
Abstract
Allele expression (AE) analysis robustly measures cis-regulatory effects. Here, we present and demonstrate the utility of a vast AE resource generated from the GTEx v8 release, containing 15,253 samples spanning 54 human tissues for a total of 431 million measurements of AE at the SNP level and 153 million measurements at the haplotype level. In addition, we develop an extension of our tool phASER that allows effect sizes of cis-regulatory variants to be estimated using haplotype-level AE data. This AE resource is the largest to date, and we are able to make haplotype-level data publicly available. We anticipate that the availability of this resource will enable future studies of regulatory variation across human tissues.
Similar content being viewed by others


Background
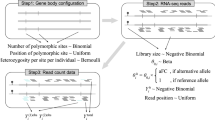
Allelic expression (AE, also known as allele-specific expression or ASE) analysis is a powerful technique that can be used to measure the expression of gene alleles relative to one another within single individuals. This makes it well suited to measure cis-acting regulatory variation using imbalance between alleles in heterozygous individuals (Fig. 1a) [1]. AE analysis can capture both common cis-regulatory variation, for example, expression quantitative trait loci (eQTLs), and rare regulatory variation [2]. It can also be used to measure allele-specific epigenetic effects such as parent of origin imprinting [3].

Capturing cis-regulatory effects with phased allelic expression data. a The presence of a heterozygous cis-regulatory variant or eQTL produces an expression-level imbalance between the two haplotypes, which can be detected using allelic expression analysis. b RNA-seq reads overlapping heterozygous SNPs in expressed regions of the gene can be used to quantify the expression of alleles relative to one another. These SNPs can be phased with each other and their counts aggregated to produce haplotype-level expression estimates, or haplotypic counts. The effects of regulatory variants can be captured by phasing them with haplotypic counts. c Spearman correlation across the 49 GTEx v8 tissues where eQTLs were called between eQTL effect size (allelic fold change, aFC) and effect size measured using AE data from the single SNP with the highest coverage (SNP AE) or haplotype-level AE generated with phASER (phASER). Results are shown with and without allelic mapping bias correction from WASP. In each tissue, only a single top significant (FDR < 5%) eQTL per gene was analyzed. p values were calculated using a Wilcoxon paired signed rank test. For boxplots, bottom whisker: Q1 − 1.5*interquartile range (IQR), top whisker: Q3 + 1.5*IQR, box: IQR, and center: median
In practice, AE analysis uses RNA-seq reads that overlap heterozygous single nucleotide polymorphisms (SNPs), where the SNP can be used to assign the read to an allele. These heterozygous SNPs capture the cumulative effects of cis-regulatory variation acting on each allele. Allelic imbalance occurs when the two alleles of a gene are expressed at different levels. The magnitude of the imbalance can be quantified by allelic fold change (aFC) [1], and the statistical significance of the imbalance can be evaluated using binomial-based statistics to account for the count-based nature of the data [4]. In some cases, these effects can be caused by the SNPs being used to measure AE themselves, for example, stop-gain variants that cause nonsense-mediated decay (NMD) [5], but often they simply capture the effects of other cis-acting variation. Traditionally, a single SNP has been used to measure AE, by taking the SNP with the highest coverage per gene. However, as a result of improvements in genome phasing, data can be aggregated across SNPs to produce estimates of AE at the haplotype level (Fig. 1b). We have previously developed a tool, phASER, which does this systematically, in a way that uses the information contained within reads to improve phasing, while preventing double counting of reads across SNPs to improve the quality of data generated [6].
In this work, we present and demonstrate the utility of an AE resource generated using the Genotype Tissue Expression (GTEx) version 8 data release comprising RNA-seq data from 54 tissues and 838 individuals, for a total of 15,253 samples [7]. We generated both SNP-level and haplotype-level AE data. While the SNP-level data is available to approved users through dbGaP, the haplotype-level data does not contain identifiable information, and we were thus able to make it publicly available on the GTEx portal. Finally, we developed an addition to phASER, called phASER-POP which makes it easy to generate population-scale, haplotype-level AE data and calculate effect sizes for regulatory variants.
Results and discussion
Both SNP-level and haplotype-level AE data were generated for each GTEx sample using current best practices, both with and without using WASP filtering [8] to reduce the mapping bias that is sometimes present in AE analysis, resulting in 4 data types per sample (Additional file 1: Fig. S1, “Data generation and availability” section in the “Methods” section). Across samples, this produced over 431 million measurements of AE at the SNP level and 153 million measurements of AE at the haplotype level. To demonstrate the ability of these data to robustly capture cis-regulatory effects and also benchmark the four data types relative to one another, we estimated eQTL effect sizes across the 49 tissues where eQTLs were mapped from AE data using allelic fold change (aFC) and compared them to those derived from eQTL mapping [7]. The effect sizes were quantified using aFC for both AE and eQTL data. To make it easier to generate aFC estimates for regulatory variants from phASER data, we developed a new add-on to the software package, phASER-POP, eliminating the need for custom scripts (Additional file 1: Fig. S2). Briefly, phASER-POP integrates genotype calls and haplotype-level AE data across individuals and phases each regulatory variant of interest (e.g., eQTL) in each individual with their AE data. It then calculates statistics, including aFC per sample, and its median across samples for individuals that are heterozygous for the variant. At the sample level, aFC is a net expression fold difference between the two haplotypes in an individual that is affected by all heterozygous regulatory variants, including other eQTLs and rare regulatory variation, and thus can differ from the expected aFC derived from eQTL mapping. However, the median aFC across all individuals in a population that is heterozygous for a given eQTL can be used as a robust estimate of its effect size [1]. The software is described in full detail in the “Methods” section.
To characterize the GTEx AE resource, we first compared aFC estimates calculated for GTEx eQTLs between SNP- and haplotype-level AE data. We found high correlations between AE and eQTL estimates, with a median Spearman rho of 0.80 across tissues for SNP-level data and 0.83 for haplotype-level data generated by phASER (Fig. 1c). Haplotype-level correlations were significantly higher than SNP-level correlations (p = 3.55e−15, Wilcoxon paired signed rank test) while at the same time producing estimates for a median of 20% more eQTLs (Additional file 1: Fig. S3). Based on this, we recommend using the haplotype-level data for most downstream analyses, as it yields more data of a higher quality. However, there are some circumstances when the SNP-level data should be used. For example, when analyzing allelic splicing, the haplotype-level data is not appropriate because it spans the entire transcript, whereas only SNPs within the exon(s) or intron(s) of interest should be analyzed. Furthermore, when analyzing transcribed variants with post-transcriptional effects on gene expression, such as stop-gain or splice variants, SNP-level AE data from the variant of interest is more straightforward to analyze.
Next, we assessed the effect of read mapping bias correction on allelic expression analysis by comparing eQTL and AE effect size correlations with and without WASP filtering. WASP filtering significantly improved correlations for both SNP- (p = 2.49e−13, median improvement 1.22%) and haplotype- (p = 3.55e−15, median improvement 1.28%) level data (Fig. 1c). Since WASP works by removing, rather than correcting reads with mapping bias, we compared the number of eQTLs for which an aFC estimate could be calculated and found only a small 3.5% reduction (Additional file 1: Fig. S3d). We therefore recommend using WASP-filtered data for most downstream analyses. This is particularly important if the aim is to identify strong signals of allelic imbalance, which can often be false positives due to mapping bias. We encourage users of the resource to assess the impact of WASP filtering for their own use case, so have included the unfiltered AE data for comparison.
Next, we characterized the WASP-filtered AE data. In the GTEx RNA-seq data, at a minimum coverage of 8 reads, samples had a median of 7,607 genes with AE data at the SNP level and 10,043 genes at the haplotype level, and this dropped as a function of increasing coverage thresholds (Additional file 1: Fig. S4). With the same coverage threshold, at the tissue level and excluding tissues with small sample sizes (N < 70) where eQTL mapping was not performed, there were a median of 18,042 genes with a median of 128 samples per gene using haplotype-level AE data, rendering the data set well-powered to detect cis-regulatory effects (Fig. 2a). The median number of samples with AE data per gene was largely dependent on tissue sample size, ranging from 39 for kidney cortex (N = 73 samples) to 321 for thyroid (N = 574 samples). The number of genes with AE data was correlated with both sample size (rho = 0.41) and the number of expressed genes (rho = 0.82), with the two cell lines having the lowest number of genes with AE data (LCLs = 15,804, fibroblasts = 16,526) and the testis having the largest number of genes with AE data (21,952) despite an intermediate sample size of 322 (Additional file 2: Table S1). This was likely driven by the number of expressed genes in testes, which was the highest across all tissues.

The GTEx v8 haplotype-level allelic expression resource. a Number of genes per tissue with haplotype-level AE data (AE genes) in at least 1 individual versus the median number of samples with data per gene. b Percentage of AE genes with significant allelic imbalance (binomial test, gene-level FDR < 5%) in at least n samples per gene using all samples (blue) or excluding samples heterozygous for any top (FDR < 5%) or independent GTEx eQTL (permutation p < 1e−4) (red). Faded points are values for individual tissues, and solid points are the median across tissues. Proportions above data points indicate the reduction in percentage of AE genes with imbalance after removing eQTL heterozygotes. A full summary of these statistics across tissues and sample thresholds is available in Additional file 3: Table S2. c The effect of the number of heterozygous variants in or proximal to gene promoters (< 10 kb upstream of TSS) on allelic imbalance stratified by minor allele frequency. Plotted values are effect estimates and 95% confidence intervals (see the “Promoter variant effect modeling” section in the “Methods” section)
Finally, we sought to demonstrate the pervasiveness of cis-regulatory effects that can be captured with this resource. We found that even strong regulatory effects, where one allele was expressed at ≥ 2x the level of the other allele, are widely present, even for protein-coding genes, with 53% of protein-coding genes showing such an effect in at least one tissue and at least 50 individuals (Additional file 1: Fig. S5). Considering all genes, we found that a median of 10,183 genes (or a median of 56% of those genes with AE data) per tissue exhibited significant allelic imbalance (binomial test, FDR < 5% at the gene level) in at least one sample, indicating the wide-spread nature of cis-regulatory effects (Fig. 2b). Removing individuals that were heterozygous for any known GTEx eQTL (“GTEx eQTLs” section in the “Methods” section) only resulted in a median reduction of 7.5% in the number of genes with significant imbalance in at least one sample, demonstrating the potential of this resource to identify additional regulatory effects, including rare regulatory effects, that are not captured in eQTL analysis. To further demonstrate this potential, we modeled allelic imbalance as a function of the minor allele frequency and number of heterozygous variants found in or proximal to gene promoters (< 10 kb upstream of TSS). As expected, we found that rare variants tended to have larger effects on allelic imbalance than common variants, with the rarest class of variants analyzed (MAF < 0.005 in GTEx) having the strongest effects (Fig. 2c).
Conclusion
In this work, we used the GTEx v8 release to produce a vast allelic expression resource, consisting of hundreds of millions of measurements. We generated SNP- and haplotype-level data, which provides better estimates of allelic expression for a greater number of genes. These data have numerous uses for the study of regulatory variation. SNP-level data from the previous v6 AE dataset [2] has been extensively used to study gene regulation, for example, to study the effects of rare regulatory variation [9], X chromosome inactivation [10], Neanderthal-introgressed regulatory variation [11], interaction between regulatory and coding variants [12], and regulatory constraint in the context of rare disease [13]. The haplotype-level v8 data presented here have similarly found broad use for studying gene regulation. For example, they have been used to replicate sex-, population-, and cell type-specific eQTLs [7, 14] as well as capture the effects of rare regulatory variants [15] and study cis-domains of lncRNA regulation [16]. By making haplotype-level AE data publicly available for the first time, we anticipate that this resource will find similarly broad use as the eQTL data it complements.
Methods
Data generation and availability
Paired-end 75-bp Illumina RNA-seq reads were aligned to hg38 using STAR [17] v2.5.3a (without allelic mapping bias correction) and v2.6.0c (with allelic mapping bias correction) in two-pass mode, and with allelic mapping bias correction enabled via the --waspOutputMode option which replicates the approach in van de Geijn et al. [8] (the full settings of the alignment pipeline are described at https://github.com/broadinstitute/gtex-pipeline). All data was generated with or without using this feature and is indicated by “_WASP_” in the file names.
SNP-level AE data was generated using the GATK ASEReadCounter tool v3.8-0-ge9d806836 with the following settings: -U ALLOW_N_CIGAR_READS -minDepth 1 --allow_potentially_misencoded_quality_scores --minMappingQuality 255 --minBaseQuality 10. Raw SNP-level data, consisting of the GATK tool output, were aggregated per subject across all tissues. Raw autosomal SNP-level data, for SNPs with ≥ 8 reads, was annotated by assigning heterozygous SNPs to genes using Gencode v26, calculating the expected null ratio for each combination of ref/alt allele [4], calculating a binomial p value by comparing to the expected null ratio, calculating a multiple hypothesis corrected p value per tissue using Benjamini-Hochberg, and flagging sites that overlapped low-mappability regions (75-mer mappability < 1 based on 75mer alignments with up to two mismatches based on the pipeline for ENCODE tracks and available on the GTEx portal), showed mapping bias in simulation [18], or had no more reads supporting two alleles than would be expected from sequencing noise alone, indicating potential genotyping errors (FDR < 1%, see Castel et al. [4] for the description of the test). The genotype warning test cannot distinguish between strong allelic expression and a true genotyping error and as a result should not be used when studying phenomena with expected mono-allelic expression (e.g., imprinting).
Haplotype-level data was generated using phASER v1.0.1 [6]. phASER was run using whole genome sequencing genotype calls that were population-phased with Shapeit v2.837 in read-backed phasing mode with whole genome sequencing reads [19]. phASER was run using all available RNA-seq libraries per subject. RNA-seq read-backed phased genotype data are provided (filename: phASER_GTEx_v8_merged.vcf.gz). Haplotypic expression was calculated using phASER Gene AE 1.2.0 and Gencode v26 gene annotations with min_haplo_maf 0.01. Haplotypic expression matrices containing all samples were generated using the “phaser_expr_matrix.py” script. This consists of a single string per sample per gene with the format “HAP_A_COUNT|HAP_B_COUNT.” One matrix was generated using only haplotypes that could be genome-wide phased such that the haplotype assignment is consistent across genes within an individual and with the phased VCF (filename: phASER_GTEx_v8_matrix.gw_phased.txt.gz). Another was generated that does not ensure genome-wide haplotype phasing across genes, which includes more counts, but makes the haplotype assignment of A/B arbitrary and unrelated across genes within an individual or the VCF (filename: phASER_GTEx_v8_matrix.txt.gz). The full settings of the haplotype-level AE pipeline are described at https://github.com/broadinstitute/gtex-pipeline/.
SNP-level data is available for authorized users via dbGaP under accession phs000424 (filenames: phe000039.v1.GTEx_v8_ASE.expression-matrixfmt-ase.c1.GRU.tar, phe000039.v1.GTEx_v8_ASE_WASP.expression-matrixfmt-ase.c1.GRU.tar) [20]. phASER-generated, haplotype-level data is available through the same dbGaP accession (folders GTEx_Analysis_v8_phASER and GTEx_Analysis_v8_phASER_WASP inside archive phe000037.v1.GTEx_v8_RNAseq.expression-data-matrixfmt.c1.GRU.tar) and on the GTEx Portal (http://gtexportal.org/).
Unless stated otherwise, all analyses were performed using only protein-coding and lncRNA genes.
Software and availability
The original phASER package produced gene-level haplotypic expression per individual [6]. We developed new additions to phASER (phASER-POP) that make it easier to analyze data across many samples, as is often done with gene expression quantifications. First, we developed a new addition to the software (phaser_expr_matrix.py) that enables the aggregation of gene-level haplotypic expression measurement files across samples to produce a single haplotypic expression matrix, where each row is a gene and each column is a sample. The values consist of a single string per sample per gene in the format “HAP_A_COUNT|HAP_B_COUNT.” This format is intended to facilitate downstream analyses of allelic expression.
Second, we developed a tool to make it easier to estimate effect sizes of regulatory variants using phASER haplotypic expression data (phaser_cis_var.py). As input, this script takes a phASER haplotype expression matrix, a phased VCF, and a list of regulatory variants (e.g., eQTLs) to calculate effect sizes for. To improve accuracy, the read-backed phased VCFs produced by phASER should be used, but first need to be combined across individuals, which can be performed using, e.g., “bcftools merge ind1.vcf.gz ind2.vcf.gz ….” Using these inputs, the tool phases each regulatory variant of interest with haplotype-level expression data in each individual. It then calculates numerous statistics, including allelic fold change (aFC) [1] per sample, and a median across samples for individuals that are heterozygous for the variant of interest. This median can be used as an estimate of regulatory variant effect size. aFC is calculated as log2((eqtl_alt_allele_haplotype_count+1)/(eqtl_ref_allele_haplotype_count+1)). The output also includes aFC estimates calculated for homozygous individuals and performs a ranksum test of absolute aFC in heterozygotes as compared to homozygotes. True regulatory variants are expected to have a significantly higher aFC in heterozygous individuals. 95% confidence intervals are included for all aFC estimates, and all underlying individual data, including haplotypic counts, are outputted.
The updated phASER package code along with extensive documentation is available through GitHub at https://github.com/secastel/phaser/tree/master/phaser_pop under the GNU General Public License v3 [21].
GTEx eQTLs
For comparison between eQTL effect size and allelic expression effect size, GTEx v8 top significant (FDR < 5%) eQTLs were used from 49 tissues [7]. This results in at most a single eQTL per gene in a given tissue. When quantifying the number of samples that are not heterozygous for a known eQTL but still show allelic imbalance, gene-level haplotypic expression levels were excluded for a sample if the individual was heterozygous for a top significant eQTL or a nominally significant (permutation p < 1e−4) independent eQTL in any of the 49 tissues.
Promoter variant effect modeling
The effects of regulatory variants in or proximal to gene promoters were modeled using haplotype-level allelic expression data. Briefly, for each individual, all heterozygous variants within 10 kb upstream of protein-coding or lincRNA gene transcription start sites (TSS) were retrieved and the median allelic imbalance for that gene across all tissues, measured using aFC, was calculated. For each individual by gene, the number of heterozygous variants (which could potentially cause allelic imbalance) falling into each of the following minor allele frequency (MAF) bins was calculated: 0.50–0.10, 0.10–0.05, 0.05–0.01, 0.01–0.005, 0.005–0. Bins were inclusive of variants whose MAF < upper bin limit and ≥ the lower bin limit. Using data from all genes by individuals, absolute aFC was modeled with a multivariate linear model (speedglm function in R) using the number of variants in each of the MAF bins as predictors. The coefficients for each of the predictors were then plotted along with their 95% confidence intervals (confint function in R) as a measure of the effect of the number of heterozygous variants in each MAF class on allelic imbalance, with a higher coefficient indicating a stronger effect (i.e., a larger allelic imbalance). Because allele frequencies were calculated within the GTEx cohort, only individuals of predominantly European ancestry (N = 699, determined by PCA) were included in the analysis, to ensure accurate allele frequency estimates. Without this filtering, population-specific variants, whose populations are not well represented in the GTEx cohort, may have inaccurate, likely underestimated allele frequencies, which can confound the analysis.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available to authorized users via dbGaP under accession phs000424 [20] and on the GTEx portal (http://gtexportal.org/). The updated phASER package code along with extensive documentation is available through GitHub at https://github.com/secastel/phaser/tree/master/phaser_pop under the GNU General Public License v3 [21].
References
Mohammadi P, Castel S, Brown A, Lappalainen T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 2017;27(11):1872–84.
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204.
Baran, Y., Subramaniam, M., Biton, A., Tukiainen, T., Tsang, E., Rivas, M., Pirinen, M., Gutierrez-Arcelus, M., Smith, K., Kukurba, K., Zhang, R., Eng, C., Torgerson, D., Urbanek, C., Consortium, t., Li, J., Rodriguez-Santana, J., Burchard, E., Seibold, M., MacArthur, D., Montgomery, S., Zaitlen, N., Lappalainen, T. (2015). The landscape of genomic imprinting across diverse adult human tissues. Genome Res 25(7), 927–936.
Castel S, Levy-Moonshine A, Mohammadi P, Banks E, Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015;16(1):195.
Rivas M, Pirinen M, Conrad D, Lek M, Tsang E, Karczewski K, Maller J, Kukurba K, DeLuca D, Fromer M, Ferreira P, Smith K, Zhang R, Zhao F, Banks E, Poplin R, Ruderfer D, Purcell S, Tukiainen T, Minikel E, Stenson P, Cooper D, Huang K, Sullivan T, Nedzel J, Consortium T, Consortium T, Bustamante C, Li J, Daly M, Guigo R, Donnelly P, Ardlie K, Sammeth M, Dermitzakis E, McCarthy M, Montgomery S, Lappalainen T, MacArthur D. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348(6235):666–9.
Castel S, Mohammadi P, Chung W, Shen Y, Lappalainen T. Rare variant phasing and haplotypic expression from RNA sequencing with phASER. Nat Commun. 2016;7(1):12817.
GTEx Consortium (2019). The GTEx Consortium atlas of genetic regulatory effects across human tissues. BioRxiv.
Geijn B, McVicker G, Gilad Y, Pritchard J. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods. 2015;12(11):1061–3.
Li X, Kim Y, Tsang EK, Davis JR, Damani FN, Chiang C, et al. The impact of rare variation on gene expression across tissues. Nature. 2017;550(7675):239–43.
Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550(7675):244–8.
McCoy RC, Wakefield J, Akey JM. Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell. 2017;168(5):916–27 e12.
Castel SE, Cervera A, Mohammadi P, Aguet F, Reverter F, Wolman A, et al. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat Genet. 2018;50(9):1327–34.
Mohammadi P, Castel SE, Cummings BB, Einson J, Sousa C, Hoffman P, et al. Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science. 2019;366(6463):351–6.
S. Kim-Hellmuth*, F. Aguet*, M. Oliva*, M. Muñoz-Aguirre, V. Wucher, S. Kasela, S.E. Castel, A.R. Hamel, A. Viñuela, A.L. Roberts, S. Mangul, X. Wen, G. Wang, A.N. Barbeira, D. Garrido-Martín, B. Nadel, Y. Zou, R. Bonazzola, J. Quan, A. Brown, A. Martinez-Perez, J.M. Soria, GTEx Consortium, G. Getz, E.T. Dermitzakis, K.S. Small, M. Stephens, H.S. Xi, H.K. Im, R. Guigó, A.V. Segrè, B.E. Stranger, K.G. Ardlie, T. Lappalainen, "Cell type specific genetic regulation of gene expression across human tissues," bioRxiv, 2019. https://doi.org/10.1101/806117.
N.M. Ferraro, B.J. Strober, J. Einson, X. Li, F. Aguet, A.N. Barbeira, S.E. Castel, J.R. Davis, A.T. Hilliard, B. Kotis, Y. Park, A.J. Scott, C. Smail, E.K. Tsang, K.G. Ardlie, T.L. Assimes, I. Hall, H.K. Im, GTEx Consortium, T. Lappalainen, P. Mohammadi, S.B. Montgomery, A. Battle, "Diverse transcriptomic signatures across human tissues identify functional rare genetic variation," bioRxiv, 2019. https://doi.org/10.1101/786053.
O.M. de Goede, N.M. Ferraro, D.C. Nachun, A.S. Rao, F. Aguet, A.N. Barbeira, S.E. Castel, S. Kim-Hellmuth, Y. Park, A.J. Scott, B.J. Strober, GTEx Consortium, C.D. Brown, X. Wen, I. M. Hall, A. Battle, T. Lappalainen, H.K. Im, K.G. Ardlie, T. Quertermous, K. Kirkegaard, S.B. Montgomery, "Long non-coding RNA gene regulation and trait associations across human tissues," bioRxiv, 2019. https://doi.org/10.1101/793091.
Dobin A, Davis C, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras T. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Panousis N, Gutierrez-Arcelus M, Dermitzakis E, Lappalainen T. Allelic mapping bias in RNA-sequencing is not a major confounder in eQTL studies. Genome Biol. 2014;15(9):467.
Delaneau O, Howie B, Cox A, Zagury J-F, Marchini J. Haplotype estimation using sequence reads. Am J Hum Genet. 2013;93(4):787–696.
Common Fund. Genotype-Tissue Expression Project (GTEx). Database of Genotypes and Phenotypes (dbGaP). phs000424. (2019).
Castel, S.E., Phasing and Allele Specific Expression from RNA-seq. Github. https://github.com/secastel/phaser. (2019).
Acknowledgements
We would like to thank members of the Lappalainen laboratory for discussions surrounding the project. We thank the GTEx donors for their contributions to science; the GTEx Laboratory, Data Analysis, and Coordinating Center (LDACC); and the GTEx analysis working group (AWG) for their work in generating the resource.
GTEx Consortium
Laboratory and Data Analysis Coordinating Center (LDACC): François Aguet1, Shankara Anand1, Kristin G Ardlie1, Stacey Gabriel1, Gad A Getz1,2,3, Aaron Graubert1, Kane Hadley1, Robert E Handsaker4,5,6, Katherine H Huang1, Seva Kashin4,5,6, Xiao Li1, Daniel G MacArthur5,7, Samuel R Meier1, Jared L Nedzel1, Duyen T Nguyen1, Ayellet V Segrè1,8, Ellen Todres1
Analysis Working Group (funded by GTEx project grants):
François Aguet1, Shankara Anand1, Kristin G Ardlie1, Brunilda Balliu9, Alvaro N Barbeira10, Alexis Battle11,12, Rodrigo Bonazzola10, Andrew Brown13,14, Christopher D Brown15, Stephane E Castel16,17, Donald F Conrad18,19, Daniel J Cotter20, Nancy Cox21, Sayantan Das22, Olivia M de Goede20, Emmanouil T Dermitzakis13,23,24, Jonah Einson25,16, Barbara E Engelhardt26,27, Eleazar Eskin28, Tiffany Y Eulalio29, Nicole M Ferraro29, Elise D Flynn16,17, Laure Fresard30, Eric R Gamazon31,32,33,21, Diego Garrido-Martín34, Nicole R Gay20, Gad A Getz1,2,3, Michael J Gloudemans29, Aaron Graubert1, Roderic Guigó34,35, Kane Hadley1, Andrew R Hamel8,1, Robert E Handsaker4,5,6, Yuan He11, Paul J Hoffman16, Farhad Hormozdiari36,1, Lei Hou37,1, Katherine H Huang1, Hae Kyung Im10, Brian Jo26,27, Silva Kasela16,17, Seva Kashin4,5,6, Manolis Kellis37,1, Sarah Kim-Hellmuth16,17,38, Alan Kwong22, Tuuli Lappalainen16,17, Xiao Li1, Xin Li30, Yanyu Liang10, Daniel G MacArthur5,7, Serghei Mangul28,39, Samuel R Meier1, Pejman Mohammadi16,17,40,41, Stephen B Montgomery30,20, Manuel Muñoz-Aguirre34,42, Daniel C Nachun30, Jared L Nedzel1, Duyen T Nguyen1, Andrew B Nobel43, Meritxell Oliva10,44, YoSon Park15,45, Yongjin Park37,1, Princy Parsana12, Abhiram S Rao46, Ferran Reverter47, John M Rouhana8,1, Chiara Sabatti48, Ashis Saha12, Ayellet V Segrè1,8, Andrew D Skol10,49, Matthew Stephens50, Barbara E Stranger10,51, Benjamin J Strober11, Nicole A Teran30, Ellen Todres1, Ana Viñuela52,13,23,24, Gao Wang50, Xiaoquan Wen22, Fred Wright53, Valentin Wucher34, Yuxin Zou54
Analysis Working Group (not funded by GTEx project grants): Pedro G Ferreira55,56,57,58, Gen Li59, Marta Melé60, Esti Yeger-Lotem61,62
Leidos Biomedical - Project Management: Mary E Barcus63, Debra Bradbury63, Tanya Krubit63, Jeffrey A McLean63, Liqun Qi63, Karna Robinson63, Nancy V Roche63, Anna M Smith63, Leslie Sobin63, David E Tabor63, Anita Undale63
Biospecimen collection source sites: Jason Bridge64, Lori E Brigham65, Barbara A Foster66, Bryan M Gillard66, Richard Hasz67, Marcus Hunter68, Christopher Johns69, Mark Johnson70, Ellen Karasik66, Gene Kopen71, William F Leinweber71, Alisa McDonald71, Michael T Moser66, Kevin Myer68, Kimberley D Ramsey66, Brian Roe68, Saboor Shad71, Jeffrey A Thomas71,70, Gary Walters70, Michael Washington70, Joseph Wheeler69
Biospecimen core resource: Scott D Jewell72, Daniel C Rohrer72, Dana R Valley72
Brain bank repository: David A Davis73, Deborah C Mash73
Pathology: Mary E Barcus63, Philip A Branton74, Leslie Sobin63
ELSI study: Laura K Barker75, Heather M Gardiner75, Maghboeba Mosavel76, Laura A Siminoff75
Genome Browser Data Integration & Visualization: Paul Flicek77, Maximilian Haeussler78, Thomas Juettemann77, W James Kent78, Christopher M Lee78, Conner C Powell78, Kate R Rosenbloom78, Magali Ruffier77, Dan Sheppard77, Kieron Taylor77, Stephen J Trevanion77, Daniel R Zerbino77
eGTEx groups: Nathan S Abell20, Joshua Akey79, Lin Chen44, Kathryn Demanelis44, Jennifer A Doherty80, Andrew P Feinberg81, Kasper D Hansen82, Peter F Hickey83, Lei Hou37,1, Farzana Jasmine44, Lihua Jiang20, Rajinder Kaul84,85, Manolis Kellis37,1, Muhammad G Kibriya44, Jin Billy Li20, Qin Li20, Shin Lin86, Sandra E Linder20, Stephen B Montgomery30,20, Meritxell Oliva10,44, Yongjin Park37,1, Brandon L Pierce44, Lindsay F Rizzardi87, Andrew D Skol10,49, Kevin S Smith30, Michael Snyder20, John Stamatoyannopoulos84,88, Barbara E Stranger10,51, Hua Tang20, Meng Wang20
NIH program management: Philip A Branton74, Latarsha J Carithers74,89, Ping Guan74, Susan E Koester90, A. Roger Little91, Helen M Moore74, Concepcion R Nierras92, Abhi K Rao74, Jimmie B Vaught74, Simona Volpi93
Affiliations
1. The Broad Institute of MIT and Harvard, Cambridge, MA, USA
2. Cancer Center and Department of Pathology, Massachusetts General Hospital, Boston, MA, USA
3. Harvard Medical School, Boston, MA, USA
4. Department of Genetics, Harvard Medical School, Boston, MA, USA
5. Program in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard University, Cambridge, MA, USA
6. Stanley Center for Psychiatric Research, Broad Institute, Cambridge, MA, USA
7. Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA
8. Ocular Genomics Institute, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, USA
9. Department of Biomathematics, University of California, Los Angeles, Los Angeles, CA, USA
10. Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, IL, USA
11. Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA
12. Department of Computer Science, Johns Hopkins University, Baltimore, MD, USA
13. Department of Genetic Medicine and Development, University of Geneva Medical School, Geneva, Switzerland
14. Population Health and Genomics, University of Dundee, Dundee, Scotland, UK
15. Department of Genetics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA
16. New York Genome Center, New York, NY, USA
17. Department of Systems Biology, Columbia University, New York, NY, USA
18. Department of Genetics, Washington University School of Medicine, St. Louis, Missouri, USA
19. Division of Genetics, Oregon National Primate Research Center, Oregon Health & Science University, Portland, OR, USA
20. Department of Genetics, Stanford University, Stanford, CA, USA
21. Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
22. Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA
23. Institute for Genetics and Genomics in Geneva (iGE3), University of Geneva, Geneva, Switzerland
24. Swiss Institute of Bioinformatics, Geneva, Switzerland
25. Department of Biomedical Informatics, Columbia University, New York, NY, USA
26. Department of Computer Science, Princeton University, Princeton, NJ, USA
27. Center for Statistics and Machine Learning, Princeton University, Princeton, NJ, USA
28. Department of Computer Science, University of California, Los Angeles, Los Angeles, CA, USA
29. Program in Biomedical Informatics, Stanford University School of Medicine, Stanford, CA, USA
30. Department of Pathology, Stanford University, Stanford, CA, USA
31. Data Science Institute, Vanderbilt University, Nashville, TN, USA
32. Clare Hall, University of Cambridge, Cambridge, UK
33. MRC Epidemiology Unit, University of Cambridge, Cambridge, UK
34. Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Barcelona, Catalonia, Spain
35. Universitat Pompeu Fabra (UPF), Barcelona, Catalonia, Spain
36. Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA, USA
37. Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA
38. Statistical Genetics, Max Planck Institute of Psychiatry, Munich, Germany
39. Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA, USA
40. Scripps Research Translational Institute, La Jolla, CA, USA
41. Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA
42. Department of Statistics and Operations Research, Universitat Politècnica de Catalunya (UPC), Barcelona, Catalonia, Spain
43. Department of Statistics and Operations Research and Department of Biostatistics, University of North Carolina, Chapel Hill, NC, USA
44. Department of Public Health Sciences, The University of Chicago, Chicago, IL, USA
45. Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA
46. Department of Bioengineering, Stanford University, Stanford, CA, USA
47. Department of Genetics, Microbiology and Statistics, University of Barcelona, Barcelona. Spain.
48. Departments of Biomedical Data Science and Statistics, Stanford University, Stanford, CA, USA
49. Department of Pathology and Laboratory Medicine, Ann & Robert H. Lurie Children's Hospital of Chicago, Chicago, IL, USA
50. Department of Human Genetics, University of Chicago, Chicago, IL, USA
51. Center for Genetic Medicine, Department of Pharmacology, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA
52. Department of Twin Research and Genetic Epidemiology, King’s College London, London, UK
53. Bioinformatics Research Center and Departments of Statistics and Biological Sciences, North Carolina State University, Raleigh, NC, USA
54. Department of Statistics, University of Chicago, Chicago, IL, USA
55. Department of Computer Sciences, Faculty of Sciences, University of Porto, Porto, Portugal
56. Instituto de Investigação e Inovação em Saúde, University of Porto, Porto, Portugal
57. Institute of Molecular Pathology and Immunology, University of Porto, Porto, Portugal
58. Laboratory of Artificial Intelligence and Decision Support, Institute for Systems and Computer Engineering, Technology and Science, Porto, Portugal
59. Columbia University Mailman School of Public Health, New York, NY, USA
60. Life Sciences Department, Barcelona Supercomputing Center, Barcelona, Spain
61. Department of Clinical Biochemistry and Pharmacology, Ben-Gurion University of the Negev, Beer-Sheva, Israel
62. National Institute for Biotechnology in the Negev, Beer-Sheva, Israel
63. Leidos Biomedical, Rockville, MD, USA
64. UNYTS, Buffalo, NY, USA
65. Washington Regional Transplant Community, Annandale, VA, USA
66. Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
67. Gift of Life Donor Program, Philadelphia, PA, USA
68. LifeGift, Houston, TX, USA
69. Center for Organ Recovery and Education, Pittsburgh, PA, USA
70. LifeNet Health, Virginia Beach, VA. USA
71. National Disease Research Interchange, Philadelphia, PA, USA
72. Van Andel Research Institute, Grand Rapids, MI, USA
73. Department of Neurology, University of Miami Miller School of Medicine, Miami, FL, USA
74. Biorepositories and Biospecimen Research Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD, USA
75. Temple University, Philadelphia, PA, USA
76. Virginia Commonwealth University, Richmond, VA, USA
77. European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, United Kingdom
78. Genomics Institute, University of California Santa Cruz, Santa Cruz, CA, USA
79. Carl Icahn Laboratory, Princeton University, Princeton, NJ, USA
80. Department of Population Health Sciences, The University of Utah, Salt Lake City, Utah, USA
81. Departments of Medicine, Biomedical Engineering, and Mental Health, Johns Hopkins University, Baltimore, MD, USA
82. Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA
83. Department of Medical Biology, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia
84. Altius Institute for Biomedical Sciences, Seattle, WA, USA
85. Division of Genetics, University of Washington, Seattle, WA, University of Washington, Seattle, WA, USA
86. Department of Cardiology, University of Washington, Seattle, WA, USA
87. HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA
88. Genome Sciences, University of Washington, Seattle, WA, USA
89. National Institute of Dental and Craniofacial Research, Bethesda, MD, USA
90. Division of Neuroscience and Basic Behavioral Science, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA
91. National Institute on Drug Abuse, Bethesda, MD, USA
92. Office of Strategic Coordination, Division of Program Coordination, Planning and Strategic Initiatives, Office of the Director, National Institutes of Health, Rockville, MD, USA
93. Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD, USA
Review history
The review history is available as Additional file 4.
Peer review information
Barbara Cheifet was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Funding
The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health (NIH) and by the National Cancer Institute (NCI), the National Human Genome Research Institute (NHGRI), the National Heart, Lung, and Blood Institute (NHLBI), the National Institute on Drug Abuse (NIDA), the National Institute of Mental Health (NIMH), and the National Institute of Neurological Disorders and Stroke (NINDS). S.E.C. was supported by NHGRI grant 1K99HG009916-01; T.L. and S.E.C. were supported by NIGMS grant R01GM122924 and NIMH grant R01MH101814; T.L. and S.E.C. were supported by NIH contract HHSN2682010000029C; T.L. was supported by NIMH grant R01MH106842 and NIH grants UM1HG008901 and 1U24DK112331. P.M. was supported by the NIH Center for Translational Science Award (CTSA) grants UL1TR002550-01 and 5UL1 TR001114-05.
Author information
Authors and Affiliations
Consortia
Contributions
S.E.C. and F.A. developed pipelines and generated the data. S.E.C. and P.M. analyzed the data. S.E.C. developed the phASER software. S.E.C., K.A., and T.L. designed the study. S.E.C., F.A., P.M., and T.L. wrote and edited the manuscript. The authors read and approved the final manuscript.
Authors’ information
Twitter handles: @stephanecastel (Stephane E. Castel); @francoisaguet (François Aguet); @pejminister (Pejman Mohammadi); @tuuliel (Tuuli Lappalainen).
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
F.A. is an inventor on a patent application related to TensorQTL; S.E.C. is a co-founder, Chief Technology Officer, and stock owner at Variant Bio; T.L. is an advisory board member of Variant Bio with equity and Goldfinch Bio.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Supplemental Figures S1-S5.
Additional file 2: Table S1
: Tissue-level summary statistics for haplotype-level AE data. Table listing sample size, number of expressed genes (defined as genes with > = 0.1 TPM in at least 1 individual), number of genes with phASER data (defined as genes with > = 8 reads in at least 1 individual), median number of samples per gene with phASER data, and if the tissue was used for GTEx v8 eQTL mapping.
Additional file 3: Table S2
: Sample-threshold and allelic imbalance statistics for haplotype-level AE data. Table where rows are each of the 49 GTEx tissues where eQTLs were called and columns list the number of genes with haplotype-level AE data at minimum number of sample thresholds from 1 to 300 (minXXX). For example, min1 lists the number of genes that have AE data from at least 1 sample. The table has three sheets, the first (all_data) presents statistics generated using all haplotype-level AE data, the second (sig_imb_fdr05), counting only cases with significant allelic imbalance (binomial test versus 50/50, gene-level FDR < 5%), and finally (sig_imb_fdr05_no_het), counting only cases with significant imbalance where the individual is not heterozygous for any top (FDR < 5%) or independent (permutation p < 1e-4) eQTLs across any GTEx tissues for the gene.
Additional file 4.
Review history.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Castel, S.E., Aguet, F., Mohammadi, P. et al. A vast resource of allelic expression data spanning human tissues. Genome Biol 21, 234 (2020). https://doi.org/10.1186/s13059-020-02122-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-020-02122-z