
Abstract
Background
Recent genome-wide studies of many species reveal the existence of a myriad of RNAs differing in size, coding potential and function. Among these are the long non-coding RNAs, some of them producing functional small peptides via the translation of short ORFs. It now appears that any kind of RNA presumably has a potential to encode small peptides. Accordingly, our team recently discovered that plant primary transcripts of microRNAs (pri-miRs) produce small regulatory peptides (miPEPs) involved in auto-regulatory feedback loops enhancing their cognate microRNA expression which in turn controls plant development. Here we investigate whether this regulatory feedback loop is present in Drosophila melanogaster.
Results
We perform a survey of ribosome profiling data and reveal that many pri-miRNAs exhibit ribosome translation marks. Focusing on miR-8, we show that pri-miR-8 can produce a miPEP-8. Functional assays performed in Drosophila reveal that miPEP-8 affects development when overexpressed or knocked down. Combining genetic and molecular approaches as well as genome-wide transcriptomic analyses, we show that miR-8 expression is independent of miPEP-8 activity and that miPEP-8 acts in parallel to miR-8 to regulate the expression of hundreds of genes.
Conclusion
Taken together, these results reveal that several Drosophila pri-miRs exhibit translation potential. Contrasting with the mechanism described in plants, these data shed light on the function of yet undescribed primary-microRNA-encoded peptides in Drosophila and their regulatory potential on genome expression.
Similar content being viewed by others


Background
More than 20 years after the first genome annotation, it is now becoming clear that the protein-centric view of gene content strongly underestimates the number of DNA regions that are expressed and fulfill important functions for development and physiology since the majority of the genome is in fact transcribed [1]. A first discovery was the importance of hundreds of small non-coding RNAs, such as microRNAs (miR), playing regulatory roles in the silencing of genes and transposable elements. More recently, genome-wide transcript profiling has disclosed the existence of numerous RNAs referred to as long non-coding RNAs (lncRNAs or lincRNAs) since they lack the classical hallmarks of protein-coding genes.
Although the functions of all lncRNAs remain largely unknown, there are several experimental cases illustrating their key role as functional RNAs in various steps of the control of genome expression [2]. In association with other molecules, lncRNAs can coordinate several physiological processes and their dysfunction impacts several pathologies, including cancer and infectious diseases. lncRNAs control genetic information, such as chromosome structure modulation, transcription, splicing, messenger RNA (mRNA) stability, mRNA availability and post-translational modifications. They also act as scaffolds, bearing interaction domains for DNA, mRNAs, miRs and proteins, depending on both their primary sequence and secondary structure [3]. In addition, while lncRNAs annotated as non-coding cannot produce large-sized proteins, they all contain myriads of short open reading frames (sORF) [4,5,6,7] and a surprising result was the discovery for a subset of them of their translation into small functional peptides [8,9,10,11].
MicroRNAs (miRs) define a class of small, non-coding RNAs able to downregulate the expression of target mRNA by binding to the 3′-ends inducing mRNA degradation and/or translation repression. Intergenic microRNAs are produced from the sequential cleavage of long precursors named primary transcripts of microRNA (pri-miRs) (frequently annotated as lncRNAs) by Drosha and Dicer into 22 nt miR duplexes associated with the RISC protein complex. Identified in a broad spectrum of living species, they are transcribed from coding genes or lncRNAs by the RNA polymerase II. MiRs are critical for normal animal development and are involved in many biological processes [12]. Due to their role in silencing, miRs, and in particular the pri-miRs they come from, have always been considered as non-coding. However, the discovery that plant pri-miRs encode small regulatory peptides (miPEPs) merely adds evidences that pri-miRs are also pre-mRNAs [13]. In plants, miPEPs specifically increase transcription of their primary transcript impacting the level of the mature miR produced and consequently affecting the control of the entire miR Gene Regulatory Network (GRN). To date, this regulation has been extended to several miRs in various plants [14,15,16,17]. In human cells, only few reports present evidences of pri-miR translation [18,19,20,21]. Pri-miR-22 host gene endogenously produces a miPEP for which the function is unknown [19]. miR-200 might produce a miPEP able to control the Vimentin, a miR200 target [18]. miPEP155 was described to control major histocompatibility complex class II-mediated antigen presentation by disrupting the HSC70-HSP90 machinery [20]. However, whether these miPEPs control the expression of their cognate miR was not investigated. More recently, miPEP133 was discovered in miR34a as a tumor suppressor localized in the mitochondria. It enhanced p53 transcriptional activation which, in turn, induces miR-34a expression [21]. In summary, whereas few human pri-miRs appear translatable, it remains not clear whether the regulation found in plants exists in animals. Here we addressed this question in Drosophila melanogaster. We show that several intergenic pri-miRs contain marks of ribosome profiling. To investigate whether Drosophila can produce miPEPs, we focused on miR-8, a previously well-characterized microRNA that sustains many developmental traits [22]. miR-8 controls organism physiology, tissue growth and survival [23,24,25,26], stem cell renewal [27,28,29,30], central nervous system development [30,31,32], signaling and developmental pathways [33,34,35,36,37,38,39,40,41]. Consequently, miR-8 loss or gain of function impinges on fly development and survival. In the pri-miR-8, we located a small ORF encoding a potential 71 amino acid peptide we called miPEP-8. We showed that this sORF is translatable in vitro and in vivo. While our attempts to reveal an auto-regulatory loop remained unsuccessful, we showed by genetic and transcriptomic approaches that miPEP-8 and miR-8 act in parallel in controlling wing development and in regulating the expression of distinct sets of genes.
Results
Translation potential of sORFs present in Drosophila pri-microRNAs
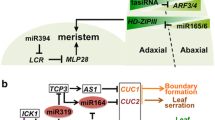
As plants pri-miRs contain sORFs (miORFs) producing functional miPEPs involved in a positive feedback loop on pri-miR expression (Fig. 1a), we first asked whether D. melanogaster pri-miRs contain significant levels of miORFs. We scored the number of sORFs present in intergenic miR genes and their pri-miR and compared this number with other classes of RNAs, coding as well as non-coding (Fig. 1b). Pri-miRs present the highest enrichment of sORFs/kb when compared with the 5′UTR of coding genes, sequences known to contain translatable short open reading frames, but contain similar amounts of sORFs when compared with lncRNAs, previously reported to be widely bound by ribosomes [43]. Ribosome profiling experiments were developed to study translatability of RNAs by scoring the sequences bound by ribosomes [7, 44, 45] including studies conducted on Drosophila [4, 46, 47]. We searched if and how many Drosophila pri-miRs were widely bound by ribosomes. Briefly, we searched in the rib-seq databases marks of ribosome binding in predicted pri-miR. To avoid difficulties for the interpretation with miRs embedded in host coding genes, we focused only on the intergenic miRs. We found many marks of ribosome profiling and identified hundreds of potentially translated sORF peptides within dozens of pri-miRs, suggesting that, as observed in plants, Drosophila pri-miRs are potentially translated (Additional file 1: Fig. S1; Additional file 2).

Translatability of pri-miR-8. a Model of miPEP regulation in plants. b Box plot representation of the number of ORFs in different classes of RNAs. 3′UTR, 5′UTR and CDS represent coding RNAs, whereas lncRNAs and pri-miRs represent non-coding RNAs. An ORF was defined as starting with an ATG and coding for a minimum of 10 amino acids. Pri-miRs reveal comparable numbers of ORFs/kb as lncRNAs. c GWIPS-vis [42] genome viewer of the Drosophila miR-8 locus. Top: genomic positions and ORFs in the three reading frames. Green bars define ATGs and red bars stop codons. Below, RNA-seq profile is shown in green and ribosome profiling is shown in red. The blue horizontal lines represent the two CR43650 non-coding RNA transcripts and potential miR-8 pri-miRs. In black is schematized the transcript we identified as detectable pri-miR-8. Bottom: miPEP-8 amino acid sequence is shown. d Western blot experiments using the anti-miPEP-8 antibody. Left panel: in vitro synthetized miPEP-8 corresponding to the constructs indicated on top. The asterisk indicates the upstream initiated peptide. The arrow indicates the miPEP-8 initiated at ATG1. Middle panel: detection of miPEP-8 in S2 cells over-expressing miPEP-8 placed in different translational contexts; Kozak (optimal); K mt (ATG mutated into TGA); mt (ATG mutated into AGT). Note that the pri-miR is translated and endogenous miPEP-8 expression is undetectable in S2 cells. Right panel: anti miPEP-8 western blot of adult Drosophila extracts and in the miR-8 deleted line Δ2/Δ2 [26] in which no miPEP-8 is detected. We noticed the presence of non-specific bands as well as additional specific bands representing possibly miPEP-8 multimeric forms or PTM modifications. Ctrl corresponds to cell extracts transfected with an empty vector
Drosophila pri-miR8 encodes a miPEP-8 translated in vivo
To further characterize the potential translation of pri-miRs, we focused on the miR-8 primary transcript (pri-miR-8). MiR-8 is an intergenic miR and is likely produced from the expression of long non-coding CR43650 spanning over the pre-miR-8 (Fig. 1c). In flybase, two CR43650 ncRNAs were predicted, a long and a short form defining putative pri-miR-8 transcripts, independently identified by two different teams and likely initiated from different promoters [48, 49]. As shown in Fig. 1c, many marks of potential translation were found along the CR43650 transcripts. We first performed 5′RACE as well as RT-PCR assays to determine which isoform was preferentially produced. While we successfully detected the short isoform in flies and S2 cells, we did not succeed in amplifying the long isoform, suggesting that the short transcript is the most abundant isoform expressed, a result confirmed by RNA-seq data generated during this study (Additional file 1: Fig. S2 and S3A). Our RNA-seq data further suggest that the long transcript might define a different transcription unit since no overlapping reads were detected in the promoter region of the short transcript (Additional file 1: Fig. S3A). In agreement with this, a GAL4 enhancer trap recapitulating miR-8 expression is inserted just upstream of the short transcript [24]. These flies express miR-8 at a level comparable to control flies (Additional file 1: Fig. S3B), showing that this insertion disrupting the co-linearity of the miR-8 locus is not detrimental for miR-8 expression. Finally, we verified that this short transcript (referred hereafter as pri-miR-8) is functional since it efficiently produces a functional mature miR-8 able to down regulate a miR-8 sensor (see below Fig. 3b, d).
We therefore looked for a potential open reading frame within the pri-miR-8 gene. Focusing on the 5′ leader sequence of pri-miR-8, we found one ORF located upstream of the pre-miR-8. This ORF is the longest ORF present 5′ to the pre-miR, which potentially encodes a miPEP of 71 amino acids in length if initiated from the first ATG (ATG1) (Fig. 1c and Additional file 1: Fig. S2A). However, the presence of a second ATG (ATG2), located downstream, gives the possibility to produce a shorter peptide. To determine whether the open reading frame is translated and which initiation codon is used, we generated and characterized specific antibodies (Additional file 1: Fig. S4). In parallel, we generated different deletion constructs and performed in vitro translation experiments using insect cell extracts. As shown in Fig. 1d (left panel), we observed an efficient translation from the longest construct (CTG) consisting in an extended genomic region of the defined 5′ leader sequence of pri-miR-8. Deletion experiments revealed a stronger and efficient translation from ATG1 but not from ATG2. A higher product, possibly initiated at an upstream codon present in the construct was also detected but was not further investigated since it was not present in pri-miR-8.
We next generated translatable and untranslatable miPEP-8 forms placed in optimal translational Kozak (K) or mutated kozak (KMT) contexts or in the miPEP-8 natural ATG1-initiated translational context. We then expressed these miPEP-8 constructs in Drosophila S2 cells (Fig. 1d, middle panel). As revealed by western blot experiments, these different constructs produced the same level of miPEP-8 when the ATG was placed in an optimal or in its natural translational context whereas no miPEP-8 was detected from mutated ATG constructs. This result reveals that the natural nucleotide context of miPEP-8 miORF is in a favorable translational context. We next questioned whether pri-miR-8 was able to produce miPEP-8. We transfected S2 cells with wild type or ATG-mutated pri-miR-8 constructs and performed western blot experiments. We observed that only the wild-type pri-miR-8 was able to produce miPEP-8. Finally, we examined whether a peptide corresponding to miPEP-8 was detectable in fly extracts by performing a western blot experiment on young adult flies. As revealed in Fig. 1d (right panel), we observed a signal co-migrating with the in vitro synthetized miPEP-8, corresponding to endogenous miPEP-8 as demonstrated by the lack of this band in miR-8 deleted Δ2 mutant flies. Sequence alignment analyses revealed that this peptide is poorly conserved among Drosophila species. Some homologies are detected in Drosophila melanogaster group but no conservation was found in more distant Drosophilae species.
Altogether, our results reveal that the pri-miR-8 transcript carries, in addition to the miR-8 sequence, at least one translated ORF located upstream of the pre-miR, able to express a miPEP-8 both in vitro and in vivo.
Expression of miPEP-8 impinges on Drosophila development
To study the function of miPEP-8, we generated flies able to express a translatable and untranslatable version of miPEP-8 (ATG mutated). miR-8 sustains many biological functions in Drosophila and either its loss of function or its over-expression leads to detrimental outcomes in cells, tissues or the whole organism [23,24,25,26,27, 29, 31,32,33,34,35,36,37, 40, 41, 50], providing a useful readout to assess miPEP-8 activity. To test our hypothesis that miPEP-8 controls miR-8 expression and modulates its activity, we used an over-expression assay. Using the miR-8 GAL4 driver, a GAL4 insertion in the endogenous miR-8 promoter reported to mimic miR-8 expression [23, 24, 33, 40], we first asked whether over-expression of miPEP-8 impinges on fly viability. As reported, driving UAS-miR-8 over-expression results in increased fly lethality [23] (Fig. 2a). Over-expression of a UAS-miPEP-8 translatable construct also affected fly viability whereas the untranslatable form did not. This indicates that, like miR-8, the translatable form of miPEP-8 is able to interfere with development, although with a weaker effect compared to miR-8.

Drosophila miPEP-8 is biologically active during development. a Lethality assay on flies over-expressing miR-8, miPEP-8 or miPEP-8mt (ATG mutated) using the miR-8 GAL4 driver. Left: details of the genetic cross and expected percentage depending on the effect (neutral, deleterious or advantageous) on Drosophila development. Right: graph indicating the percentage of hatched flies over-expressing the different constructs. White flies (w) crossed with the driver line were used as a control. Expressing miR-8 resulted in developmental lethality since less than 20% of flies hatched (expected value 50%). A significant decrease occurred following miPEP-8 over-expression but not with the untranslatable miPEP-8mt construct. Number of independent crosses: for w and miR-8 n = 23; for miPEP-8 wt and mt n = 24. b Same as in a except the constructs were expressed in wings using the MS1096 driver and the phenotypes scored on wing size. Number of wings analyzed: for w n = 20; for miR-8 n = 27; for miPEP-8 wt and mt n = 27. * or ns: Significant differences are indicated relative to white recipient flies. AU arbitrary units
By loss or gain of function experiments, miR-8 was shown to induce a « small wing » phenotype [24, 26, 40]. We therefore questioned whether over-expression of miPEP-8 also induced a wing phenotype (Fig. 2b). Using the wing driver line MS1096, miR-8 over-expression induced several wing defects, from a reduced size to the loss of wing vein, sensory organs, miss shaped, depending on the transgene/promoter strength [40, 41]. Quantifying the wing size appeared the most reliable criteria, and we compared this phenotype with the phenotype observed with miPEP-8 over-expression. Consistently with our above result, miPEP-8 over-expression induced a slight, albeit significant wing size reduction, revealing yet again a weaker activity compared with miR-8 (Fig. 2b). Importantly, miPEP-8-induced wing reduction was dependent on the integrity of the translation codon since the same construct with the mutated ATG did not induce any phenotype (Fig. 2b).
Altogether, these experiments show that miPEP-8 appears to be biologically active but induces a milder phenotype compared to miR-8.
In Drosophila, miR-8 expression is independent of miPEP-8 expression
In plants, miPEPs positively auto-regulate the expression of their own miR by regulating the expression of their cognate pri-miR. To test whether miPEP-8 regulates miR-8 expression in Drosophila, we monitored the level of pri-miR-8 and miR-8 through quantitative PCR experiments on S2 cells and on flies expressing the translatable form of miPEP-8. We first set up experimental conditions in S2 cells by transfecting the pri-miR-8 construct and quantified the level of the exogenous pri-miR and mature miR produced. When pri-miR-8 was over-expressed, both over-expression of pri-miR-8 and miR-8 was detectable (Fig. 3b). We then over-expressed miPEP-8 and quantified the endogenous level of pri-miR-8 and mature miR-8. Whereas we unambiguously detected the over-expression of the miPEP-8 construct, we did not see any change in the levels of endogenous pri-miR-8 or mature miR-8 expression (Fig. 3c). As observed in S2 cells, no change in pri-miR-8 or miR-8 levels was observed in flies upon miPEP-8 expression using the miR-8 GAL4 driver (Additional file 1: Fig. S5).

Pri-miR-8 expression is independent of miPEP-8 control/activity. a schematic representation of constructs tested on miR-8 expression and activity levels. Arrows locate the primers used in the qPCR experiments determining miPEP and pri-miR relative expression levels. b the characterized pri-miR-8 produces a mature miR-8. S2 cells were transfected with a vector expressing pri-miR-8. Left: detection of the over-expression level of pri-miR-8 by qPCR. Right: detection of the over-expression level of mature miR-8 using the same RNA samples, n = 11 c miPEP-8 lacks repressive activity towards miR-8 expression. Left: level of miPEP-8 over-expression. Middle and right panels: quantification of pri-miR-8 and mature miR-8 in miPEP-8 over-expressing cells compared to control transfected cells (ctrl). n = 13 for the ctrl and 14 for miPEP-8. d, e Insensitivity of miR-8 sensor to miPEP-8 over-expression in S2 transfected cells (n = 16) (d) or in wing imaginal discs when miPEP-8 is expressed under the ptc-GAL4 promoter (e). In d, a miR-8 construct (n = 12) [17] was used as a positive control repressing the miR-8 luciferase sensor [20]. Of note, pri-miR-8 (n = 21) also repressed the miR-8 luciferase sensor. In e, first panel to the left: ptc GAL4 crossed with a UAS mCherry. Second panel to the left: expression pattern of the GFP miR-8 sensor alone. Scale bars (white) indicate 100 μm. A repressive activity is observed with miR-8 expressed in the ptc domain but not with miPEP-8. A representative disc is shown out of ten analyzed
We next questioned the potential regulatory role of miPEP-8 on miR-8 expression by testing miPEP-8 overexpression on endogenous miR-8 activity level in the presence or absence of. One way of challenging this question is the use of a sensor of miR-8 activity, whether endogenous or resulting from over-expression. Thus, we designed a miR-8 luciferase reporter, bearing a 3′UTR from the escargot gene (esg), previously shown directly regulated by miR-8 [27]. Over-expression of pri-miR-8 in S2 cells was able to repress the miR-8 sensor to the same extent as miR-8 (Fig. 3d, left panel), hence validating our miR-8 sensor. As mentioned above, this also indicates that the pri-miR-8 construct is able to generate a functional and mature miR-8. Clearly, however, over-expression of miPEP-8 did not reveal any modulation of the luciferase reporter (Fig. 3d right panel). We performed similar experiments in vivo using a miR-8 GFP sensor in wing imaginal discs where miR-8 was previously shown to be functional [33]. Expressing miR-8 under the patched (ptc) GAL4 promoter led to the repression of the GFP in the ptc domain (Fig. 3e ptc > miR8 panel). Consistently, miPEP-8 over-expression had no effect on the miR-8 GFP sensor in vivo (Fig. 3e ptc > miPEP-8 panel). Altogether, these results indicate that miPEP-8 is not able to control miR-8 expression or activity for that matter (see more below). We obtained similar conclusions on endogenous miR-8 target in S2 cells and in wing discs (Additional file 1: Fig. S6).
Finally, we asked whether the miPEP-regulation of pri-miR observed in plants is system specific by testing whether a plant pri-miR, upregulated by its miPEP in plant cells, could be upregulated in Drosophila cells. Reciprocally, we tested whether miPEP-8 is able to upregulate its pri-miR-8 in plant cells. To that end, we expressed the plant Arabidopsis thaliana pri-miR165a and miPEP-165a in S2 cells using the actin promoter and measured the level of pri-miR produced in the absence or presence of the miPEP. Reciprocally, the Drosophila pri-miR-8 and its miPEP-8 were cloned in plant expression vectors and agroinfiltrated in Nicotiana benthamiana leaves as performed previously [13]. Whereas the upregulation of the A. thaliana pri-miR-165a by miPEP-165a was observed in N. benthamiana, we did not detect any upregulation, but rather a downregulation of pri-miR-165a in Drosophila S2 cells (Additional file 1: Fig. S7). Reciprocally, we could detect a slight but significant increase of pri-miR-8 expression upon miPEP-8 over-expression in N. bentamiana leaves, suggesting that a difference of regulation occurs between plant cells and insect cells (Additional file 1: Fig. S7). Although, miR-8 expression appears to be miPEP-8 independent in Drosophila, these results further suggest that, like for plants miPEPs, animal miPEPs might nonetheless have the potential of autoregulating the expression of their cognate pri-miR.
Endogenous miPEP-8 alteration reveals in vivo activity
To investigate the functional requirement of miPEP-8 in Drosophila, we tried several times to edit the miPEP-8 in flies using CRISPR/Cas9, but unsuccessfully. In contrast, it was possible from the first attempt to delete the entire miR-8 locus, showing that the failure to obtain a specific miPEP-8 edited line is not due to trivial technical problems. We therefore created a specific P landing platform in place of pri-miR-8 transcript to perform Knock In strategies (Fig. 4a and Additional file 1: Fig. S8). This edited line exhibits the previously miR-8 reported phenotypes [26], including a strong developmental lethality with only few escaping flies exhibiting a reduced size (including wings (Fig. 4c and Additional file 1: Fig. S8)) and leg defects (not shown). We further knocked in the wild type pri-miR-8 and the pri-miR-8 miPEP-8 untranslatable form (mt) at the P landing site and analyzed the outcomes. For both constructs, we observed a nearly total rescue since the theoretical expected 33.3% homozygotes (and 66.6% of CyO flies) in the progeny was almost reached (Fig. 4b left panel). These rescued flies appeared phenotypically normal and re-expressed miR-8 at levels close to miR-8 endogenous expression (Fig. 4b right panel). Both pri-miR-8 constructs restored significantly wing sizes when compared to the ΔmiR-8 CRISPR line (Fig. 4c). Interestingly, a significant difference was observed in wings between the pri-miR-8 wild type construct and the pri-miR-8 mt in which the miPEP-8 translatability was disrupted (Fig. 4c).

targeting miPEP-8 in vivo in Drosophila induces a wing phenotype. a Strategy for endogenous miPEP-8 edition. The pri-miR-8 gene region was deleted by CRISPR and a P landing site was created. Wild type and miPEP-8 ATG mutated pri-miR-8 in pattB were inserted at the P landing site. b Similar rescue efficiency was observed in at least three independent transgenic lines (left panel). qPCR on mature miR-8 in wild type and mutant (mt) pri-miR-8 Knock In (KI) lines showed similar miR-8 levels (n = 4), (right panel). c Wing phenotype in miR-8 deletion edited line. The pri-miR-8 miPEP-8 mutated (mt) shows a reduced wing size compared to the wild type pri-miR-8. (n = 15 and 28 respectively). d–f Analyses in miPEP-8 mutant identified in DGRP polymorphism. d miR-8 level determined by qPCR in white recipient flies (w) and in white flies carrying the miPEP-8 truncated form (miPEP-8alt), (n = 6 and 8, respectively). e, f Wing size determination in different genetic contexts. miPEP-8alt homozygotes or over miR-8 deficiencies revealed significant reduced wing size relative to the white recipient flies (w, n = 19; miPEP-8alt, n = 21; miPEP-8alt/miR-8 deletions, n = 40). Expressing miPEP-8 rescued the wing phenotype of miPEP-8alt flies relative to sibling flies not expressing miPEP-8 (n = 18 and 28, respectively). Significant (*) or nonsignificant (ns) differences are indicated either relative to white recipient flies or between the two groups
As a second approach, we took advantage of a polymorphism mutation detected in Drosophila Gene Reference Panel (DGRP) lines generating a premature stop codon leading to a 24 amino acid C-terminal miPEP-8 truncation called miPEP-8alt [51]. We outcrossed the miPEP-8alt DGRP line into white background and analyzed the consequence of the miPEP-8alt mutation. Whereas no significant difference was observed for miR-8 level between these the two miPEPs variants (Fig. 4d), flies homozygous for miPEP-8alt exhibit a significant wing size reduction when compared with the white flies expressing the miPEP-8 (Fig. 4e). We further analyzed the resulting wing phenotypes in different genetic contexts. The phenotype is also present when miPEP-8alt mutation was tested over a deletion of the miR-8 gene (the Δ2 and the ΔmiR-8 CRISPR line generated in this study) (Fig. 4e), suggesting that the observed phenotype is a consequence of miPEP-8 loss of function. To test this, we performed rescue experiments by expressing miPEP-8 using the miR-8 GAL4 driver line in miPEP-8alt/ΔmiR-8 background (Fig. 4f). Flies expressing miPEP-8 in miPEP-8alt/ΔmiR-8 CRISPR restored the wing size phenotype contrasting with the sibling control flies carrying no miPEP-8 transgene (absence of expression of wild type miPEP-8) (Fig. 4f).
Therefore, altogether, these experiments revealed an in vivo miPEP-8 function.
The function of miPEP-8 is uncoupled from miR-8 expression and activity
The above experiments suggest that in Drosophila, miPEP-8 is not involved in a positive auto-regulatory feedback loop as observed in plants. However, due to the similarities of the phenotypes observed between miR-8 and miPEP-8, we questioned whether miPEP-8 could be involved in the miR-8 pathway through another mechanism or whether it acts in parallel of miR-8. As both miR-8 and miPEP-8 affected wing formation, we developed a genetic assay to test whether miPEP-8 acts through miR-8 using a previously validated miR-8 sponge, which titrates miR-8 hence rescuing miR-8-induced phenotypes [29, 33]. Using this rescue assay, we asked whether the miR-8 sponge could also compensate the miPEP-8 induced phenotype. Co-expressing miR-8 together with a miR sponge scramble (as a control and to maintain the number of UAS transgenes identical) using the MS1096 GAL4 driver led to wing size reduction. This phenotype was efficiently rescued by co-expressing miR-8 with the effective miR-8 sponge (Fig. 5). In contrast, when miPEP-8 was co-expressed with the miR-8 sponge, no compensation of the miPEP-8-induced wing reduction was observed. Therefore, this result strongly suggests that miPEP-8 acts in parallel of miR-8.

Uncoupled activity of miR-8 and miPEP-8. a Rescue assay of miR-8- or miPEP-8-induced wing phenotype in flies co-over-expressing miR-8 or miPEP-8 along with a miR-8 sponge (miR-8sp) or a miR-8 scramble (miR-8scr). Only miR-8-sp (and not miR-8scr) compensates for miR-8-induced wing size reduction, hence efficiently titrating miR-8, while it has no effect on miPEP-8-induced wing phenotype. b Quantification of a. “ctrl” (MS1096/+) n = 19; “mir-8; mir-8scr” n = 20; “mir-8; mir-8sp” n = 21; “miPEP-8; mir-8scr” n = 23; “miPEP-8; mir-8sp” n = 19. * p < 0.05
We thus reasoned that the effect of miPEP-8 on wing development could be linked to the modulation of gene expression independent of miR-8. To identify these putative miPEP-8-regulated genes and to compare them with the miR-8-regulated transcriptome, we overexpressed miR-8 or miPEP-8 in S2 cells and performed RNA-seq 48 h after transfection (Fig. 6; Additional file 3). Clearly, the transcriptomes appeared different (Fig. 6a; Additional file 1: Fig. S9). The assays performed on miR-8 over-expressing cells successfully retrieved previously identified miR-8 targets both at the RNA (Additional file 1: Fig. S9A and Additional file 2) and protein level (Additional file 1: Fig. S6), hence validating our experimental conditions. GO term enrichment identified biological pathways fitting with miR-8 activity such as “regulation of organism or cell growth and differentiation”, “wing development”, “apoptosis” and “regulation of actin cytoskeleton” (Additional file 1: Fig. S10). As for miPEP-8 controlled genes, strikingly, the majority of them were miPEP-8 specific (76%) (Fig. 6b, c) since only 24% appeared co-regulated (Fig. 6b, e). In both cases, we found activated and repressed genes (Fig. 6a, c–e). Remarkably, miPEP-8-modulated genes were frequently more strongly modulated than miR-8-modulated genes (Fig. 6c, d). Increasing the Fold change (FC > 1.5) led to a decrease of the number of genes but the respective proportions and conclusions remained unchanged (Additional file 1: Fig. S9B). Our analyses of GO term enrichment clearly identified shared functions for miR-8 and miPEP-8 (Additional file 1: Fig. S10A and S11A; Additional file 4), some of which being related to wing morphogenesis (such as cell junction organization actin filament-based processes, epithelial cell morphogenesis, cell differentiation) or developmental processes (such as neurogenesis, cell migration, embryonic morphogenesis) (Additional file 4). However, miR-8 and miPEP-8 also exhibit specific biological functions such as snRNA modification and leucine metabolic process for miR-8 or K48 linked ubiquitination and chromatin-mediated maintenance of transcription for miPEP-8 (Additional file 1: Fig. S10B and S11B).

miR-8 and miPEP-8 control distinct set of genes. a Heatmap representing the RNA-seq results obtained from S2 cells over-expressing either miR-8 or miPEP-8. Significant sets of genes are modulated in response to mirR-8 or miPEP-8 over-expression, when compared to control transfected cells (ctrl). N = 5. b Venn diagram representing the miR-8 versus miPEP-8 modulated genes. c–e Different subgroups are distinguished; miPEP-8 specific (c), miR-8 specific (d) and co-regulated by miPEP-8 and miR-8 (e)
Altogether, these experiments suggest that miR-8 and miPEP-8 independently control similar biological processes, while regulating functions specific to one or the other.
Discussion
In the present study, we investigated whether a small ORF present in Drosophila pri-miR-8 was capable of producing a miPEP-8 and we propose that animal miPEPs are able to act in parallel of their corresponding miRs. Several studies performed in a broad range of organisms have revealed the prevalence of translated small/short open reading frames (smORFs/sORFs) [7, 44, 45, 52,53,54,55,56,57]. Although sORF peptides were initially identified as being encoded by unusual long non-coding RNAs, to date, it turns out that many classes of RNAs can produce these peptides. Therefore, sORF-encoded peptides (SEPs) are emerging as an unexplored reservoir of putative regulators. However, while a growing body of evidence further supports the importance of sORFs and associated peptides in development, physiology and diseases [8, 55, 58, 59], the number of SEPs that have been characterized so far still remains limited. Therefore, the current challenge resides in deciphering the full repertoire of their functions and molecular modes of action, an issue largely dependent on experimental approaches.
We show with several experimental data that a miPEP-8 is indeed produced from Drosophila pri-miR-8. First, we found a signal of ribosome binding in the pri-miR of several microRNAs and in particular, miR-8. Second, we show that the initiation codon of the miORF present within pri-miR-8 is in a favorable translational context. Third, after having generated specific antibodies, we detected a peptide co-migrating with in vitro translated miPEP-8 in fly extracts. Fourth, forced expression or loss of function of this peptide led to a significant developmental phenotype in Drosophila and induced significant variations of cellular gene expression. Therefore, the poor conservation detected among Drosophila species indicates that this sORF-encoded peptide differs from the few conserved ones characterized so far and shed light on it by its recent invention.
Here, we tackled the question of whether the miPEP auto-regulatory function was identical to that of plants. While we did not detect any auto-regulatory loop (miPEP increasing the expression of its own pri-miR and miR), we observed that the action of miPEP-8 is uncoupled from miR-8 regulation. On the one hand, our data suggest that this peptide could control similar developmental outcomes or developmental pathways and share the regulation of identical subsets of genes. In this context, we further analyzed whether we could detect a significant miPEP-8 activity in other miR-8 developmental processes such as intestinal stem cell differentiation [27] and eye morphogenesis [37] (data not shown). However, no significant activity was detected, suggesting that, in the experimental conditions tested, miPEP-8 does not act in all miR-8 developmental pathways. Such an example was observed in S2 cells in which miR-8 is expressed at detectable levels whereas endogenous miPEP-8 is not. On the other hand, we reveal that miPEP-8 likely has a regulatory function all of its own, independently of miR-8. Indeed, our RNA-seq data indicates that miPEP-8 regulates specific genes and biological processes (i.e. independent of miR-8 activity). This also occurs in vivo since few candidates of the top list of miPEP-8 specific regulated genes identified in S2 cells are also modulated in miPEP-8 loss of function in adult flies (Additional file 1: Fig. S12). Future loss of function and expression pattern analyses throughout development should bring further insight into miPEP-8-specific regulatory functions.
Is the uncoupling of miPEP activity from miR regulation a general feature of animal miR genes? The study performed here suggests that the mechanisms involved in animals might be different from the miPEP auto-regulatory mechanism observed in plants. As such, a recent study on human miR155 revealed an activity for a miPEP155 that is not correlated to miR155 control [20] and on human miR34 where a miPEP133 mitochondrial function impinging on p53 activity was reported [21]. In light of these results, of course, we cannot affirm that the mechanisms described here are common to all Drosophila miR genes. It remains possible that some of them might be auto-regulated by their miPEPs as described in plants. In addition, since ribosome occupancy were not found in all Drosophila miR genes, it remains possible that some pri-miRs are unable to produce miPEPs. Therefore, additional studies will be required to determine whether miPEP-dependent pri-miR auto-regulation is specific or widespread among miR genes.
Is the pri-miR coding capacity conserved throughout the animal kingdom? In a search for non-coding RNAs able to express sORF-encoded peptides, Razooki and co-workers found that human miR-22 host gene (pri-miR-22) produces a potential miPEP-22 that is induced during viral infection [19]. sORFs have also recently been identified in miR-200a and miR-200b pri-miRs, the human orthologues of the Drosophila miR-8. Like miR-200a and miR-200b, miPEP-200a and b over-expression in prostate cancer cells inhibits migration of these cells by regulating the vimentin-mediated pathway, suggesting that the miPEP-coding function of pri-miRs is present in humans [18]. Accordingly, most recently, micropeptides encoded by MIR155HG and MIR34HG were described to be involved in autoimmune inflammation by controlling antigen presentation [20] and mitochondrial function respectively [21] via their interaction with different HSP proteins. It is interesting to note however, that these miPEPs appear to be involved in infections/pathologic-conditions, hence suggesting that revealing miPEP function might be largely dependent on the biological context.
Ribosome-associated lncRNAs has been considered to constitute a hallmark of protein translation. Here we found a signal of ribosome binding in the pri-miR of several Drosophila miRs genes. Furthermore, we showed that the initiation codon of the miORF present within pri-mR-8 is in a favorable translational context. Indeed, after having generated specific antibodies, we detected a peptide co-migrating with in vitro translated miPEP-8 in fly extracts. However, an alternative possibility proposed by others is that ribosome marks illustrate a mechanism for cellular control of lncRNA levels through ribosome degradation-promoting activity [57, 60]. It will be of interest to investigate further whether the short ORFs present in pri-miRs are able to influence their regulation by controlling their stability and degradation as it has been shown for other coding genes. Finally, the molecules that give rise to miR-8/miPEP-8 are probably not the same ones since Drosha processing would separate the ORF from the poly(A) tail and thereby cause rapid decapping and degradation of the ORF-containing fragment. In light with these considerations, it is difficult to conclude on a pervasive coding capacity of pri-miRs in Drosophila. Future work will determine both in plants and animals whether all of them are sources of miPEPs and to what extent their auto-regulatory capacity and/or modes of action are diverse and specific.
Conclusion
Many studies performed recently have led to functional characterization of a handful of additional SEPs in the plant and animal kingdom. Illustrating the diversity of functions of these new players, these SEPs were identified from different sources of RNAs and play different roles [9, 61, 62]. Among these, contrasting with their initial definition as non-coding, pioneer works in plants showed that even precursors transcript of miRs produces SEPs involved in an autoregulatory feedback loop. By addressing the conservation of this mechanism in animals, our findings combined with others confirm that miR-encoded genes probably represent evolutionary conserved bi-functional RNAs carrying coding and non-coding functions. However, contrasting with the mechanism described in plants, our data shed light on the diverse functions fulfilled by microRNA-encoded-peptides despite their poor conservation among Drosophila species.
Methods
Fly strains and genetics
Drosophila flies were maintained on standard cornmeal-yeast medium (Dutscher). Experiments were performed at 25 °C when miR-8 GAL4 (NP5247) was used as driver. For the experiments of wing phenotype of flies expressing transgenes under the control of MS1096 Gal4, crosses were placed at 28 °C. UAS-pri-miR-8 and UAS-miPEP-8 transgenic lines were inserted in attP86F site through PhiC31-mediated integration. Injections were performed by Bestgene Inc. (USA). Generating pri-miR-8 fly founder line: pri-miR-8 fly founder line was designed and generated by inDroso Functional Genomics (Rennes, France) using CRISPR/Cas9. The pri-miR-8 fly founder line was generated by excising from position 16830745 to 16831521 on Chromosome 2R arm and replacing it by an attP::loxP::3xP3-dsRED::loxP cassette (Additional file 1: Fig. S8). The two following guide RNA sequences were used to cut on either side of the pri-miR-8: CACATATG|CAACGGAAAGAG and GTTGGTGG|TACTGAAGGTTA. The edited region was verified by sequencing. The two pri-miR-8 constructs in pattB were inserted at the ΔmiR-8 created P site. Three independent transformants were used for analyses. The same strategy was used to generate the driver miR-8 GAL4, ΔmiR-8. The miPEP-8 alternative form creating a premature stop codon in miPEP-8 was derived from the DGRP-859 line, outcrossed into white recipient flies and kept over CyO. Experiments are the sum of at least 3 independent crosses. n indicates the number of individuals analyzed. For wing measurements, young flies (2–5 days) of the appropriate genotypes were stored in ethanol. For analysis of wings, females adult wings were removed in wash buffer (PBS and 0.1% Triton X-100) and mounted on a slide in 80% glycerol in PBS as described [63]. Wings or wing discs images were acquired on a Zeiss Axiozoom stereomicroscope. Measurements of wing size were performed using ImageJ software.
Molecular methods
For cloning procedure, miPEP-8, miR-8 or pri-miR-8 plasmids were constructed from PCR amplification of genomic DNA, gene synthesis or by RNA reverse transcription from S2 cells or adult Drosophila RNA and cloned in pUAS-attB vector constructs using the In-fusion HD cloning kit (Takarabio) according to the manufacturer specification. All constructs were verified by sequencing. For quantitative PCR experiments, total RNA was isolated from young adult fruit flies (2–5 days) or S2 cells using TRI Reagent (Sigma) according to the manufacturer specifications, followed by RQ1DNase treatment (promega) according the manufacturer specifications. The cDNA template was synthesized using SuperScript III (Invitrogen) with oligo-dT18 as anchor primers. Quantitative real-time PCR was performed on the LightCycler 480 Instument II (Roche Life Science) using LightCycler480 SYBR GREEN I master (Roche Life Science). The mRNA abundance of the examined genes was estimated by qPCR. For the endogenous pri-miR or coding genes, RP49 and tubulin genes were used as reference genes and used for normalization. For quantifying mature miRNA, stem loop PCR conditions were set up and the small RNAs U14 and Sno442 were used as reference. Datas presented are the same whatever the reference gene used. When the S2 cells are transiently transfected, the co-transfected pActin-GAL4 vector (Addgene # 24344) was used to monitor transfection efficiency. Typically, each experiment presented was performed with four replicates processed independently and was repeated in time at least three times. All experiments were taken into account and “n” indicate the total number of biological replicates used for analyses. Primers used in the qPCR are listed in the Additional file 1: Fig. S13.
RNA analysis
For Rib-seq analyses, Dmel primiR three frames translations have been performed with transeq (Emboss suite 6.6.0). A homemade script written in Perl was generated to compare the resulting translated peptides to the ribo-seq sORF-encoded peptides described in [4, 46, 47]. RNA-seq was processed by genewiz (Germany). Each dataset contains five independent biological replicates of control miR-8 and miPEP-8 over-expressing S2 cells RNA-seq. The reads were subjected to standard quality control (QC) and filtered according to the following parameters: (1) trimming and cleaning reads that aligned to primers and/or adaptors, (2) reads with over 50% of low-quality bases (quality value ≤15) in one read and (3) reads with over 10% unknown bases (N bases). We used Trimmomatic software (v0.36) to remove primers and bad quality reads. After filtering, we removed short reads (parameters were used with default values). Gene and PSI lists for each dataset were compared to identify common events between them. For RNAseq analysis, htseq-count files were analyzed using the version 3.24.3 of package EdgeR [64], in order to normalize raw counts by “trimmed mean of M-values” (TMM), and test differential expression using the negative binomial distribution. RNA-seq analysis: raw p values were adjusted with the Benjamini–Hochberg procedure to control the false discovery rate (FDR). A gene was declared differentially expressed if it is adjusted p value ≤0.05. Heat map parameters applied: row-by-row normalization by standardization (mean and standard deviation). GO term analysis was performed with PANTHER (http://pantherdb.org/) [65]. Sashimi plots were created with IGV (Integrative Genomics Viewer, https://igv.org/) and GWIPS-viz [42]. Statistical analyses were performed using the version 3.5.2 of R software and Bioconductor packages. For QPCR analysis, the version 1.3-1 of package Agricolae was used.
Cell culture and western blot and luciferase assays
Drosophila S2 cells were maintained in Schneider’s medium (Invitrogen) supplemented with 10% fetal bovine serum (Sigma), 50 U/ml penicillin and 50 μg/ml streptomycin (Invitrogen) at 25 °C. For western blot experiments, miPEPs sequences cloned into pF25A ICE T7 Flexi vector were expressed in vitro using TnT® T7 Insect Cell Extract Protein Expression System (Promega). For cells extracts and Drosophila extracts, we directly freeze them in nitrogen just before western blot. Proteins were prepared in Laemli buffer (63 mM Tris HCl pH 7.5, 2% SDS, 5% 2-mercaptoethanol) and run on SDS-PAGE according to [13]. Primary antibodies used for western were as follows: rabbit anti-miPEP-8 were raised against the sequence KQSDKQNSKERKKNTQI (generated and affinity purified by Agro-bio, France), mouse anti-GAPDH (ThermoFisher AM4300), rabbit anti-Sra-1 (1/1000, provided by A. Giangrande, IGBMC CNRS, France), mouse anti-peanut (1/100, DSHB, USA) and rabbit anti-ABP-1 (1:250, provided by Michael Kessels Jena University Hospital, Germany). HRP-conjugated secondary antibodies are from Santa Cruz Biotechnology (1/10000 sc-516102). For luciferase assays, in each experiment, S2 cells were transfected in quadruplicate, in 24-wells plates (700,000 cells/well) using FuGene HD transfection Reagent (Promega). Experiments were repeated timely independently at least 3 times. After 48 h of transfection, cells were washed with PBS and lysed with 100 μL passive lysis buffer (Dual Luciferase Reporter Assay System, Promega). Firefly luciferase (FL) and Renilla luciferase (RL) activities were then quantified with Dual Luciferase Reporter Assay (Promega) using 50 μl of reagents/well and measure using a Greiner luminometer instrument.
Statistical analyses
Statistical analyses were performed using GraphPad Prism and illustrated as follow: *p value< 0.05, **p value< 0.01, ***p value< 0.001 and ****p value< 0.0001. In all experiments, results represent mean ± s.e.m. n represents the number of biological independent replicates. Normality test was first performed using D’Agostino Pearson test. If the distribution is Gaussian and in order to detect a global difference between all groups, one-way ANOVA was performed using one-way analyses of variances followed by Bartlett’s test for equal variance and Bonferroni’s multiple comparison tests. In other cases, when variance or sample sizes are not equal, non-parametric analyses were performed using Kruskal-Wallis test to detect a global difference between all groups followed by comparisons between two groups performed using adjustments for multiple comparisons. When only two groups were compared, a Mann-Whitney test was performed.
Availability of data and materials
The RNAseq datasets generated during the current study are available in the NCBI Gene Expression Omnibus (GEO) repository under the accession number BioProject ID PRJNA645280 [66] [https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA645280&o=acc_s%3Aa].
References
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94.
Gardini A, Shiekhattar R. The many faces of long noncoding RNAs. FEBS J. 2015;282:1647–57.
Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. 2018;19:143–57.
Aspden JL, Eyre-Walker YC, Phillips RJ, Amin U, Mumtaz MA, Brocard M, Couso JP. Extensive translation of small open reading frames revealed by poly-Ribo-Seq. Elife. 2014;3:e03528.
Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, Giraldez AJ. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–93.
de Andres-Pablo A, Morillon A, Wery M. LncRNAs, lost in translation or licence to regulate? Curr Genet. 2017;63:29–33.
Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802.
Plaza S, Menschaert G, Payre F. In search of lost small peptides. Annu Rev Cell Dev Biol. 2017;33:391–416.
Rathore A, Martinez TF, Chu Q, Saghatelian A. Small, but mighty? Searching for human microproteins and their potential for understanding health and disease. Expert Rev Proteomics. 2018;15:963–5.
Ruiz-Orera J, Alba MM. Translation of small open reading frames: roles in regulation and evolutionary innovation. Trends Genet. 2019;35:186–98.
Yeasmin F, Yada T, Akimitsu N. Micropeptides encoded in transcripts previously identified as long noncoding RNAs: a new chapter in transcriptomics and proteomics. Front Genet. 2018;9:144.
O'Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). 2018;9:402.
Lauressergues D, Couzigou JM, Clemente HS, Martinez Y, Dunand C, Becard G, Combier JP. Primary transcripts of microRNAs encode regulatory peptides. Nature. 2015;520:90–3.
Couzigou JM, Andre O, Guillotin B, Alexandre M, Combier JP. Use of microRNA-encoded peptide miPEP172c to stimulate nodulation in soybean. New Phytol. 2016;211:379–81.
Sharma A, Badola PK, Bhatia C, Sharma D, Trivedi PK. Primary transcript of miR858 encodes regulatory peptide and controls flavonoid biosynthesis and development in Arabidopsis. Nat Plants. 2020;6:1262–74.
Chen QJ, Deng BH, Gao J, Zhao ZY, Chen ZL, Song SR, Wang L, Zhao LP, Xu WP, Zhang CX, Ma C, Wang SP. A miRNA-Encoded Small Peptide, vvi-miPEP171d1, Regulates Adventitious Root Formation. Plant Physiol. 2020;183:656–70.
Zhang QL, Su LY, Zhang ST, Xu XP, Chen XH, Li X, Jiang MQ, Huang SQ, Chen YK, Zhang ZH, Lai ZX, Lin YL. Analyses of microRNA166 gene structure, expression, and function during the early stage of somatic embryogenesis in Dimocarpus longan Lour. Plant Physiol Biochem. 2020;147:205–14.
Fang J, Morsalin S, Rao V, Reddy ES. Decoding of non-coding DNA and non-coding RNA: pri-micro RNA-encoded novel peptides regulate migration of cancer cells. J Pharm Sci Pharmacol. 2017;3:23–7.
Razooky BS, Obermayer B, O'May JB, Tarakhovsky A. Viral infection identifies micropeptides differentially regulated in smORF-containing lncRNAs. Genes (Basel). 2017;8:206.
Niu LM, Lou FZ, Sun Y, Sun LB, Cai XJ, Liu ZY, Zhou H, Wang H, Wang ZK, Bai J, Yin QQ, Zhang JX, Chen LJ, Peng DH, Xu ZY, Gao YY, Tang SB, Fan L, Wang HL. A micropeptide encoded by lncRNA MIR155HG suppresses autoimmune inflammation via modulating antigen presentation. Sci Adv. 2020;6:eaaz2059.
Kang M, Tang B, Li JX, Zhou ZY, Liu K, Wang RS, Jiang ZY, Bi FF, Patrick D, Kim D, Mitra AK, Yang-Hartwich Y. Identification of miPEP133 as a novel tumor-suppressor microprotein encoded by miR-34a pri-miRNA. Mol Cancer. 2020;19:143.
Sander M, Herranz H. MicroRNAs in Drosophila cancer models. Adv Exp Med Biol. 2019;1167:157–73.
Jin H, Kim VN, Hyun S. Conserved microRNA miR-8 controls body size in response to steroid signaling in Drosophila. Genes Dev. 2012;26:1427–32.
Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell. 2007;131:136–45.
Umegawachi T, Yoshida H, Koshida H, Yamada M, Ohkawa Y, Sato T, Suyama M, Krause HM, Yamaguchi M. Control of tissue size and development by a regulatory element in the yorkie 3'UTR. Am J Cancer Res. 2017;7:673–87.
Hyun S, Lee JH, Jin H, Nam J, Namkoong B, Lee G, Chung J, Kim VN. Conserved MicroRNA miR-8/miR-200 and its target USH/FOG2 control growth by regulating PI3K. Cell. 2009;139:1096–108.
Antonello ZA, Reiff T, Ballesta-Illan E, Dominguez M. Robust intestinal homeostasis relies on cellular plasticity in enteroblasts mediated by miR-8-Escargot switch. EMBO J. 2015;34:2025–41.
Antonello ZA, Reiff T, Dominguez M. Mesenchymal to epithelial transition during tissue homeostasis and regeneration: patching up the Drosophila midgut epithelium. Fly (Austin). 2015;9:132–7.
Boukhatmi H, Bray S. A population of adult satellite-like cells in Drosophila is maintained through a switch in RNA-isoforms. Elife. 2018;7:e35954
Morante J, Vallejo DM, Desplan C, Dominguez M. Conserved miR-8/miR-200 defines a glial niche that controls neuroepithelial expansion and neuroblast transition. Dev Cell. 2013;27:174–87.
Loya CM, McNeill EM, Bao H, Zhang B, Van Vactor D. miR-8 controls synapse structure by repression of the actin regulator enabled. Development. 2014;141:1864–74.
Lu CS, Zhai B, Mauss A, Landgraf M, Gygi S, Van Vactor D. MicroRNA-8 promotes robust motor axon targeting by coordinate regulation of cell adhesion molecules during synapse development. Philos Trans R Soc Lond Ser B Biol Sci. 2014;369:20130517.
Bolin K, Rachmaninoff N, Moncada K, Pula K, Kennell J, Buttitta L. miR-8 modulates cytoskeletal regulators to influence cell survival and epithelial organization in Drosophila wings. Dev Biol. 2016;412:83–98.
Choi IK, Hyun S. Conserved microRNA miR-8 in fat body regulates innate immune homeostasis in Drosophila. Dev Comp Immunol. 2012;37:50–4.
Etebari K, Asgari S. Conserved microRNA miR-8 blocks activation of the Toll pathway by upregulating Serpin 27 transcripts. RNA Biol. 2013;10:1356–64.
Kennell JA, Cadigan KM, Shakhmantsir I, Waldron EJ. The microRNA miR-8 is a positive regulator of pigmentation and eclosion in Drosophila. Dev Dyn. 2012;241:161–8.
Kennell JA, Gerin I, MacDougald OA, Cadigan KM. The microRNA miR-8 is a conserved negative regulator of Wnt signaling. Proc Natl Acad Sci U S A. 2008;105:15417–22.
Lee GJ, Hyun S. Multiple targets of the microRNA miR-8 contribute to immune homeostasis in Drosophila. Dev Comp Immunol. 2014;45:245–51.
Lee GJ, Jun JW, Hyun S. MicroRNA miR-8 regulates multiple growth factor hormones produced from Drosophila fat cells. Insect Mol Biol. 2015;24:311–8.
Sander M, Eichenlaub T, Herranz H. Oncogenic cooperation between Yorkie and the conserved microRNA miR-8 in the wing disc of Drosophila. Development. 2018;145:dev153817.
Vallejo DM, Caparros E, Dominguez M. Targeting Notch signalling by the conserved miR-8/200 microRNA family in development and cancer cells. EMBO J. 2011;30:756–69.
Michel AM, Fox GM, Kiran A, De Bo C, O'Connor PB, Heaphy SM, Mullan JP, Donohue CA, Higgins DG, Baranov PV. GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res. 2014;42:D859–64.
Zeng C, Fukunaga T, Hamada M. Identification and analysis of ribosome-associated lncRNAs using ribosome profiling data. BMC Genomics. 2018;19:414.
Hsu PY, Calviello L, Wu HL, Li FW, Rothfels CJ, Ohler U, Benfey PN. Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc Natl Acad Sci U S A. 2016;113:E7126–35.
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23.
Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife. 2013;2:e01179.
Kronja I, Whitfield ZJ, Yuan BB, Dzeyk K, Kirkpatrick J, Krijgsveld J, Orr-Weaver TL. Quantitative proteomics reveals the dynamics of protein changes during Drosophila oocyte maturation and the oocyte-to-embryo transition. Proc Natl Acad Sci U S A. 2014;111:16023–8.
Enderle D, Beisel C, Stadler MB, Gerstung M, Athri P, Paro R. Polycomb preferentially targets stalled promoters of coding and noncoding transcripts. Genome Res. 2011;21:216–26.
Qian J, Zhang Z, Liang J, Ge Q, Duan X, Ma F, Li F. The full-length transcripts and promoter analysis of intergenic microRNAs in Drosophila melanogaster. Genomics. 2011;97:294–303.
Lucas KJ, Roy S, Ha J, Gervaise AL, Kokoza VA, Raikhel AS. MicroRNA-8 targets the Wingless signaling pathway in the female mosquito fat body to regulate reproductive processes. Proc Natl Acad Sci U S A. 2015;112:1440–5.
Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu DH, Casillas S, Han Y, Magwire MM, Cridland JM, Richardson MF, Anholt RRH, Barron M, Bess C, Blankenburg KP, Carbone MA, Castellano D, Chaboub L, Duncan L, Harris Z, Javaid M, Jayaseelan JC, Jhangiani SN, Jordan KW, Lara F, Lawrence F, Lee SL, Librado P, Linheiro RS, Lyman RF, et al. The Drosophila melanogaster genetic reference panel. Nature. 2012;482:173–8.
Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176.
Ji Z, Song R, Regev A, Struhl K. Many lncRNAs, 5’UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife. 2015;4:e08890.
Ladoukakis E, Pereira V, Magny EG, Eyre-Walker A, Couso JP. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011;12:R118.
Pauli A, Valen E, Schier AF. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 2015;37:103–12.
Raj A, Wang SH, Shim H, Harpak A, Li YI, Engelmann B, Stephens M, Gilad Y, Pritchard JK. Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling. Elife. 2016;5:e13328.
Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, Geisler S, Hu W, Coller J, Baker KE. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–66.
Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet. 2014;15:193–204.
Saghatelian A, Couso JP. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat Chem Biol. 2015;11:909–16.
Carlevaro-Fita J, Rahim A, Guigo R, Vardy LA, Johnson R. Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA. 2016;22:867–82.
Chugunova A, Loseva E, Mazin P, Mitina A, Navalayeu T, Bilan D, Vishnyakova P, Marey M, Golovina A, Serebryakova M, Pletnev P, Rubtsova M, Mair W, Vanyushkina A, Khaitovich P, Belousov V, Vysokikh M, Sergiev P, Dontsova O. LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism. Proc Natl Acad Sci U S A. 2019;116:4940–5.
Khitun A, Ness TJ, Slavoff SA. Small open reading frames and cellular stress responses. Mol Omics 2019;15:108–16.
Gault WJ, Olguin P, Weber U, Mlodzik M. Drosophila CK1-gamma, gilgamesh, controls PCP-mediated morphogenesis through regulation of vesicle trafficking. J Cell Biol. 2012;196:605–21.
McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97.
Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–66.
Montigny A, Tavormina P, Duboe C, San Clémente H, Aguilar M, Valenti P, Lauressergues D, Combier JP, Plaza S: Data for « Drosophila primary microRNA-8 encodes a microRNA encoded peptide acting in parallel of miR-8 ». 2020. Available from https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA645280&o=acc_s%3Aa. Accessed 31 Mar 2021.
Acknowledgements
We thank C. Dozier and the « Peptides & small RNAs » team members for critical reading and their helpful comments on the manuscript. We thank H. Boukhatmi, A. Giangrande, S. Cohen and F. Juge for sharing flies, constructs and antibodies. We thank the FRAIB RIO Imaging facility for microscopy. We acknowledge the Bloomington Drosophila Stock Center (NIH P40OD018537) and the Drosophila Stock Center, KYOTO Stock Center (DGRC) in Kyoto Institute of Technology, for providing fly stocks, the Developmental Studies Hybridoma Bank (DSHB) for antibodies and DNASU and Addgene for plasmids.
Review history
The review history is available as Additional file 6.
Peer review information
Anahita Bishop was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Funding
This work was funded by the French ANR project BiomiPEP (ANR-16-CE12-0018-01) and the Association de Recherche sur le Cancer (ARC PGA1RF2018206987). It was carried out in the Laboratoire de Recherche en Sciences Végétales, which belongs to the Laboratoire d’Excellence entitled TULIP (ANR-10-LABX-41). PT was supported by a PhD fellowship from the ANR BiomiPEP and AM from the ARC, respectively.
Author information
Authors and Affiliations
Contributions
SP and JPC conceived the project. SP designed the research. AM, PT, CD, PV, DL and SP performed the experiments. HSC and MA performed bioinformatics analyzes. SP, AM, PT and JPC wrote the paper. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Figures S1 to S13.
Additional file 2.
list of sORF peptides defined from Rib-seq analyses.
Additional file 3.
Normalized RNA-seq values from S2 cells.
Additional file 4.
Protein ANalysis THrough Evolutionary Relationships (PANTHER) Overrepresentation Test.
Additional file 5.
Uncropped Western blots.
Additional file 6.
Review history.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Montigny, A., Tavormina, P., Duboe, C. et al. Drosophila primary microRNA-8 encodes a microRNA-encoded peptide acting in parallel of miR-8. Genome Biol 22, 118 (2021). https://doi.org/10.1186/s13059-021-02345-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-021-02345-8