
Abstract
Background
Pubertal growth patterns correlate with future health outcomes. However, the genetic mechanisms mediating growth trajectories remain largely unknown. Here, we modeled longitudinal height growth with Super-Imposition by Translation And Rotation (SITAR) growth curve analysis on ~ 56,000 trans-ancestry samples with repeated height measurements from age 5 years to adulthood. We performed genetic analysis on six phenotypes representing the magnitude, timing, and intensity of the pubertal growth spurt. To investigate the lifelong impact of genetic variants associated with pubertal growth trajectories, we performed genetic correlation analyses and phenome-wide association studies in the Penn Medicine BioBank and the UK Biobank.
Results
Large-scale growth modeling enables an unprecedented view of adolescent growth across contemporary and 20th-century pediatric cohorts. We identify 26 genome-wide significant loci and leverage trans-ancestry data to perform fine-mapping. Our data reveals genetic relationships between pediatric height growth and health across the life course, with different growth trajectories correlated with different outcomes. For instance, a faster tempo of pubertal growth correlates with higher bone mineral density, HOMA-IR, fasting insulin, type 2 diabetes, and lung cancer, whereas being taller at early puberty, taller across puberty, and having quicker pubertal growth were associated with higher risk for atrial fibrillation.
Conclusion
We report novel genetic associations with the tempo of pubertal growth and find that genetic determinants of growth are correlated with reproductive, glycemic, respiratory, and cardiac traits in adulthood. These results aid in identifying specific growth trajectories impacting lifelong health and show that there may not be a single “optimal” pubertal growth pattern.
Similar content being viewed by others
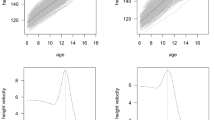

Introduction
A distinct feature of human growth is the pubertal growth spurt, characterized by accelerated height growth. The characteristics of the growth spurt can differ substantially even among healthy children [1]. The timing of the onset of the pubertal growth spurt, the total amount of growth, and the duration of growth vary and are influenced by both genetic and environmental factors [2]. For instance, twin studies place the heritability of height growth during adolescence at about 75% [2, 3]. Furthermore, the secular trend of advancing pubertal timing (i.e., pubertal onset beginning at a younger age) in girls over the twentieth century has also been observed in boys by measuring peak height growth velocity (PHV) [3], the amount of height gained during the most rapid phase of height growth during puberty.
The timing and duration of the pubertal growth spurt affect the attainment of an individual’s final height, which is associated with, and a causal factor for, many adult health outcomes [4]. Thus, it is important to establish whether specific growth trajectory features impact these health risks. Indeed, epidemiological or observational studies have shown evidence that growth patterns during childhood are associated with health outcomes later in life, including adverse cardiovascular health [5], cancer [6, 7], bone outcomes such as lower bone mineral density in later life—a risk factor for osteoporosis [8, 9], type 2 diabetes [10,11,12], and respiratory health [13].
As noted above, variation in pubertal growth is highly heritable [2], but the specific genetic factors underlying pubertal growth trajectories remain largely unknown. In our previous study, we identified 10 genome-wide significant loci [14] using relatively simple phenotypes targeting the take-off phase of the growth spurt (the age in mid-childhood when growth velocity begins rising after falling since infancy), the total amount of pubertal growth, and the amount of late pubertal growth, which roughly marks the timing of PHV. The identified genetic signals influencing pubertal growth were also associated with pubertal timing, adiposity, and height growth potential. Half of the signals (5 out of 10) impacted height growth evenly across childhood and pubertal growth, while the other half were pubertal growth-specific, supporting the idea that some genetic factors contribute only during the growth spurt [2, 15,16,17]. We have also shown previously that independent signals (rs7759938 and rs314277) at one genetic locus (near LIN28B) can influence either postnatal growth from birth until adulthood or specifically pubertal growth [18], with the pubertal growth signal also being the strongest pubertal timing association in GWAS of age at menarche [19]. These signals have distinct associations with adult adiposity-related traits, with the puberty-associated signal also associated with adult height but not adiposity, whereas a second adult height-increasing signal also associated with increased weight and hip circumference [20].
To better capture the dynamic nature of the pubertal growth spurt, we expanded our previous work by utilizing longitudinal modeling of repeated height measurements across childhood and adolescence. Additionally, in contrast to our previous work, where we only included participants of European ancestry, we now include subjects from diverse ancestral backgrounds. Next, we uncovered genetic variants associated with longitudinal height growth and assessed the genetic associations between pubertal growth trajectories and later-life health traits.
Results
Modeled peak height velocity and age at peak height velocity across cohorts
We performed Super-Imposition by Translation And Rotation (SITAR) growth curve analysis [21] in up to 56,659 samples (53.3% female; 41,468 European, 7852 African American, 2714 Asian, 2387 Native American, and 2238 Hispanic; Additional file 1: Table S1), including height measurements between age 5 and 20 years. SITAR estimates three random effects comparing each individual’s growth curve to the population mean, as well as predicted age at peak height velocity (APHV) and peak height velocity (PHV) for each overall population. Note that PHV in SITAR is the instantaneous peak velocity, which is greater than the peak annual velocity. We performed SITAR modeling separately in females and males separately in each contributing cohort. Overall, APHV across cohorts was consistent with the average ages reported previously (11.5 years in girls and 13.5 years in boys [22]), and non-European cohorts had earlier APHV than Europeans. There was a significant linear relationship between APHV (in years) and PHV (in cm/year) largely explained by sex (sex-adjusted P = 1.2 × 10−15; Fig. 1).

Mean peak height velocity (cm/year) vs. mean age at peak height velocity (years) for cohorts of diverse ancestral backgrounds. Yellow and purple lines represent a linear model fit to APHV ~ PHV × sex. The shaded bars represent the standard deviation of the linear regression. The samples included 19 cohorts of European ancestry, 3 African American cohorts, 2 East Asian cohorts, and 5 American Native or Hispanic cohorts
Genome-wide association
We performed a series of genome-wide association study (GWAS) analyses on six phenotypes: three simple height or height difference phenotypes, as previously assessed [14], and the three SITAR fixed effects (Fig. 2): (I) the take-off phase of the pubertal growth spurt (height at age 10 in girls and 12 in boys); (II) total pubertal growth, between ages 8 and adult (> 19 years); and (III) late pubertal growth, between ages 14 and adult (> 19 years). Additionally, we included three phenotypes derived from SITAR longitudinal modeling: (IV) a-size; (V) b-timing; and (VI) c-intensity. We ran ancestry-specific meta-analyses in the European, African American, Asian, and Native American/Hispanic ancestry groups, as well as a trans-ancestry meta-analysis followed by credible set analysis (Table 1, Additional file 1: Table S2). Twin studies suggest that height growth is most heritable during adolescence (up to 0.83 in boys and 0.76 in girls) [2, 23]; using Linkage Disequilibrium Score Regression (LDSC) [24], we calculated the SNP-heritability (h2SNP) of the six phenotypes and found five to have a genetic component: 10F/12 M = 0.32 (SE = 0.02); 8–adult = 0.22 (0.03); 14–adult = 0.21 (0.04); a-size = 0.15 (0.02); c-intensity = 0.13 (0.02); Additional file 1: Table S3).

Six phenotypes assessed by GWAS. The top panel shows a typical growth curve for boys (blue) and girls (red), with age (years) on the x-axis and height gain (cm/year) on the y-axis. First, we included three simple height or height-difference phenotypes as previously assessed [14]: (I) The take-off phase of the pubertal growth spurt (height at age 10 in girls and 12 in boys); (II) total pubertal growth, between ages 8 and adult; and (III) late pubertal growth, between ages 14 and adult. Additionally, we included three phenotypes derived from SITAR longitudinal modeling: (IV) a-size; (V) b-timing; and (VI) c-intensity. The black line represents the mean population growth curve for a cohort (by sex). Each individual gets a random effect for the three parameters; for example, if a subject is taller than their peers (upper red line), they get a positive value for a-size, while a shorter individual (lower line in panel IV) gets a negative value. A subject who enters their growth spurt earlier than the mean (left line in panel V) gets a negative value for b-timing, while a subject growing later (right line in panel V) gets a positive value. Finally, a subject who grows faster than the population mean (steeper line in panel VI) receives a positive value for c-intensity, while a subject growing slower (shallower line in panel VI) gets a negative value for c-intensity
Twenty-six loci achieved genome-wide significance (P < 5 × 10−8) in the trans-ancestral meta-analysis (Table 1). Four loci were significant for both height at 10F/12 M and a-size, and we noted that the LIN28B locus was associated with height difference age 14–adult, b-timing, and c-intensity. We then performed credible set analyses, and for 10 signals we reduced the 95% credible set to fewer than 10 SNPs (Additional file 1: Table S2). For three loci, the credible set was distilled down to a single SNP. Three example regional plots are shown in Additional file 2: Fig. S1.
We subsequently extracted all associations with other traits from previously published GWAS for these loci using PhenoScanner [25] (Additional file 1: Tables S4 and S5). All but three signals ((1) nearest to ZNF365, (2) nearest to the gene for hemoglobin beta, HBB, and (3) nearest to LINC00520) were genome-wide significantly associated with adult height, and all but four signals (the three above as well as at LIN28B) were associated with the UK Biobank phenotype “comparative height at age 10.” We then clustered the genome-wide significant loci based on their associations with the three SITAR parameters (Additional file 2: Fig. S2A) and found that most loci behaved similarly, again with the exception of the LIN28B and HBB loci. We observed that the LIN28B signal was associated with the timing and velocity of the pubertal growth spurt, as well as late pubertal growth, with a resulting impact on adult stature, but there was no effect on growth prior to puberty [14, 18].
The other outlying locus, near HBB, was associated with delayed b-timing (rs16911905, PTrans-Ancestry = 1.32 × 10−9), but there was no evidence for association with childhood or adult stature based on previously published GWAS efforts. Investigating further, we found that the only cohort showing association was in the Children’s Hospital of Philadelphia African American cohort. The strongest association was at rs334 (beta (SE) = 0.342 (0.03), P = 5.80 × 10−27) (Additional file 2: Fig. S2B). rs334 is associated with sickle cell anemia, and children with sickle cell anemia have decreased pubertal growth velocity [26]. rs334 is only common in African populations, in which sickle cell anemia is most prevalent, so it did not pass quality control for the trans-ancestral analysis. When we conditioned for sickle cell anemia in the Children’s Hospital of Philadelphia African American cohort, we found that the signal was almost completely ablated (P = 0.03) (Additional file 2: Fig. S2C). Thus, this analysis likely picked up an association driven by children with sickle cell anemia in this cohort, who display reduced pubertal growth.
Prioritized genes
Most GWAS signals are intergenic. Thus, we combined several approaches to identify the most likely effector gene for each locus. We looked for skeletal and body size aberrations in human Mendelian disease using OMIM and in mice knockout experiments (JAX, IMPC) and annotated the genes at each locus with Gene Ontology biological process terms. Altogether, we identified likely causal genes at 23 loci (Additional file 1: Table S6). These include several genes that result in severe ossification and/or skeletal abnormalities when dysregulated in mice, such as PRKG2, ARID5B, SOCS2, SMAD3, PKD1, and GDF5; of these, rare mutations in PRKG2 [27, 28], SMAD3 [29,30,31], and GDF5 [32,33,34] are also associated with rare Mendelian disorders in humans with skeletal phenotypes. GO-term-based pathway analysis revealed a significant enrichment of height at 10F/12 M with “regulation of chondrocyte differentiation” (Bonferroni-corrected P = 0.006).
Genetic relationship between pubertal growth and anthropometric traits
Next, we explored the genetic correlation between pubertal growth traits and anthropometric and health-related outcomes in the European ancestry-specific results. The relationship between pubertal timing (e.g., its onset) and intensity (e.g., the speed of progression through puberty) remains controversial, with studies reporting discrepant findings [3, 4]; here, we found a negative genetic correlation between the SITAR-derived b-timing and c-intensity parameters (rg = − 0.12; Additional file 1: Table S7; Additional file 2: Fig. S3), as well as a highly significant negative genetic correlation between c-intensity and age at menarche (rg = − 0.52, P = 6.72 × 10−23), indicating that genetic determinants of later pubertal onset also favor a slower tempo of growth.
We then investigated anthropometric traits across the life course. In particular, the relationship between pubertal growth and BMI has been unclear. Here, c-intensity was not genetically correlated with childhood obesity or adult BMI (Fig. 3A), whereas taller height at 10/12 years was genetically correlated with higher childhood and adult BMI. Furthermore, more growth 8-to-adult and 14-to-adult were negatively genetically correlated with childhood and adult BMI. C-intensity was, however, positively genetically correlated with adult waist circumference, meaning that faster pubertal growth tempo correlated with larger adult waist circumference, as did taller a-size and height at 10F/12 M.

Genetic correlation (rg) between pubertal growth and anthropometric traits. A Correlation with adiposity traits. B Correlation with body size traits. 10F/12 M, height at age 10 years in girls and age 12 years in boys; 14–adult, height difference between age 14 years and adult; 8–adult, height difference between age 8 years and adult; a-size, SITAR-derived height across the growth trajectory; c-intensity, SITAR-derived tempo of the pubertal growth spurt
We observed a strong positive genetic correlation between c-intensity and adult height (rg = 0.54, P = 1.12 × 10−23), as well as between the other pubertal growth parameters and height across the life course (Fig. 3B) [23, 35]. Additionally, while b-timing would be expected to be correlated with the timing of puberty (not assessed here due to low heritability), we found that taller height at 10F/12 M, and higher c-intensity of the growth spurt, were genetically correlated with earlier age at menarche. Meanwhile, later age at menarche was genetically correlated with more growth from 8–adult and 14–adult, which is expected since adolescents who develop later have a longer period of growth.
Relationship between the genetics of pubertal growth and later-life health outcomes
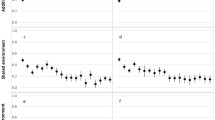
Variation in growth during puberty is known to be associated with later-life health outcomes [5, 6, 8,9,10,11,12,13]. Here, analysis of both the genome-wide significant signals and genome-wide genetic correlations pointed towards pleiotropy between pubertal growth and health outcomes. With LDSC, which compares genome-wide association data across the genome between pairs of traits, we observed genetic correlations with bone mineral density, cardiovascular traits such as atrial fibrillation and coronary artery disease, glycemic traits, lung function and lung cancer, neurological, psychiatric, and intelligence traits, and overall well-being (Fig. 4).

Genetic correlation (rg) of pubertal growth with adult health outcomes. A Bone and glycemic outcomes, including femoral neck and lumbar spine bone mineral density and three glycemic traits. B Lung cancer and lung function outcomes. C Cardiac and lipid outcomes. D Neuropsychiatric outcomes and overall wellbeing (parents’ age at death). LDL, low-density lipoprotein; HDL, high-density lipoprotein; HOMA-IR, homeostatic model assessment for insulin resistance; PEF, peak expiratory flow; FVC, forced vital capacity; FEV1, forced exhalation volume in 1 s; ALS, Amyotrophic lateral sclerosis. 10F/12 M, height at age 10 years in girls and age 12 years in boys; 14–adult, height difference between age 14 years and adult; 8–adult, height difference between age 8 years and adult; a-size, SITAR-derived height across the growth trajectory; c-intensity, SITAR-derived tempo of the pubertal growth spurt
With evidence of a genetic relationship between pubertal growth and later health outcomes, we next generated polygenic risk scores (PRS) from sets of genetic variants (see the “Methods” section, Additional file 1: Table S8) and performed phenome-wide association (PheWAS) scans on individual-level data in the Penn Medicine Biobank (PMBB) and the UK Biobank. In the PMBB, no phenotypes passed Bonferroni multiple testing correction. However, 140 traits showed a nominal association (P < 0.05, Additional file 1: Table S9). We performed validation analyses in the UK Biobank, selecting 37 traits that demonstrated either significant LDSC-based genetic correlations or nominally significant associations in the PMBB. Although the proportions of phenotypic variance explained by the PRS were small, three phenotypes remained significantly associated with one or more PRS after multiple test correction in the UK Biobank: atrial fibrillation, type 2 diabetes, and adult BMI (Table 2, Additional file 1: Table S10). Notably, additional cardiac traits were nominally associated with the PRS.
Discussion
In this study, we assessed children and adolescents of multiple ancestries with repeated height measurements and genetic data to gain a better understanding of how pubertal height growth relates to health across the life course. First, we performed longitudinal modeling by sex and ancestry with SITAR, a validated method for producing more precise and less biased estimates of APHV and PHV [36, 37]. Previously, SITAR has been used to estimate these parameters in individual cohorts [38, 39]. Here, we compared these estimates across cohort studies of different ancestral backgrounds collected over a range of years, from the 1930s to the present day. Studies of growth in healthy children have reported a mean APHV at around 11.5 years in girls and 13.5 years in boys [22]. Overall, the SITAR-modeled APHV values are consistent, with variation in mean age by ancestral background (non-European earlier than European), sex (females earlier than males), and the era of cohort collection (contemporary cohorts earlier than those collected decades ago).
The relationship between pubertal timing and intensity is controversial, with studies reaching various conclusions. For example, Marceau et al. [40] found a relationship between timing and intensity for boys, but not girls, whereas German et al. [41] observed a negative correlation between pubertal onset and progression in girls. Our genetic correlation results support the latter, with a strong negative correlation between age at menarche and c-intensity. However, in Fig. 1, we see a weaker positive correlation between APHV and PHV. Additionally, higher childhood BMI is an established risk factor for earlier pubertal onset [42], but its relationship with the intensity of puberty is less clear than with pubertal timing. German et al. found that higher childhood BMI did not correlate with intensity, but instead with earlier pubertal timing. Our results corroborate these findings.
For final adult height, our findings diverge from previous studies that have found that the timing or intensity of pubertal development is unrelated to final stature [43, 44]. These previous findings could be due to small sample sizes. Here, with longitudinal data on > 50,000 adolescents, we show that genetic determinants of pubertal growth not only impact final stature, but measures of body size from birth to adulthood.
Utilizing genetic data, we performed multi-ancestry GWAS of pubertal growth. While the loci we identified are mostly known height loci, we report an association with the tempo (c-intensity) of pubertal growth at LIN28B, adding to its known associations with pubertal timing and childhood growth trajectories [14, 18].
While epidemiological studies have observed relationships between puberty and adult health outcomes, our large sample size and genetic data allowed us to explore genetic relationships using genetic correlation analyses and PRS in two large biobanks (PMBB and UKBiobank). Our results support genetic relationships between pubertal growth and a range of adult-health traits. For instance, previous studies have identified later puberty and slower growth velocity as risk factors for later-life decline in bone density [9] and increased fracture risk [8]. Here, we observed a similar genetic relationship between slower c-intensity of growth and lower adult bone mineral density. For glycemic traits, previous studies showed that accelerated childhood growth associated with increased type 2 diabetes risk [10, 11], which corroborates with our observation of a positive correlation between c-intensity and type 2 diabetes.
The most consistent relationship was that of pubertal growth with cardiac traits. Shorter adult stature and less growth from age 7 to 13 years are risk factors for coronary heart disease [45], and we observed a similar genetic correlation between diminished growth 8–adult and 14–adult and coronary heart disease. To the contrary, greater pubertal growth was positively correlated with atrial fibrillation, flutter, and dysrhythmias. Both birthweight [46] and adult height [4] are known causal risk factors for atrial fibrillation, and several studies show relationships between increasing height and atrial fibrillation incidence [47,48,49,50]. These findings support the idea that body height has a lifelong impact on atrial fibrillation risk.
Our study does have some limitations. In our data, the b-timing parameter was not heritable, although the timing of puberty is well-established as a heritable trait; thus, we were unable to perform adult heath genetic correlation analyses with this trait. This could be due to our use of age rather than log(age) in the SITAR modeling, but we do detect well-established loci associated with pubertal timing despite the low heritability; in the future, studies running SITAR modeling with log(age) may provide more accurate estimates for b-timing. Furthermore, we did not study gene by environment interactions, which could be important given that pubertal timing is strongly affected by environmental factors; this analysis was outside the scope of the current study Additionally, not all cohorts were able to collect height data annually or bi-annually; thus, estimation of APHV in these cohorts may not be as precise. However, annual height measurements have been shown to estimate APHV as accurately as more frequent measurements [51]. A fuller understanding of the complex relationship between the genetic variation that impacts pubertal timing and adult health outcomes would be achieved by adding other factors (e.g., cumulative estimates of environmental risk factor exposure) to statistical models, which could be pursued in future investigations. Finally, the sample sizes for some of the ancestry groups remain small. In the future, we hope additional non-European datasets with longitudinal height measurements will become available.
Conclusions
Here, we present the first trans-ancestry genetic study of childhood and adolescent growth. Large-scale growth modeling data allowed an unprecedented view of APV and PHV across contemporary and 20th-century pediatric cohorts. Our data supports genetic relationships between pediatric height growth and health across the life course, with different growth trajectories correlated with different outcomes. Being taller at early puberty associated with less growth across puberty; conversely, measures of growth from birth until adulthood were genetically correlated. Meanwhile, being shorter at age 10/12 for girls/boys correlated with a slower intensity of pubertal growth and later age at menarche. In terms of adult health outcomes, a faster intensity of pubertal growth correlated with higher BMD, HOMA-IR, fasting insulin, and T2D, and lung cancer, and being taller at early puberty and taller across puberty as well as having quicker pubertal growth tempo were associated with higher risk for atrial fibrillation. These results show that there may not be a single “optimal” pubertal growth pattern and highlight the importance of adolescent growth for later life health.
Methods
Contributing studies and phenotypes
Twenty-two cohorts contributed to this study (Additional file 1: Table S1), some with more than one ancestral background. These included up to 41,468 samples of European ancestry (EUR) in 19 cohorts, 7852 African American samples (AFR) in 3 cohorts, 2714 East Asian (EAS) in 2 cohorts, and 4625 American Native or Hispanic (AMR) in 5 cohorts. In total, we included data from 26,478 boys and 30,181 girls aged 5–18 years. Height was measured using standard practices [6]. Details on the range, average, and median number of measurements per cohort is provided in Supplementary Table 1.
Each cohort individually modeled height growth using SITAR following a standard protocol.
For cohorts with sparse height measurements, modeling was performed together with the BMDCS or ALSPAC cohorts as a reference (see Additional file 1: Table S1). The SITAR random effects a-size, b-timing, and c-intensity were used as input phenotypes for GWAS, in addition to three simple height or height difference phenotypes: standardized height at age 10 in girls or age 12 in boys, standardized height difference between age 8 and adult (> 19 years), and standardized height difference between age 14 and adult (> 19 years) (these phenotypes are previously described [14]).
Genome-wide association and meta-analysis
Each cohort individually performed genotyping and GWAS. Genotyping was performed on Illumina or Affymetrix genotyping arrays with centrally recommended post-genotyping quality control (QC). This included a sample call rate < 0.95, autosomal heterozygosity rate > 3 standard deviation from mean, SNP call rate < 0.98, mismatching reported and genotype-based sex, Hardy–Weinberg equilibrium (HWE) p < 1 × 10−6, and MAF < 0.01. We also excluded SNPs with high duplicate discordance rates and monomorphic SNPs. When possible, imputation was performed against the Haplotype Reference Consortium v.1.0 or 1.1 [52]; other population-specific panels were used for specific studies (SISU v2 for HBCS; Pima Indian-specific panel for the Southwest American Indians; 1000 Genomes for SCORM, NSHD, CLHNS). Following imputation, it was recommended to only remove monomorphic SNPs and to not filter on any other criteria as pre-meta-analysis QC would be performed centrally. Next, each cohort performed GWAS using cohort-specific covariates (Additional file 1: Table S1). Post-imputation QC was then performed centrally for all studies and included filters for MAF < 0.05, HWE, and imputation quality (excluding INFO < 0.4). EasyQC [53] was used to perform cohort-specific QC and meta-level QC. Ancestry-specific meta-analysis was then performed with GWAMA using 2 rounds of genomic control to correct for population.
Trans-ancestral meta-analysis
Meta-Regression of Multi-Ethnic Genetic Association (MR-Mega) [54] was used to perform meta-analysis on all cohorts to account for differences in cohort ancestry. A female only, male only, and all sex combined meta-analysis was performed for each of the six phenotypes. Post meta-analysis SNPs were filtered out if they had MAF < 0.05 or were present in less than 50% of the cohorts. Manhattan plots and QQ plots are shown in Additional file 2: Fig. S4 and S5, respectively. Lambda values for each GWAS are given in Additional file 1: Table S11.
Credible set analysis
The script credible_set_analysis.py was used to calculate the 95% credible sets for every genome-wide significant locus. The sum of the posterior probabilities was calculated from a sorted list of the most significant Bayes’ factors until the cumulative sum was equal to or greater than 0.95. This set of SNPs was then considered the 95% credible set.
Functional annotation (FUMA, OpenTargets, Phenoscanner)
Trans-ancestry summary statistics were uploaded to Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA GWAS; https://fuma.ctglab.nl/) to provide detailed annotation of the GWAS results [55]. PhenoScanner V2 (http://www.phenoscanner.medschl.cam.ac.uk/) was used to look up significant sentinel signals and their proxies (r2 = 0.8) to gather all statistically significant GWAS associations with other traits and diseases [25, 56]. OpenTargets (https://www.genetics.opentargets.org/) was further used to gather functional information on sentinel SNPs and target genes. Additionally, functional information on potential effector genes was gathered from the Online Mendelian Inheritance in Man (OMIM) database (https://www.ncbi.nlm.nih.gov/omim), the International Mouse Phenotyping Consortium (IMPC) database (https://www.mousephenotype.org/), and the Mouse Genome Informatics database (http://www.informatics.jax.org/).
Genetic correlations
LD Score Regression [24], either on http://ldsc.broadinstitute.org/ or using python scripts downloaded from GitHub (https://github.com/bulik/ldsc), was used to perform genetic correlation analyses between the pubertal growth phenotypes (European meta-analyses) and outcome traits.
PMBB PheWAS
The Penn Medicine BioBank (PMBB) recruits participants through the University of Pennsylvania Health system. At the time of medical appointment, participants give informed consent to access their electronic health records and donate a blood sample for DNA analysis. Genotyping, imputation, and phenotyping of this sample have been described in detail previously [57]. For the analysis presented here, we used genotype and phenotype data for 10,182 European ancestry individuals.
As the primary phenotypes for polygenic risk scores are not readily available in the PMBB cohort due to it being an adult sample, we identified the best performing PRS in a leave-one-out analysis, and then used that PRS in PMBB. First, we repeated the meta-analysis in European ancestry cohorts only, leaving out the CHOP Center for Applied Genomics (CAG) cohort. SNPs were then restricted to a common set present in all three datasets: the meta-analysis, CAG, and PMBB: 1 = 5,070,548, 2 = 5,065,379, 3 = 5,035,856, 4 = 5,035,192, 5 = 5,034,759, 6 = 5,035,034. Multiple candidate polygenic risk scores at nine p-value thresholds (× 10−6, 1 × 10−5, 1 × 10−4, 1 × 10−3, 0.01, 0.05, 0.1, 0.5, 1) were calculated in European individuals from the CAG cohort using PRSice, with 2,500 randomly selected European ancestry samples from CAG used to estimate LD (r2 > 0.1). Each candidate PRS for each phenotype was then tested against the corresponding phenotype in the CAG cohort in order to find the optimal p-value threshold. The SNPs included in the candidate PRS that produced the strongest association for each phenotype were then used to create the PRS in the PMBB cohort (Additional file 1: Table S8).
In the PMBB, PRS were created in 10,182 European ancestry individuals in PLINK 1.9 using the SNPs from the best performing PRS in CAG, and the weights derived from the leave-one-out meta-analysis. PRS were standardized with mean = 0 and SD = 1. A PheWAS was performed using logistic regression models with the PRS as the independent variable, phecodes as the dependent variable, and age, sex, and the first 10 principal components (PCs) as covariates. Phecodes with > 100 cases were tested (N = 512).
UKBB PheWAS
We used the UK Biobank (UKBB) cohort with imputed genotype and electronic health record data to replicate nominally significant PMBB PheWAS findings and LDSC genetic correlations. Details on genotyping, imputation, and phenotyping in UKBB have been published elsewhere [58]. The best performing PRS—as described above in the CAG cohort—was constructed and simultaneously tested for their associations with 57 binary (N = 421,679) and 26 continuous phenotypes (Nmax = 486,248) using PRSice-2 [59]. Disease status was defined using ICD10 codes from hospital admission data released in March 2020 and only outcomes with > 100 cases were included. Binary phenotypes were tested in logistic regression while continuous phenotypes in linear regression models. Covariates included age, sex, genotyping array, and the first six genetic principal components in all models.
Availability of data and materials
The SITAR code used in this study is available at https://github.com/DCousminer/EGG_SITAR and https://doi.org/10.5281/zenodo.10214403 Credible set analysis was performed using https://github.com/edm1/Credible-set-analysis/blob/master/credible_set_analysis.py.
GWAS summary stats will be deposited in the GWAS catalog database at publication.
Change history
21 May 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13059-024-03276-w
References
Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. 2001;86:2364–8.
Silventoinen K, et al. Genetic regulation of growth from birth to 18 years of age: The Swedish young male twins study. Am J Hum Biol. 2008;20:292–8.
Ohlsson C, et al. Secular Trends in Pubertal Growth Acceleration in Swedish Boys Born From 1947 to 1996. JAMA Pediatr. 2019;173:860–5.
Lai FY, et al. Adult height and risk of 50 diseases: a combined epidemiological and genetic analysis. BMC Med. 2018;16:187.
Antonisamy B, et al. Weight Gain and Height Growth during Infancy, Childhood, and Adolescence as Predictors of Adult Cardiovascular Risk. J Pediatr. 2017;180:53–61.e3.
Okasha M, Gunnell D, Holly J, Davey Smith G. Childhood growth and adult cancer. Best Pract Res Clin Endocrinol Metab. 2002;16:225–41.
Lope V, et al. Perinatal and childhood factors and risk of breast cancer subtypes in adulthood. Cancer Epidemiol. 2016;40:22–30.
Mikkola TM, et al. Childhood growth predicts higher bone mass and greater bone area in early old age: findings among a subgroup of women from the Helsinki Birth Cohort Study. Osteoporos Int. 2017;28:2717–22.
Cole TJ, et al. Using Super-Imposition by Translation And Rotation (SITAR) to relate pubertal growth to bone health in later life: the Medical Research Council (MRC) National Survey of Health and Development. Int J Epidemiol. 2016;45:1125–34.
Eriksson JG, et al. Effects of size at birth and childhood growth on the insulin resistance syndrome in elderly individuals. Diabetologia. 2002;45:342–8.
Bjerregaard LG, Jensen BW, Baker JL. Height at Ages 7–13 Years in Relation to Developing Type 2 Diabetes Throughout Adult Life. Paediatr Perinat Epidemiol. 2017;31:284–92.
Ohlsson C, Bygdell M, Nethander M, Kindblom JM. Early puberty and risk for type 2 diabetes in men. Diabetologia. 2020. https://doi.org/10.1007/s00125-020-05121-8.
Suresh S, O’Callaghan M, Sly PD, Mamun AA. Impact of Childhood Anthropometry Trends on Adult Lung Function. Chest. 2015;147:1118–26.
Cousminer DL, et al. Genome-wide association and longitudinal analyses reveal genetic loci linking pubertal height growth, pubertal timing and childhood adiposity. Hum Mol Genet. 2013;22:2735–47.
Fischbein S, Pedersen NL. Multivariate analysis of genetic and environmental influences for longitudinal height and weight data. Acta Genet Med Gemellol (Roma). 1987;36:171–80.
Phillips K, Matheny AP. Quantitative genetic analysis of longitudinal trends in height: preliminary results from the Louisville Twin Study. Acta Genet Med Gemellol (Roma). 1990;39:143–63.
Silventoinen K, et al. Changing genetic architecture of body mass index from infancy to early adulthood: an individual based pooled analysis of 25 twin cohorts. Int J Obes (Lond). 2022;46:1901–9.
Widén E, et al. Distinct variants at LIN28B influence growth in height from birth to adulthood. Am J Hum Genet. 2010;86:773–82.
Day FR, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. 2017;49:834–41.
Leinonen JT, et al. Association of LIN28B with adult adiposity-related traits in females. PLoS ONE. 2012;7: e48785.
Cole TJ, Donaldson MDC, Ben-Shlomo Y. SITAR—a useful instrument for growth curve analysis. Int J Epidemiol. 2010;39:1558–66.
Abbassi V. Growth and normal puberty. Pediatrics. 1998;102:507–11.
Jelenkovic A, et al. Genetic and environmental influences on height from infancy to early adulthood: An individual-based pooled analysis of 45 twin cohorts. Sci Rep. 2016;6:28496.
Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Kamat MA, et al. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics. 2019;35:4851–3.
Rhodes M, et al. Growth patterns in children with sickle cell anemia during puberty. Pediatr Blood Cancer. 2009;53:635–41.
Díaz-González F, et al. Biallelic cGMP-dependent type II protein kinase gene (PRKG2) variants cause a novel acromesomelic dysplasia. J Med Genet. 2022;59:28–38.
Pagnamenta AT, et al. Variable skeletal phenotypes associated with biallelic variants in PRKG2. J Med Genet. 2022;59:947–50.
Regalado ES, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–6.
van de Laar IMBH, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49:47–57.
Zhang W, et al. A Novel Mutation of SMAD3 Identified in a Chinese Family with Aneurysms-Osteoarthritis Syndrome. Biomed Res Int. 2015;2015: 968135.
Kania K, et al. Regulation of Gdf5 expression in joint remodelling, repair and osteoarthritis. Sci Rep. 2020;10:157.
Wang X, et al. A novel mutation in GDF5 causes autosomal dominant symphalangism in two Chinese families. Am J Med Genet A. 2006;140A:1846–53.
Yang W, et al. Novel point mutations in GDF5 associated with two distinct limb malformations in Chinese: brachydactyly type C and proximal symphalangism. J Hum Genet. 2008;53:368–74.
Jelenkovic A, et al. Associations between birth size and later height from infancy through adulthood: An individual based pooled analysis of 28 twin cohorts participating in the CODATwins project. Early Hum Dev. 2018;120:53–60.
Cao Z, Hui LL, Wong MY. New approaches to obtaining individual peak height velocity and age at peak height velocity from the SITAR model. Comput Methods Programs Biomed. 2018;163:79–85.
Simpkin AJ, Sayers A, Gilthorpe MS, Heron J, Tilling K. Modelling height in adolescence: a comparison of methods for estimating the age at peak height velocity. Ann Hum Biol. 2017;44:715–22.
Frysz M, Howe LD, Tobias JH, Paternoster L. Using SITAR (SuperImposition by Translation and Rotation) to estimate age at peak height velocity in Avon Longitudinal Study of Parents and Children. Wellcome Open Res. 2018;3:90.
Aris IM, et al. Association of mode of delivery with offspring pubertal development in Project Viva: a prospective pre-birth cohort study in the USA. Hum Reprod. 2021;37:54–65.
Marceau K, Ram N, Houts RM, Grimm KJ, Susman EJ. Individual differences in boys’ and girls’ timing and tempo of puberty: Modeling development with nonlinear growth models. Dev Psychol. 2011;47:1389–409.
German A, Shmoish M, Hochberg Z. Predicting pubertal development by infantile and childhood height, BMI, and adiposity rebound. Pediatr Res. 2015;78:445–50.
Aksglaede L, Juul A, Olsen LW, Sørensen TIA. Age at Puberty and the Emerging Obesity Epidemic. PLoS ONE. 2009;4: e8450.
Llop-Viñolas D, et al. Onset of puberty at eight years of age in girls determines a specific tempo of puberty but does not affect adult height. Acta Paediatr. 2004;93:874–9.
Pantsiotou S, et al. Maturational tempo differences in relation to the timing of the onset of puberty in girls. Acta Paediatr. 2008;97:217–20.
Nelson CP, et al. Genetically determined height and coronary artery disease. N Engl J Med. 2015;372:1608–18.
Kember RL, et al. Genetically Determined Birthweight Associates With Atrial Fibrillation: A Mendelian Randomization Study. Circ Genom Precis Med. 2020;13: e002553.
Marott JL, et al. Increasing population height and risk of incident atrial fibrillation: the Copenhagen City Heart Study. Eur Heart J. 2018;39:4012–9.
Berge T, et al. Prevalence of atrial fibrillation and cardiovascular risk factors in a 63–65 years old general population cohort: the Akershus Cardiac Examination (ACE) 1950 Study. BMJ Open. 2018;8: e021704.
Andersen K, Rasmussen F, Neovius M, Tynelius P, Sundström J. Body size and risk of atrial fibrillation: a cohort study of 1.1 million young men. J Int Med. 2018;283:346–55.
Crump C, Sundquist J, Winkleby MA, Sundquist K. Height, Weight, and Aerobic Fitness Level in Relation to the Risk of Atrial Fibrillation. Am J Epidemiol. 2018;187:417–26.
Cole TJ. Optimal design for longitudinal studies to estimate pubertal height growth in individuals. Ann Hum Biol. 2018;45:314–20.
the Haplotype Reference Consortium. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Winkler TW, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–212.
Mägi R, et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum Mol Genet. 2017;26:3639–50.
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826.
Staley JR, et al. PhenoScanner: a database of human genotype–phenotype associations. Bioinformatics. 2016;32:3207–9.
Kember RL, et al. Polygenic Risk of Psychiatric Disorders Exhibits Cross-trait Associations in Electronic Health Record Data From European Ancestry Individuals. Biol Psychiat. 2021;89:236–45.
Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9.
Choi SW, O’Reilly PF. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience. 2019;8:giz082.
Review history
The review history is available as Additional file 3.
Peer review information
Tim Sands was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Funding
Cohort funding can be found in the Supplementary Data. S.F.A.G. is funded by R01 HD056465 and the Daniel B. Burke Endowed Chair for Diabetes Research. K.A.B is funded by the National Institutes of Health (T32HL129982). M.McC is a Wellcome Trust Investigator (funding through 212259/Z/18/Z). K.P. is funded by an Academy of Finland research fellowship (no. 322112).
J.P. is supported by the Medical Research Council (Unit programs: MC_UU_12015/2, MC_UU_00006/2). R.M.F. is supported by a Wellcome Senior Research Fellowship (WT220390). S.F. is supported by an MRC (UK) Programme grant (G0802782).
I.P. and Z.B. are funded by the Diabetes UK (BDA number: 20/0006307), the European Union’s Horizon 2020 research and innovation programme (LONGITOOLS, H2020-SC1-2019–874739). I.P. is supported by Agence Nationale de la Recherche (PreciDIAB, ANR-18-IBHU-0001), by the European Union through the “Fonds européen de développement regional” (FEDER), by the “Conseil Régional des Hauts-de-France” (Hauts-de-France Regional Council) and by the “Métropole Européenne de Lille” (MEL, European Metropolis of Lille).
Author information
Authors and Affiliations
Consortia
Contributions
JPB performed the meta-analysis and follow-up experiments. RLK performed Penn Biobank analyses. AU and ZB performed UK Biobank analyses. AA, IMA, JAB, AB, ZC, J-FC, NMD, DF-O, MB, RF, AG, AH, J-JH, CI, SK, JLe, EL, L-PL, AM, NP, RMS, CTH, DPS, ET, SV, KHW, CAW, AW, KG performed individual cohort analyses. LAH, CC, MC, PE, SF, CF-B, WJG, VG, RLH, MKa, HK, AK, JTG, JK, MKä, CL, JLa, NRL, FDM, NM, HN, EO, KP, XS, Y-YT, LJB, TvB, LSA, DIB, ET, MG, JGE, JFF, FDG, TH, RH, M-FH, J-CH, VWVJ, MRJ, TL, DAM, DM, KLM, JM, SO, CEP, JRBP, OR, FR, S-MS, SS, JAS, MS, TIAS, NJT, MT, GW, EH, CP, MIM, RMF, EW, HH, IP, BFV, and BSZ were involved in sample collection and/or cohort management. JPB, RLK, DLC, SFAG, AU, ZB, IP, BFV, AC, JTG, AK, JK, SM, JAM, and BSZ contributed to data interpretation. DLC, JPB, RLK, AU, ZB, and SFAG wrote the paper with review by all authors. DLC and SFAG designed the experiments and supervised the work. All authors read and approved of the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Diana L. Cousminer is an employee of and shareholder of GlaxoSmithKline (GSK). Mark McCarthy is employed by Genentech and a holder of Roche stock. John Perry is an employee and shareholder of Adrestia Therapeutics. Christine Frithioff-Bøjsøe is currently employed at Novo Nordisk. This work is unrelated to any activities at these companies.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: The spelling of author name of Zhanna Balkhiyarova has been corrected.
Supplementary Information
Additional file 2.
Figures S1-S5, Cohort funding and acknowledgments.
Additional file 3.
Peer review history
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bradfield, J.P., Kember, R.L., Ulrich, A. et al. Trans-ancestral genome-wide association study of longitudinal pubertal height growth and shared heritability with adult health outcomes. Genome Biol 25, 22 (2024). https://doi.org/10.1186/s13059-023-03136-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-023-03136-z