the metagenes is revealed with enrichment analysis. A joint analysis of the figure and results from enrichment analysis suggest that donor 1 may suffer a poor progression of GBM, donor 6 may experience more severe hypoxia than other donors, and donor 10 has a stronger immune response and a lower level of gliogenesis and axonogenesis than other donors. Technical details of enrichment analysis and result interpretation are provided (Additional file 1: section S2). However, due to a lack of clinical information on these donors to validate our findings, further investigation is needed to explore our findings.
Comparative study of scParser on cell type annotation
In addition to the interpretability empowered by gene modules presented above, scParser also helps annotate cell types. To evaluate its ability in annotating cell populations, we compared it against eight methods in cell identity annotation, including Seurat V3 [12], LIGER [78], Harmony [9], BBKNN [8], ComBat [79], FastMNN [11], scINSIGHT [29], and Scanorama [13].
Practically, we implemented the clustering procedure in SCANPY [80] and Seurat [12] for all methods for a fair comparison, where we performed cluster analysis on the low-rank embeddings from each method using Louvain clustering [81]. We considered the number of cell types provided by the original data as the expected number of clusters. In practice, we adjusted the resolution until the expected number of clusters was attained. Then, we assessed the clustering performance of the methods with adjusted Rand index (ARI), adjusted mutual information (AMI), normalized mutual information (NMI), and homogeneity score. In the comparison, we considered the cell types annotated in original studies as true labels and ignored the unannotated observations. To clarify, the cell types provided in the original studies are annotated by differential analysis with marker genes. For visualization, we employed UMAP [82] to obtain two-dimensional embeddings and plotted them against annotated cell types.
Here we compared the performance of scParser against that of the eight methods on the COVID-19, T2D, and Smoking datasets (Fig. 5A) and the performance of batch scParser on cell type annotation on the Immune and GBM datasets (Fig. 5B). Details of the five datasets are provided in the “Datasets” section.

Comparative study of scParser against other eight methods in cell type annotation. A The clustering performance of scParser was compared to that of the other eight methods in terms of ARI, AMI, NMI, and Homogeneity score with the COVID-19, T2D, and Smoking datasets. The words “COVID-19”, “T2D”, and “Smoking” stand for the COVID-19, T2D, and Smoking datasets, respectively. B The clustering performance of batch scParser is compared to that of the other eight methods in terms of ARI, AMI, NMI, and Homogeneity score on the GBM and Immune datasets. The performance of Combat and scINSIGHT are not shown due to the scalability issue
In general, across the four different evaluation metrics, scParser achieved the start-of-the-art clustering performance (Fig. 5). In terms of the T2D dataset, it outperforms all other methods in the dataset in terms of ARI, NMI, and AMI (Fig. 5A). In the COVID-19 and Smoking datasets, scParser achieved cluster performances comparable to the state-of-the-art methods (Fig. 5A). Moreover, batch scParser outperforms all other methods in terms of ARI (Fig. 5B). Regarding the other three evaluation metrics, the performance of batch scParser is similar to that of other methods (Fig. 5B). In the application of scINSIGHT to the other four datasets except for the COVID-19 dataset, it failed to yield results in two days, so we stopped the program for time consideration.
In our visual comparison of scParser with the three methods (LIGER, Seurat, and Harmony) recommended for scRNA-seq data integration by [83], we found that four methods demonstrate a similar performance on the COVID-19 dataset (Additional file 1: Fig. S3A). In the analysis of the T2D dataset, Harmony and scParser outperform the other two methods, and scParser seems to perform better in separating A (alpha) and B (beta) cells (Additional file 1: Fig. S3B). Specifically, LIGER cannot well distinguish PP cells from B cells, and Seurat fails to distinguish PP cells from a subpopulation of A cells (Additional file 1: Fig. S3B). Moreover, quantitative measurement of these methods in separating A, B, and PP cells with silhouette scores also confirms our observation, with the silhouette scores equal to 0.26, 0.22, 0.23, and 0.14 for scParser, LIGER, Seurat, and Harmony, respectively. All methods perform similarly in the Smoking dataset (Additional file 1: Fig. S3C), and this is also indicated by Fig. 5A. Batch scParser achieved a similar performance in cell type annotation, compared with the other three methods (Additional file 1: Fig. S3D and S3E).
We noticed that the clustering performance for different methods varies across different datasets when different evaluation metrics are used (Fig. 5). Hence, for each evaluation metric above, we followed the idea from the previous study [84] to calculate the averaged ranking for each method across the five datasets as its overall performance (Additional file 1: section S3). The idea of the overall ranking follows the recommended practice for ranking methods in performance comparison from the study [85]. scParser demonstrates the highest average ranking across the five datasets for all four evaluation metrics (Additional file 1: section S3), suggesting that scParser has a superior overall performance over other methods in cell clustering.
In summary, scParser achieves state-of-the-art performance in cell type annotation in each application and demonstrates superior overall performance over other methods in cell clustering. The batch-fitting strategy improves the scalability of scParser without impairing its performance in cell-type annotation. Further experiments also show scParser demonstrates satisfactory performance in batch effect correction (Additional file 1: section S3).
Computational time and memory usage
scParser is a computationally efficient framework for interpretable studies of scRNA-seq datasets. The computational time for scParser with the whole data-fitting strategy on the COVID-19 data, the T2D dataset, and the Smoking dataset is ~20 min, ~1 h, and ~2.5 h, respectively, and the memory consumption of scParser for the applications is less than 5 G. For the batch scParser with 20 batches, it takes roughly 6 h and 12 G memory to complete the analyses with the GBM and Immune datasets.
Discussion
The interpretability and broad applicability of scParser in scRNA-seq data analysis have been demonstrated through comprehensive applications. Specifically, scParser was applied to three scRNA-seq datasets, and batch scParser was employed on two other scRNA-seq datasets of over 200,000 cells. The outputs from these applications enable various interpretative data analyses. Firstly, scParser enables connecting gene expression to diabetes status via biologically meaningful gene modules by path analysis and revealing changes in the expression of genes across COVID-19 infection outcomes and GBM stages. Moreover, enrichment analyses of the most variable genes across biological conditions reveal the effect of COVID-19 outcomes and different GBM stages on cells. Additionally, the ability of scParser to discern subtle changes in gene expression across adjacent time points with the iPSC dataset [86] has been demonstrated (Additional file 1: section S5). Secondly, it captures the heterogeneous effects of biological conditions on cell populations via biologically meaningful gene modules. Hence, scParser pinpoints the biological context in cell subpopulations that contribute to disease pathogenesis via the gene modules by path analysis. Moreover, putative phenotype- or disease-critical cell populations are identified by comparing the adjusted expression profiles (provided by scParser) for cell populations across phenotypes or disease status. Thirdly, donor representations from scParser characterize the molecular aspects of donors with GBM. Fourthly, scParser is computationally efficient in analyzing scRNA-seq data. Empirical studies show that scParser completes analyzing scRNA-seq data with different cell numbers and gene numbers in a reasonable time (Additional file 1: section S7). Additionally, scParser is robust to the sparsity due to dropout events in scRNA-seq data (Additional file 1: section S8). Lastly, scParser has a favorable performance in cell clustering and demonstrates a satisfactory performance on batch effect correction.
In brief, scParser has the following virtues: (1) scParser is a general, flexible, and scalable framework for scRNA-seq data analysis, in which the heterogeneous variation from biological conditions is modeled via biologically meaningful gene modules. In scParser, biological conditions can be defined upon study design and research question; (2) two fitting strategies offered by scParser make it tailored for analyzing scRNA-seq data of various sample sizes; (3) Empowered by gene modules, scParser can boost the signal of disease-associated genes; (4) the extended scParser models the heterogeneous effects of biological conditions on different cell populations, thus providing us with insights on the contributing role in disease pathology; (5) the output from scParser enables various interpretative analyses via biologically meaningful gene modules; and (6) scParser demonstrates unique properties, compared to the standard Seurat pipeline and scINSIGHT [29] (Additional file 1: section 15).
We noticed that \(D,P\) in the vanilla scParser are not identifiable. The issue can be alleviated by the following practices. On the one hand, the ridge penalty on \(D\) and \(P\) can relieve it. In practice, the hyperparameter \({\lambda }_{1}\) for the ridge penalty for \(P,D\) across our applications ranges from 10 to 50, suggesting that the elements of \(P,D\) are shrunk towards 0. Moreover, the Frobenius norm of \(D\) is larger than that of \(P\) (\({N}_{1}>{N}_{2}\)), so the ridge penalty pushes the elements of \(D\) closer to 0, compared to \(D\). On the other hand, we initialize \(D\) to 0 and always update \(P\) before the update for \(D\) in model fitting. Therefore, intuitively, the update for \(D\) is computed on the residuals of \({X}^{P}P\). We think that this will further alleviate the identifiability of \(D\) and \(P\).
The scRNA-seq technology has been widely used to understand gene regulation across heterogeneous biological systems at the single-cell level [1, 3,4,5, 7, 87]. However, most existing computational methods for scRNA-seq data analysis focus on data integration and do not model the variation from biological conditions. Therefore, their ability to understand the heterogeneous effects of biological conditions at the single-cell level is limited. scParser offers an efficient and scalable solution to fill the gap in scRNA-seq data analysis. With a rapid rise in the scale of scRNA-seq data, scParser further incorporates batch-fitting strategy to accommodate scRNA-seq data of arbitrary sample size. Its wide applicability in biomedical data analysis has been demonstrated through comprehensive applications. With these virtues, scParser will benefit researchers in the biomedical field. Further extensions include analysis of other single-cell omics.
Conclusions
This article proposed a computational framework, scParser, for integrative scRNA-seq data analysis. scParser captures variation from heterogeneous biological conditions with matrix factorization and decomposes the cellular variation with sparse representation learning simultaneously. The gene modules empowered by scParser connect gene expression to disease status via gene modules. The extended scParser models the heterogeneous effects of biological conditions on different cell populations, thus pinpointing the relevant biological context/processes through which cell subpopulations contribute to the disease pathogenesis and identifying the putative cell populations associated with biological conditions. The outputs from scParser provide informative inputs for various interpretative analyses. Its wide applicability in biomedical data analysis has been demonstrated through comprehensive applications.
Methods
Here we propose a novel statistical approach, scParser, to decompose cellular variations in scRNA-Seq samples with considering heterogeneous variations from multiple biological conditions (e.g., donor, tissue, phenotype) in low-rank latent spaces to facilitate downstream analysis. In scParser, we integrate matrix factorization to capture variation from biological conditions with sparse representation learning to obtain embeddings of cells. In the employment of sparse representation learning, we introduce elastic-net regularization to encourage sparsity on the low-rank embeddings of cells to facilitate cell clustering and norm constraint to ensure each dimension with equal scale.
The scParser model and its extension
For illustration, let \({Z}^{N\times M}\) denote the matrix of log-normalized scRNA-Seq expression levels of \(N\) samples of \(M\) genes. The \(N\) samples can originate from several biological conditions (e.g., individuals, phenotypes, tissues, disease phases, or different time points). Here we use donor and phenotype for demonstration. The samples come from \({N}_{1}\) donors with \({N}_{2}\) phenotypes. Thus, the expression level of gene \(m\) for cell \(i\) from donor \(j\) with phenotype \(t\), \({z}_{im}\), can be modeled as
where \({d}_{j},{p}_{t},{v}_{m}\) are vectors of length \({K}_{1}\), \({s}_{i},{g}_{m}\) are vectors of length \({K}_{2}\). The donor and phenotype information of the samples is known. For simplicity, we name the formulation above as vanilla scParser. The rationale behind our modeling is that biological conditions (e.g., donor and phenotype) affect activities of the common transcription factors (TFs) or biological processes/signals, which in turn affect the expression of genes. In practice, we also observed that increasing model complexity by modeling variation from donor and phenotype in two separate latent spaces does not lead to explaining more variations. Therefore, we capture variation from donors and phenotypes in a shared low-rank latent space. An extra benefit of this practice is that it can reduce model complexity and improve optimization efficiency without impairing model interpretability. Intuitively, after modeling the heterogeneous biological variation across biosamples, we capture the cellular variation across biosamples in another low-rank latent space. This practice allows us to model the cellular variation more independently and also increases model flexibility, compared to restricting our modeling in only one latent space. In our implementation, we considered the option to restrict \({v}_{m}\) and \({g}_{m}\) to be the same. Thus, the last term on the right of Eq. 2 helps obtain the low-rank embeddings of cells. In scParser, biological conditions can be defined based on the research questions and study design, and the number of biological conditions can be further increased.
The objective function for Eq. 2 is formulated as
In the equation, \({g}_{mk}\) is the \(k\)-th element of \({g}_{m}\), and \(c\) is a constant (usually 1). We introduce the elastic net penalty on the cell representation \({s}_{i}\) to encourage sparsity to facilitate cell clustering. Moreover, the norm constraint is imposed to ensure the same scale of each latent dimension in decomposing cellular variation. Equation 3 can be represented with matrix operation as follows:
where \({X}^{D},{X}^{P}\) are indicator matrices of \(N\) rows and \({N}_{1}\) and \({N}_{2}\) columns, respectively, which are the dummy variables for \(N\) samples. \({D}^{{N}_{1}\times {K}_{1}},{P}^{{N}_{2}\times {K}_{1}},{V}^{{K}_{1}\times M}\) are matrices of latent representations of \({N}_{1}\) donors, \({N}_{2}\) phenotypes, and \(M\) genes, respectively, and denote \({S}^{N\times {K}_{2}},{G}^{{K}_{2}\times M}\) latent representations for \(N\) cells and \(M\) genes after modeling variation from donor and phenotype. \(c\) is a constant, restricting the scale of \({G}_{k}\), the \(k\)-th row of matrix \(G\). Each row of \({V}^{K\times M}\) represents a gene module, encoding certain biological processes, where the genes can be up-regulated and down-regulated. For a given gene module, the positive loadings of the genes stand for the up-regulation of corresponding genes, and the negative loadings represent the down-regulation of corresponding genes.
The above formulation can be extended to explore the heterogeneous effects of biological conditions on different cell populations after cell type annotation with \(S\) from Eq. 4. To exploit this idea, we model the expression level of gene \(m\) in cell population \(k\), which comes from sample \(i\) obtained from donor \(j\) with phenotype \(t\), \({z}_{im}\) as
where \({d}_{j}, {w}_{kt},{v}_{m}\) are vectors of length \(K\), and \({w}_{kt}\) is the latent representation for cell population \(k\) from donors with phenotype \(t\), which captures the interactive effect between phenotypes and cell populations while controlling variation from donors. In Eq. 5, the biological condition and cell populations can be defined based on research questions. The extension above provides a useful tool to explore the heterogeneous effects of biological conditions on different cell populations, which can be annotated with other methods. Throughout this study, we name the new formulation as extended scParser.
The objective function for Eq. 5 with matrix notation is defined as
Here \({X}^{W}\) and \({W}^{{(N}_{2}*{\text{N}}_{\text{c}})\times {K}_{1}}\) denote the indicator matrix of \(N\) rows and \({N}_{2}*{N}_{c}\) columns and latent representations for \({N}_{c}\) cell populations under \({N}_{2}\) biological conditions, respectively. Other notations are the same as in Eq. 4.
Model fitting
Alternating block coordinate descent (BCD) is employed to optimize Eqs. 4 and 6. Practically, each time we update a set of independent parameters with all other parameters fixed. In each iteration of BCD, we update each set of parameters of our model sequentially and repeat the process until the stopping criteria are met. Note that we always update \(P\) before the update for \(D\) in practice.
Optimize with the whole data strategy
With the objective function defined by Eqs. 4 and 6, we can easily derive closed forms for updating \({d}_{j},{p}_{t},{v}_{m}\). Specifically, we have the following update for \({v}_{m}\)
For the objective function defined by Eq. 4, \(U={X}^{D}D+{X}^{P}P\) and \(\widetilde{Z}=Z-SG\); for the objective function is defined by Eq. 6, \(U={X}^{D}D+{X}^{W}W\) and \(\tilde{Z}\) is equal to \(Z\). Under both situations, \(\tilde{Z}_m\) is the \(m\)-th column of \(\tilde{Z}\). Similarly, the update for \({d}_{j}\) is
where \(\tilde{Z}=Z-SG-{X}^{P}PV\) and \(\tilde{Z}=Z-{X}^{W}WV\) for the objective function defined by Eq. 4 and Eq. 6, respectively, \({B}_{j}\) is the set of indices of cells from donor \(j\), and \({N}_{j}\) is the number of elements in \({B}_{j}\). Likewise, the update for \({p}_{t}\) is
where \(\tilde{Z}=Z-SG-{X}^{D}DV\) for the objective function defined by Eq. 4, \({B}_{t}\) is the set of indices of cells from donors with phenotype \(t\), and \({N}_{t}\) is the number of elements in \({B}_{t}\). Finally, the update for \({w}_{kt}\) is
where \(\tilde{Z}=Z-{X}^{D}DV\) for the objective function defined by Eq. 6, \({B}_{kt}\) is the set of indices of cells of cell type \(k\) from donors with phenotype \(t\), and \({N}_{kt}\) is the number of elements in \({B}_{kt}\).
When optimizing Eq. 4 with respect to \(G\), the Lagrange dual proposed in the study [88] is employed. The Lagrange dual for our problem is of the following form
Here \(W={\tilde{Z}}^{\intercal }S\), \(\tilde{Z}=Z-{X}^{D}DV-{X}^{P}PV\), \(Q={S}^{\intercal }S\), and \(\Psi\) is a \({K}_{2}\times {K}_{2}\) diagonal matrix with dual variables \(\psi\) expanding along its diagonal. By taking derivative with respect to \(G\), we have
Then, by substituting Eq. 8 into Eq. 7, we have the following dual for our problem
The gradient \(\nabla\) and Hessian \(H\) for \(\mathcal{D}\left(\overrightarrow{\psi }\right)\) with respect to \(\overrightarrow{\psi }\) can be derived as follows:
Newton’s method is used to optimize our dual problem with respect to \(\Psi\). Thus, the update for \(\Psi\) at iteration \(t\) can be written as
where \({\Psi }^{\left(t-1\right)},{H}^{\left(t-1\right)},{\nabla }^{\left(t-1\right)}\) are the diagonal matrix of \(\psi\), gradient, and Hessian matrix at iteration \(t-1\), respectively. In practice, we alternatively compute the updates for \(G,\Psi\) until the sum of the squared difference in \(\Psi\) between two consecutive iterations is less than a predefined threshold (\({10}^{-4}\) is used in our studies). Further details on the derivation of Lagrange dual are provided (Additional file 1: section S10).
When optimizing Eq. 4 with respect to \({s}_{i}\), the \(i\)-th row of \(S\), our objective function can be simplified to
Here \(\tilde{Z}=Z-{X}^{D}DV-{X}^{P}PV\), and \({\tilde{Z}}_{i}\) is the vector of the \(i\)-th row of \(\tilde{Z}\). To solve the problem, random coordinate descent (RCD) with strong rules is employed, which was proposed in our previous study [89]. Since the subproblems defined for each row of \(S\) are independent, we update \({s}_{i}\) in parallel in practice.
Theoretically, we proved that the BCD proposed above guarantees to reduce our objective defined by Eq. 4 at each iteration and that it converges to the local optimum of the objective in a finite number of steps. In our empirical study on its convergence speed, scParser has a satisfactory convergence speed in analyzing scRNA-Seq data. Details on the proof and empirical study are provided (Additional file 1: section S6).
Optimize with the batch-fitting strategy
When the sample size of data is huge, optimizing scParser with the whole data is memory-demanding and computationally intensive. To relieve this issue, we propose a batch-fitting strategy to optimize scParser. Practically, we split the whole data into several batches and optimized our object function with one batch each time to lower the memory consumption and computation burden.
The key to perform batch optimization in scParser is to propose a surrogate that asymptotically converges to the solution to Eq. 4. As inspired by one previous study [90], we define the following surrogate for our objective
Here \(k\) is the number of batches, \({I}_{j}\) denotes the set of indices of cells from batch \(j\), and \({P}_{{I}_{j}},{D}_{{I}_{j}},{S}_{{I}_{j}}\) denote the latent representations for phenotypes, donors, and cells, respectively, that are obtained for batch \(j\). Technically, the surrogate for Eq. 6 is a subproblem of Eq. 10, so we focus on optimizing our objective defined by Eq. 10 and do not diverge to discuss the technical details in optimizing the surrogate for Eq. 6. Algorithm 1 below is proposed to optimize the surrogate defined by Eq. 10.

Algorithm 1 Batch scParser
In Algorithm 1, we noticed that \({A}_{k},{B}_{k},{E}_{k},{F}_{k}\) carry information for the same batch from all previous iterations. Actually, the information from early iterations is outdated. Mairal et al. [90] suggested removing the old information from the matrices to accelerate convergence. Owning to the design of our algorithm, we use the following equations to exploit this idea:
Here \(D^{\prime}_{k},P^{\prime}_{k},S^{\prime}_{k}\) are the matrices for donor, phenotype, and cell representations, respectively, for batch \(k\) at iteration \(t-1\). With a slight abuse of notation, \({\tilde{Z}^{\prime}}_{{I}_{k}}\) in the 2nd and 4th lines of the above equation is computed with the equations in lines 6 and 12 in Algorithm 1, respectively.
In practice, we carried out an additional experiment to show that scParser can handle the extreme case in which the data is divided by samples with another batch-fitting strategy, which has been incorporated into our software scParser (Additional file 1: section S4). In the experiment, the performance of batch scParser with random shuffling is slightly better compared to that of batch scParser with batch assigned according to the samples. Therefore, it is recommended to employ scParser with random batch assignment, as it is slightly more computationally efficient and less memory demanding than scParser with the by-sample batch assignment.
Initialization, hyperparameter tuning, and the stopping criteria
In scParser, all latent variables (\(P,V,S,G\)) except \(D\) are initiated from the normal distribution \(N\left(0,0.001\right)\), and \(D\) is initialized to a zero matrix. For initiating \({s}_{i}\) in Eq. 10 in optimization, we use the solution for \({s}_{i}\) from the previous iteration as a warm start.
For model selection in scParser, grid search is utilized to select hyperparameters \({\lambda }_{1},{\lambda }_{2},\alpha ,{K}_{1},{K}_{2}\). When the number of observations is huge (e.g., \(\ge\) 500,000), we randomly and evenly draw a small proportion (e.g., 0.1) of cells from scRNA-Seq samples as a dataset for model selection. Then, we randomly draw \(10\%\) of elements from the dataset as a test set. For each combination of candidate hyperparameters, we run alternating BCD several iterations (e.g., 20) and choose the one with the best performance in terms of root-mean-square error (RMSE) on the test set.
The robustness of scParser to the hyperparameters \({\lambda }_{1},{\lambda }_{2},\alpha\) has been demonstrated by additional experiments (Additional file 1: section S9). In the experiments, we observed that scParser is insensitive to \({\lambda }_{1}\) and favors \(\alpha =1\). Thus, we set \(\alpha\) equal to 1 to simplify model selection. Meanwhile, we found that our two hyperparameters \({K}_{1}\) and \({\lambda }_{1}\) are somewhat redundant since one can increase \({K}_{1}\) and \({\lambda }_{1}\) simultaneously without changing model complexity. Therefore, we set \({K}_{1}={K}_{2}\).
The detailed procedure for model selection is as follows. First, we set \({\lambda }_{1},{\lambda }_{2}\) to a small number (e.g., \(0.01\)) to avoid singularity in matrix inverse, \(\alpha\) is fixed to \(1\), and choose the ranks \({K}_{1}={K}_{2}\) from the sequence from 10 to 40 with step size 2 with grid search. After choosing the ranks, we define a broad parameter grid for \({\lambda }_{1},{\lambda }_{2}\) and perform a grid search. One may also refine the parameter grid based on the performance of the parameter grid on the test set. Finally, we select the parameters \({\lambda }_{1},{\lambda }_{2}\) with the best performance on the test set and run scParser with the selected parameters on the whole data matrix until the stopping criteria are met.
In our study, the stopping criteria are defined as
where \({\mathcal{L}}_{i}\left(\cdot \right)\) is the loss at iteration \(i\). To reduce computational burden, we calculated it every 10 iterations. \(\sigma\) is a predefined threshold and set to \({10}^{-7}\) in our experiments.
Cell clustering and cell type annotation
Practically, we implemented the clustering procedure in SCANPY [80] and Seurat [12] for all methods for a fair comparison. For scParser, we computed the neighborhood graph of cells using SCANPY [80] with the low-rank embeddings from scParser with the parameter n_neighbors set to 20 and default settings for other parameters and performed Louvain clustering [81] directly on the neighborhood graph. In the clustering, we considered the number of cell types provided by the original data to be the expected number of clusters. Note that the cell types provided by original studies are annotated by marker genes with differential expression analysis.
In searching for a desired number of clusters, we first defined two parameters, min_resolution (usually set to 0) and max_resolution (usually set to 1), and ran the Louvain clustering algorithm with the resolution parameter equal to max_resolution. If the number of clusters obtained equals the expected cluster number, we stop searching and report the clustering result for further analysis; otherwise, we update the resolution parameters according to the following strategy and perform Louvain clustering again with the updated max_resolution. If the number of clusters we obtain is greater than expected, we redefine the max_resolution to be (min_resolution+ max_resolution)/2; if the number of clusters we obtained is less than expected, we set the value of min_resolution to be max_resolution and double the value of max_resolution. We recursively adjust the resolution parameter of the Louvain clustering algorithm until the expected number of clusters is attained.
The cell types of clusters are annotated with the cell types provided by original studies. If there are multiple cell types for one cluster, the cell type of the cluster is determined by the major cell types. For cell type annotation visualization, we further embedded the graph in two dimensions using UMAP [82] and plotted the two-dimensional embeddings against cell types annotated by original studies.
Availability of data and materials
The software presented in the paper is publicly available on GitHub (https://github.com/kai0511/scParser.git) and the Zenodo repository [91] (https://doi.org/10.5281/zenodo.12743914). The software is distributed under the MIT open-source license [92]. In our applications, the raw scRNA-seq data for the T2DM dataset can be accessed via GSE101207 [93] and directly downloaded from https://www.ebi.ac.uk/gxa/sc/experiments/E-HCAD-31/downloads. The raw scRNA-seq data for the Smoking dataset can be accessed via GSE134174 [94] and also downloaded from https://www.ebi.ac.uk/gxa/sc/experiments/E-CURD-114/downloads. The raw scRNA-seq data for the COVID-19 dataset can be accessed via E-MTAB-9221 [95] and downloaded from https://www.ebi.ac.uk/gxa/sc/experiments/E-MTAB-9221/downloads. The processed scRNA-seq data for the Immune dataset [96] was downloaded at https://www.tissueimmunecellatlas.org. The raw scRNA-seq data for the GBM dataset can be downloaded from https://singlecell.broadinstitute.org/single_cell/study/SCP1985/ or via GSE182109. The raw scRNA-seq data for the iPSC dataset can be downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175634 or accessed via GSE175634. We considered gene expression of cells from three time points (days 7, 11, and 15) in our application to the iPSC dataset.
Change history
04 September 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13059-024-03378-5
References
Domínguez Conde C, Xu C, Jarvis LB, Rainbow DB, Wells SB, Gomes T, et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science. 2022;376(6594):eabl5197.
Fang Z, Weng C, Li H, Tao R, Mai W, Liu X, et al. Single-cell heterogeneity analysis and CRISPR screen identify key β-cell-specific disease genes. Cell Rep. 2019;26(11):3132,3144. e7.
Silvin A, Chapuis N, Dunsmore G, Goubet A, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182(6):1401,1418. e18.
Abdelfattah N, Kumar P, Wang C, Leu J, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun. 2022;13(1):767.
Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat Commun. 2020;11(1):1–21.
Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177(7):1873,1887. e17.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984,997. e24.
Polański K, Young MD, Miao Z, Meyer KB, Teichmann SA, Park J. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics. 2020;36(3):964–5.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–96.
Ming J, Lin Z, Zhao J, Wan X, Tabula Microcebus Consortium Consortium TTM, Ezran C, Liu S, Yang C, et al. FIRM: flexible integration of single-cell RNA-sequencing data for large-scale multi-tissue cell atlas datasets. Briefings in bioinformatics. 2022;23(5):bbac167.
Haghverdi L, Lun AT, Morgan MD, Marioni JC. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 2018;36(5):421–7.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Hie B, Bryson B, Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat Biotechnol. 2019;37(6):685–91.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888,1902. e21.
Qian K, Fu S, Li H, Li WV. scINSIGHT for interpreting single-cell gene expression from biologically heterogeneous data. Genome Biol. 2022;23(1):82.
Insulin.; 2024 [updated -05-15T00:45:19Z; Cited May 29, 2024. Available from: https://en.wikipedia.org/w/index.php?title=Insulin&oldid=1223895142.
Arystarkhova E, Liu YB, Salazar C, Stanojevic V, Clifford RJ, Kaplan JH, et al. Hyperplasia of pancreatic beta cells and improved glucose tolerance in mice deficient in the FXYD2 subunit of Na K-ATPase. J Biol Chem. 2013;288(10):7077–85.
Bearrows SC, Bauchle CJ, Becker M, Haldeman JM, Swaminathan S, Stephens SB. Chromogranin B regulates early-stage insulin granule trafficking from the Golgi in pancreatic islet β-cells. J Cell Sci. 2019;132(13):jcs231373.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9.
Ozcan U, Cao Q, Yilmaz E, Lee A, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–61.
Transcription factor Jun.; 2023 [updated -11-29T02:38:58Z; Cited May 29, 2024]. Available from: https://en.wikipedia.org/w/index.php?title=Transcription_factor_Jun&oldid=1187414653.
Protein c-Fos.; 2024 [updated -05-15T16:23:27Z; cited May 29, 2024]. Available from: https://en.wikipedia.org/w/index.php?title=Protein_c-Fos&oldid=1223992566.
Klymenko O, Huehn M, Wilhelm J, Wasnick R, Shalashova I, Ruppert C, et al. Regulation and role of the ER stress transcription factor CHOP in alveolar epithelial type-II cells. J Mol Med. 2019;97:973–90.
Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29(1):42–61.
Jagadeesh KA, Dey KK, Montoro DT, Mohan R, Gazal S, Engreitz JM, et al. Identifying disease-critical cell types and cellular processes by integrating single-cell RNA-sequencing and human genetics. Nat Genet. 2022;54(10):1479–92.
Zhang Y, Wang S, Xia H, Guo J, He K, Huang C, et al. Identification of monocytes associated with severe COVID-19 in the PBMCs of severely infected patients through single-cell transcriptome sequencing. Engineering. 2022;17:161–9.
Kvedaraite E, Hertwig L, Sinha I, Ponzetta A, Hed Myrberg I, Lourda M, et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc Natl Acad Sci. 2021;118(6):e2018587118.
Muhammad JS, ElGhazali G, Shafarin J, Mohammad MG, Abu-Qiyas A, Hamad M. SARS-CoV-2-induced hypomethylation of the ferritin heavy chain (FTH1) gene underlies serum hyperferritinemia in severe COVID-19 patients. Biochem Biophys Res Commun. 2022;631:138–45.
Yang X, Yu Y, Xu J, Shu H, Liu H, Wu Y, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–81.
Koscielny G, An P, Carvalho-Silva D, Cham JA, Fumis L, Gasparyan R, et al. Open Targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 2017;45(D1):D985-94.
André S, Picard M, Cezar R, Roux-Dalvai F, Alleaume-Butaux A, Soundaramourty C, et al. T cell apoptosis characterizes severe Covid-19 disease. Cell Death Differ. 2022;29(8):1486–99. https://doi.org/10.1038/s41418-022-00936-x.
Wu T, Li Y, Liu B, Zhang S, Wu L, Zhu X, et al. Expression of ferritin light chain (FTL) is elevated in glioblastoma, and FTL silencing inhibits glioblastoma cell proliferation via the GADD45/JNK pathway. PloS One. 2016;11(2):e0149361.
Ravi V, Madhankumar AB, Abraham T, Slagle-Webb B, Connor JR. Liposomal delivery of ferritin heavy chain 1 (FTH1) siRNA in patient xenograft derived glioblastoma initiating cells suggests different sensitivities to radiation and distinct survival mechanisms. PLoS One. 2019;14(9):e0221952.
Said HM, Hagemann C, Stojic J, Schoemig B, Vince GH, Flentje M, et al. GAPDH is not regulated in human glioblastoma under hypoxic conditions. BMC Mol Biol. 2007;8(1):1–13.
Zottel A, Novak M, Šamec N, Majc B, Colja S, Katrašnik M, et al. Anti-vimentin nanobody decreases glioblastoma cell invasion in vitro and in vivo. Cancers. 2023;15(3):573.
Dludla PV, Mabhida SE, Ziqubu K, Nkambule BB, Mazibuko-Mbeje SE, Hanser S, et al. Pancreatic β-cell dysfunction in type 2 diabetes: implications of inflammation and oxidative stress. World J Diabetes. 2023;14(3):130.
Retinol binding protein 4.; 2024 [updated -05-09T03:09:47Z; cited May 29, 2024]. Available from: https://en.wikipedia.org/w/index.php?title=Retinol_binding_protein_4&oldid=1222977796.
Proprotein convertase 1.; 2023 [updated -08-26T15:33:20Z; cited May 29, 2024]. Available from: https://en.wikipedia.org/w/index.php?title=Proprotein_convertase_1&oldid=1172358418.
Amylin.; 2024 [updated -05-06T04:41:36Z; cited May 29, 2024]. Available from: https://en.wikipedia.org/w/index.php?title=Amylin&oldid=1222474905.
Broch M, Vendrell J, Ricart W, Richart C, Fernández-Real J. Circulating retinol-binding protein-4, insulin sensitivity, insulin secretion, and insulin disposition index in obese and nonobese subjects. Diabetes Care. 2007;30(7):1802–6.
Boutari C, Pappas PD, Theodoridis TD, Vavilis D. Humanin and diabetes mellitus: a review of in vitro and in vivo studies. World J Diabetes. 2022;13(3):213.
Alsters SI, Goldstone AP, Buxton JL, Zekavati A, Sosinsky A, Yiorkas AM, et al. Truncating homozygous mutation of carboxypeptidase E (CPE) in a morbidly obese female with type 2 diabetes mellitus, intellectual disability and hypogonadotrophic hypogonadism. PloS One. 2015;10(6):e0131417.
Huang R, Bai X, Li X, Zhao L, Xia M. Retinol binding protein 4 impairs pancreatic beta-cell function, leading to the development of type 2 diabetes. Diabetes. 2018;67(Supplement_1):1826–P. https://doi.org/10.2337/db18-1826-P.
Flamez D, Roland I, Berton A, Kutlu B, Dufrane D, Beckers MC, et al. A genomic-based approach identifies FXYD domain containing ion transport regulator 2 (FXYD2) γa as a pancreatic beta cell-specific biomarker. Diabetologia. 2010;53:1372–83.
Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–13.
Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014;19(3):512–26.
Kilicarslan M, de Weijer BA, Sjödin KS, Aryal P, Ter Horst KW, Cakir H, et al. RBP4 increases lipolysis in human adipocytes and is associated with increased lipolysis and hepatic insulin resistance in obese women. FASEB J. 2020;34(5):6099.
Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436(7049):356–62.
Morton GJ, Schwartz MW. The NPY/AgRP neuron and energy homeostasis. Int J Obes. 2001;25(5):S56-62.
Loh K, Zhang L, Brandon A, Wang Q, Begg D, Qi Y, et al. Insulin controls food intake and energy balance via NPY neurons. Mol Metab. 2017;6(6):574–84.
Carreras-Badosa G, Remesar X, Prats-Puig A, Xargay-Torrent S, Lizarraga-Mollinedo E, de Zegher F, et al. Dlk1 expression relates to visceral fat expansion and insulin resistance in male and female rats with postnatal catch-up growth. Pediatr Res. 2019;86(2):195–201.
Traustadóttir GÁ, Lagoni LV, Ankerstjerne LBS, Bisgaard HC, Jensen CH, Andersen DC. The imprinted gene Delta like non-canonical Notch ligand 1 (Dlk1) is conserved in mammals, and serves a growth modulatory role during tissue development and regeneration through Notch dependent and independent mechanisms. Cytokine Growth Factor Rev. 2019;46:17–27.
Petry CJ, Burling KA, Barker P, Hughes IA, Ong KK, Dunger DB. Pregnancy serum DLK1 concentrations are associated with indices of insulin resistance and secretion. J Clin Endocrinol Metab. 2021;106(6):e2413-22.
Gomes LG, Cunha-Silva M, Crespo RP, Ramos CO, Montenegro LR, Canton A, et al. DLK1 is a novel link between reproduction and metabolism. J Clin Endocrinol Metab. 2019;104(6):2112–20.
Heni M, Haupt A, Schäfer SA, Ketterer C, Thamer C, Machicao F, et al. Association of obesity risk SNPs in PCSK1 with insulin sensitivity and proinsulin conversion. BMC Med Genet. 2010;11:1–8.
Leak TS, Keene KL, Langefeld CD, Gallagher CJ, Mychaleckyj JC, Freedman BI, et al. Association of the proprotein convertase subtilisin/kexin-type 2 (PCSK2) gene with type 2 diabetes in an African American population. Mol Genet Metab. 2007;92(1–2):145–50.
Matsuoka T, Artner I, Henderson E, Means A, Sander M, Stein R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci. 2004;101(9):2930–3.
Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. 2007;50:348–58.
Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25(12):4969–76.
Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, et al. Age-dependent pancreatic gene regulation reveals mechanisms governing human β cell function. Cell Metab. 2016;23(5):909–20.
Hachiya T, Komaki S, Hasegawa Y, Ohmomo H, Tanno K, Hozawa A, et al. Genome-wide meta-analysis in Japanese populations identifies novel variants at the TMC6–TMC8 and SIX3–SIX2 loci associated with HbA1c. Sci Rep. 2017;7(1):16147.
Spracklen CN, Horikoshi M, Kim YJ, Lin K, Bragg F, Moon S, et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature. 2020;582(7811):240–5.
Arden C, Hampson LJ, Huang GC, Shaw JA, Aldibbiat A, Holliman G, et al. A role for PFK-2/FBPase-2, as distinct from fructose 2, 6-bisphosphate, in regulation of insulin secretion in pancreatic β-cells. Biochem J. 2008;411(1):41–51.
Muller YL, Piaggi P, Hanson RL, Kobes S, Bhutta S, Abdussamad M, et al. A cis-eQTL in PFKFB2 is associated with diabetic nephropathy, adiposity and insulin secretion in American Indians. Hum Mol Genet. 2015;24(10):2985–96.
Harold KM, Matsuzaki S, Pranay A, Loveland BL, Batushansky A, Mendez Garcia MF, et al. Loss of cardiac PFKFB2 drives metabolic, functional, and electrophysiological remodeling in the heart. J Am Heart Assoc. 2024;13(7):e033676. https://doi.org/10.1161/JAHA.123.033676.
Solimena M, Schulte AM, Marselli L, Ehehalt F, Richter D, Kleeberg M, et al. Systems biology of the IMIDIA biobank from organ donors and pancreatectomised patients defines a novel transcriptomic signature of islets from individuals with type 2 diabetes. Diabetologia. 2018;61:641–57.
Miranda MA, Macias-Velasco JF, Lawson HA. Pancreatic β-cell heterogeneity in health and diabetes: classes, sources, and subtypes. Am J Physiol-Endocrinol Metab. 2021. https://doi.org/10.1152/ajpendo.00649.2020.
Tewari M, Wolf FW, Seldin MF, O’Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol (Baltimore, Md: 1950). 1995;154(4):1699–706.
Ramljak D, Vukoja M, Curlin M, Vukojevic K, Barbaric M, Glamoclija U, et al. Early response of CD8 T cells in COVID-19 patients. J Pers Med. 2021;11(12):1291.
Shin K, Jeon I, Kim B, Kim I, Park Y, Koh C, et al. Monocyte-derived dendritic cells dictate the memory differentiation of CD8 T cells during acute infection. Front Immunol. 2019;10:1887.
Chakarov S, Fazilleau N. Monocyte-derived dendritic cells promote T follicular helper cell differentiation. EMBO Mol Med. 2014;6(5):590–603.
Chu K, Batista NV, Girard M, Watts TH. Monocyte-derived cells in tissue-resident memory T cell formation. J Immunol. 2020;204(3):477–85.
Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl Sci Rev. 2020;7(6):998–1002.
Junqueira C, Crespo Â, Ranjbar S, De Lacerda LB, Lewandrowski M, Ingber J, et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature. 2022;606(7914):576–84.
Pulliam JR, van Schalkwyk C, Govender N, von Gottberg A, Cohen C, Groome MJ, et al. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science. 2022;376(6593):eabn4947.
Feng C, Shi J, Fan Q, Wang Y, Huang H, Chen F, et al. Protective humoral and cellular immune responses to SARS-CoV-2 persist up to 1 year after recovery. Nat Commun. 2021;12(1):4984.
Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181(7):1489,1501. e15.
Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177(7):1873,1887. e17.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27.
Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):1–5.
Blondel VD, Guillaume J, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech. 2008;2008(10):P10008.
Becht E, McInnes L, Healy J, Dutertre C, Kwok IW, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2019;37(1):38–44.
Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020;21(1):1–32.
Luecken MD, Büttner M, Chaichoompu K, Danese A, Interlandi M, Müller MF, et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods. 2022;19(1):41–50.
Maier-Hein L, Eisenmann M, Reinke A, Onogur S, Stankovic M, Scholz P, et al. Why rankings of biomedical image analysis competitions should be interpreted with care. Nat Commun. 2018;9(1):5217.
Elorbany R, Popp JM, Rhodes K, Strober BJ, Barr K, Qi G, et al. Single-cell sequencing reveals lineage-specific dynamic genetic regulation of gene expression during human cardiomyocyte differentiation. PLoS Genet. 2022;18(1):e1009666.
Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, et al. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci. 2015;18(5):637–46.
Lee H, Battle A, Raina R, Ng A. Efficient sparse coding algorithms. Adv Neural Inform Process Syst. 2006;19:801–8. https://dl.acm.org/doi/10.5555/2976456.2976557.
Zhao K, Huang S, Lin C, Sham PC, So H, Lin Z. INSIDER: interpretable sparse matrix decomposition for RNA expression data analysis. Plos Genet. 2024;20(3):e1011189.
Mairal J, Bach F, Ponce J, Sapiro G. Online dictionary learning for sparse coding. Proceedings of the 26th annual international conference on machine learning; ; 2009.
KAI, ZHAO, HonCheong, SO, Zhixiang LIN. scParser: sparse representation learning for scalable single-cell RNA sequencing data analysis. Zenodo. 2024. https://doi.org/10.5281/zenodo.12743914.
HonCheong SO KAI ZHAO Zhixiang LIN. scParser: sparse representation learning for scalable single-cell RNA sequencing data analysis. Github. 2024. https://github.com/kai0511/scParser.git.
Zhou Fang, Chen Weng, Haiyan Li, et al. Single-cell heterogeneity analysis and CRISPR screen identify key β-cell-specific disease genes. Datasets. Gene Expression Omnibus. 2019. https://www.ebi.ac.uk/gxa/sc/experiments/E-HCAD-31/downloads.
Katherine C. Goldfarbmuren, Nathan D. Jackson, Satria P. Sajuthi, et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Datasets. Gene Expression Omnibus. 2020. https://www.ebi.ac.uk/gxa/sc/experiments/E-MTAB-9221/downloads.
Aymeric Silvin, Nicolas Chapuis, Garett Dunsmore, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Datasets. Gene Expression Omnibus. 2020. https://www.ebi.ac.uk/gxa/sc/experiments/E-MTAB-9221/downloads.
C. Domínguez Conde, C. Xu, L. B. Jarvis, et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Datasets. Gene Expression Omnibus. 2022. https://www.tissueimmunecellatlas.org.
Brunet J, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci. 2004;101(12):4164–9.
Liang W, Fang J, Zhou S, Hu W, Yang Z, Li Z, et al. The role of ubiquitin-specific peptidases in glioma progression. Biomed Pharmacother. 2022;146:112585.
Lin Y, Liao K, Miao Y, Qian Z, Fang Z, Yang X, et al. Role of asparagine endopeptidase in mediating wild-type p53 inactivation of glioblastoma. JNCI. 2020;112(4):343–55.
Zhang Z, Sun H, Mariappan R, Chen X, Chen X, Jain MS, et al. scMoMaT jointly performs single cell mosaic integration and multi-modal bio-marker detection. Nat Commun. 2023;14(1):384.
Gao C, Liu J, Kriebel AR, Preissl S, Luo C, Castanon R, et al. Iterative single-cell multi-omic integration using online learning. Nat Biotechnol. 2021;39(8):1000–7.
McGrath BT, Tsan YC, Salvi S, Ghali N, Martin DM, Hannibal M, et al. Aberrant extracellular matrix and cardiac development in models lacking the PR-DUB component ASXL3. bioRxiv. 2022:2022.07.14.500124. https://doi.org/10.1101/2022.07.14.500124.
Kiewitz R, Lyons GE, Schäfer BW, Heizmann CW. Transcriptional regulation of S100A1 and expression during mouse heart development. Biochimica et Biophysica Acta (BBA)-Mol Cell Res. 2000;1498(2–3):207–19.
Reboll MR, Korf-Klingebiel M, Klede S, Polten F, Brinkmann E, Reimann I, et al. EMC10 (endoplasmic reticulum membrane protein complex subunit 10) is a bone marrow–derived angiogenic growth factor promoting tissue repair after myocardial infarction. Circulation. 2017;136(19):1809–23.
Albelda SM. Endothelial and epithelial cell adhesion molecules. Am J Respir Cell Mol Biol. 1991;4(3):195–203.
Tibshirani R, Bien J, Friedman J, Hastie T, Simon N, Taylor J, et al. Strong rules for discarding predictors in lasso-type problems. J Royal Stat Soc Series B. 2012;74(2):245–66.
Carboxypeptidase E.; 2024 [updated -01-13T17:57:37Z; Cited May 29, 2024]. Available from: https://en.wikipedia.org/w/index.php?title=Carboxypeptidase_E&oldid=1195397704.
Pancreatic beta cell mass biomarker. Merck and Co Inc, assignee. JP. https://patents.google.com/patent/JP2011522224A/en.
Moreno-Navarrete JM, Novelle MG, Catalán V, Ortega F, Moreno M, Gomez-Ambrosi J, et al. Insulin resistance modulates iron-related proteins in adipose tissue. Diabetes Care. 2014;37(4):1092–100.
Suckale J, Solimena M. The insulin secretory granule as a signaling hub. Trends Endocrinol Metab. 2010;21(10):599–609.
Bearrows SC, Bauchle CJ, Becker M, Haldeman JM, Swaminathan S, Stephens SB. Chromogranin B regulates early-stage insulin granule trafficking from the Golgi in pancreatic islet β-cells. J Cell Sci. 2019;132(13):jcs231373.
Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47(W1):W212-24.
Bohuslavova R, Fabriciova V, Lebrón-Mora L, Malfatti J, Smolik O, Valihrach L, et al. ISL1 controls pancreatic alpha cell fate and beta cell maturation. Cell Biosci. 2023;13(1):53.
Gosmain Y, Katz LS, Masson MH, Cheyssac C, Poisson C, Philippe J. Pax6 is crucial for β-cell function, insulin biosynthesis, and glucose-induced insulin secretion. Mol Endocrinol. 2012;26(4):696–709.
Ahlqvist E, Turrini F, Lang ST, Taneera J, Zhou Y, Almgren P, et al. A common variant upstream of the PAX6 gene influences islet function in man. Diabetologia. 2012;55:94–104.
Cho YS, Chen C, Hu C, Long J, Hee Ong RT, Sim X, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2012;44(1):67–72.
Scavuzzo MA, Hill MC, Chmielowiec J, Yang D, Teaw J, Sheng K, et al. Endocrine lineage biases arise in temporally distinct endocrine progenitors during pancreatic morphogenesis. Nat Commun. 2018;9(1):3356.
Sen P, Kaur H. In Silico transcriptional analysis of asymptomatic and severe COVID-19 patients reveals the susceptibility of severe patients to other comorbidities and non-viral pathological conditions. Hum Gene. 2023;35:201135.
Gurshaney S, Morales-Alvarez A, Ezhakunnel K, Manalo A, Huynh T, Abe J, et al. Metabolic dysregulation impairs lymphocyte function during severe SARS-CoV-2 infection. Commun Biol. 2023;6(1):374.
Simón-Fuentes M, Ríos I, Herrero C, Lasala F, Labiod N, Luczkowiak J, et al. MAFB shapes human monocyte–derived macrophage response to SARS-CoV-2 and controls severe COVID-19 biomarker expression. JCI Insight. 2023;8(24):e172862. https://doi.org/10.1172/jci.insight.172862.
Zhang Y, Li H, Zeng T, Chen L, Li Z, Huang T, et al. Identifying transcriptomic signatures and rules for SARS-CoV-2 infection. Front Cell Dev Biol. 2021;8:627302.
Ziegler CG, Miao VN, Owings AH, Navia AW, Tang Y, Bromley JD, et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell. 2021;184(18):4713,4733. e22.
Soto ME, Fuentevilla-Álvarez G, Palacios-Chavarría A, Vázquez RRV, Herrera-Bello H, Moreno-Castañeda L, et al. Impact on the clinical evolution of patients with COVID-19 pneumonia and the participation of the NFE2L2/KEAP1 polymorphisms in regulating SARS-CoV-2 infection. Int J Mol Sci. 2022;24(1):415.
Wu X, Liu K, Li S, Ren W, Wang W, Shang Y, et al. Integrated bioinformatics analysis of dendritic cells hub genes reveal potential early tuberculosis diagnostic markers. BMC Med Genom. 2023;16(1):214.
Kalfaoglu B, Almeida-Santos J, Satou Y, Ono M. T-cell hyperactivation and paralysis in severe COVID-19 infection revealed by single-cell analysis. Front Immunol. 2020;11:589380.
Tang H, Wei P, Duell EJ, Risch HA, Olson SH, Bueno-de-Mesquita HB, et al. Axonal guidance signaling pathway interacting with smoking in modifying the risk of pancreatic cancer: a gene-and pathway-based interaction analysis of GWAS data. Carcinogenesis. 2014;35(5):1039–45.
Nimmakayala RK, Seshacharyulu P, Lakshmanan I, Rachagani S, Chugh S, Karmakar S, et al. Cigarette smoke induces stem cell features of pancreatic cancer cells via PAF1. Gastroenterology. 2018;155(3):892,908. e6.
Elangovan IM, Vaz M, Tamatam CR, Potteti HR, Reddy NM, Reddy SP. FOSL1 promotes Kras-induced lung cancer through amphiregulin and cell survival gene regulation. Am J Respir Cell Mol Biol. 2018;58(5):625–35.
Martos SN, Campbell MR, Lozoya OA, Wang X, Bennett BD, Thompson IJ, et al. Single-cell analyses identify dysfunctional CD16 CD8 T cells in smokers. Cell Rep Med. 2020;1(4). https://doi.org/10.1016/j.xcrm.2020.100054.
Hoang TT, Lee Y, McCartney DL, Kersten ET, Page CM, Hulls PM, et al. Comprehensive evaluation of smoking exposures and their interactions on DNA methylation. EBioMedicine. 2024;100. https://doi.org/10.1016/j.ebiom.2023.104956.
Zhang S, Zhao S, Qi Y, Li B, Wang H, Pan Z, et al. SPI1-induced downregulation of FTO promotes GBM progression by regulating pri-miR-10a processing in an m6A-dependent manner. Mol Ther-Nucleic Acids. 2022;27:699–717.
Lei J, Zhou M, Zhang F, Wu K, Liu S, Niu H. Interferon regulatory factor transcript levels correlate with clinical outcomes in human glioma. Aging (Albany NY). 2021;13(8):12086.
Kosti A, Chiou J, Guardia GD, Lei X, Balinda H, Landry T, et al. ELF4 is a critical component of a miRNA-transcription factor network and is a bridge regulator of glioblastoma receptor signaling and lipid dynamics. Neuro-oncology. 2023;25(3):459–70.
Bozdag S, Li A, Riddick G, Kotliarov Y, Baysan M, Iwamoto FM, et al. Age-specific signatures of glioblastoma at the genomic, genetic, and epigenetic levels. PloS One. 2013;8(4):e62982.
Greenwood HC, Bloom SR, Murphy KG. Peptides and their potential role in the treatment of diabetes and obesity. Rev Diabetic Stud. 2011;8(3):355–68. https://doi.org/10.1900/RDS.2011.8.355.
Brandt SJ, Götz A, Tschöp MH, Müller TD. Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides. 2018;100:190–201.
Kenche H, Baty CJ, Vedagiri K, Shapiro SD, Blumental-Perry A. Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J. 2013;27(3):965–77.
Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS One. 2008;3(2):e1651.
Stein-O’Brien GL, Arora R, Culhane AC, Favorov AV, Garmire LX, Greene CS, et al. Enter the matrix: factorization uncovers knowledge from omics. Trends Genet. 2018;34(10):790-805.
Acknowledgements
We thank our colleague, Prof. Ben DAI, for the helpful discussions.
Funding
This work has been supported by the Chinese University of Hong Kong startup grant (4930181), the Chinese University of Hong Kong Science Faculty’s Direct Grant for Research 2022/2023, the Chinese University of Hong Kong Science Faculty’s Collaborative Research Impact Matching Scheme (CRIMS 4620033), the Hong Kong Research Grant Council (GRF 14301120, GRF 14300923), the KIZ-CUHK Joint Laboratory of Bioresources and Molecular Research of Common Diseases, Kunming Institute of Zoology and The Chinese University of Hong Kong, China, Hong Kong Branch of the Chinese Academy of Sciences Center for Excellence in Animal Evolution and Genetics, The Chinese University of Hong Kong, and the Lo Kwee Seong Biomedical Research Fund from The Chinese University of Hong Kong.
Author information
Authors and Affiliations
Contributions
ZK, S HC, and L ZX conceived the study and designed the experiments. ZK was responsible for the software implementation and applications. ZK wrote the paper. ZK, S HC, and L ZX revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Peer review information
Andrew Cosgrove was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Review history
The review history is available as Additional file 3.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Erroneous symbols were corrected in equation 3, 4 and 10.
Supplementary Information
13059_2024_3345_MOESM2_ESM.xlsx
Additional file 2: Tables S1–S2. It provides the distribution of proportions of cell populations across diabetes status, and the p-values for two-sidedt-tests to compare the expression levels of the eight genes associated with T2DM between normal controls and diabetes across all six cell populations identified by scParser in its application to the T2DM dataset.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, K., So, HC. & Lin, Z. scParser: sparse representation learning for scalable single-cell RNA sequencing data analysis. Genome Biol 25, 223 (2024). https://doi.org/10.1186/s13059-024-03345-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-024-03345-0