
Abstract
We are entering an era of epigenome engineering. The precision manipulation of chromatin and epigenetic modifications provides new ways to interrogate their influence on genome and cell function and to harness these changes for applications. We review the design and state of epigenome editing tools, highlighting the unique regulatory properties afforded by these systems.
Similar content being viewed by others

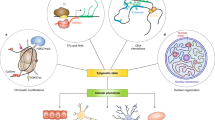
Introduction
Chromatin is decorated by a large array of biochemical modifications made to DNA and histone proteins [1]. These modifications—and the broader organizational structure of chromatin—provide an important additional layer of information that is superimposed upon genome sequence and thus are widely referred to as the epigenome. Given its physical association with genomic material, the epigenome has been suggested to play key roles in regulating genome structure and function, including the timing, strength, and memory of gene expression [2–4]. The epigenome is thought to help control which genes are expressed in a given context, for example, to produce the gene expression patterns that underlie the many different cellular phenotypes that arise during an organism’s development. Because many modifications are heritably maintained, the epigenome is also believed to be key in determining how these gene expression patterns are subsequently maintained for the life of an organism. Moreover, a large body of evidence suggests that the epigenome is inappropriately altered in many human diseases, including most cancers [5–8].
Yet, there remains much that we do not understand about the function of the epigenome. Recently, with the advent of genomic techniques, there has been remarkable progress in our ability to map epigenomic modifications at a global scale and to correlate them with gene expression. While the roles of many chromatin modifications remain unclear, some important patterns have begun to emerge in which epigenome states have come to define key signatures of gene regulation, cell activity, and even disease states [2, 3]. Despite these significant advances, many questions remain unresolved, especially concerning the cause and consequence of chromatin marks with respect to gene expression and other regulatory processes. Thus, the stage is set for the development of new methods that can selectively manipulate and probe the epigenome. Tools that can be used to edit chromatin modifications at specific locations and times will deepen our functional understanding of the epigenome, for example, by allowing researchers to directly interrogate the relationship between the epigenome and transcriptional control. They will also provide opportunities to transform the increasingly precise genome-wide maps that have been generated for developmental and disease states into therapeutics and other benefits for human health.
At the center of these new efforts are the programmable DNA-targeting technologies behind the genome engineering revolution: zinc fingers (ZFs), transcription activator-like effectors (TALEs), and the CRISPR/Cas systems. These technologies are now being utilized for targeted epigenome editing through the recruitment of functional domains to DNA sequences of interest (Fig. 1). Chromatin is, however, an incredibly complex and dynamic regulatory system, which offers both unique opportunities and challenges for this class of technologies. Here, we review the current state of epigenome engineering. Specifically, we discuss new tools and approaches that have allowed researchers to address, interrogate, and reprogram four key features of chromatin: (1) the biochemical diversity of chromatin modifications, (2) the combinatorial and context-dependent nature of chromatin modifications, (3) the memory and long-term stability of modifications, and (4) the potential for long-range spatial regulation (Fig. 1). Throughout, we highlight key design considerations and challenges and suggest strategies for addressing them. We pose ways in which these functional tools can be expanded to help to answer fundamental questions about gene and cellular regulation and we tackle a range of application spaces. Finally, we note that synthetic control over chromatin provides new capabilities in the field of synthetic biology, the engineering of functional biological systems from genetically encoded “parts”. New possibilities include engineering higher-order transcriptional control in cells and programming cellular memory states through the manipulation of epigenetic marks. The development of engineered readers, writers, and erasers that can effectively process the reversible modifications made to chromatin will expand the synthetic biology toolkit available for establishing synthetic linkages in cellular networks, enabling a better understanding of the function of these networks and control of complex cellular behaviors (Fig. 1) [9, 10].

Epigenome engineering is the selective manipulation of chromatin and epigenetic modifications in the genome. a Epigenetic modifications provide a rich set of capabilities and challenges for engineering, including 1) a large biochemical diversity, 2) a preponderance of combinatorial interactions, 3) the potential for long-term memory, and 4) the ability to regulate genes over large spatial ranges. b Programmable DNA-binding domains, which have been used extensively in genome engineering applications and are now being harnessed to design epigenome engineering tools. Epigenetic editors are fusions of a DNA-binding module (zinc fingers (ZFs), transcription activator-like effectors (TALEs) or CRISPR-Cas9) to one or more chromatin regulator (CR) modules. Each ZF domain recognizes ~3–4 nucleotide sequences, whereas each TALE domain recognizes a single nucleotide. The Cas9 protein is directed to its target site by an engineered guide RNA (gRNA) that binds genomic sequences via Watson–Crick base pairing. dCas9 nuclease-null Cas9 protein. c The manipulation of chromatin and epigenetic modifications can be understood in terms of reader/writer/eraser schemes. Molecular writers and erasers serve to catalyze the transfer and removal of chemical marks on target histone residues. The mark is then interpreted by readers, which function to recruit and/or alter functionality. Inspired by and adapted from [9]
Biochemical diversity: selecting modifications and substrates
To explore and exploit the functional roles of DNA and histone modifications, new tools are being developed to selectively alter chromatin biochemistry at specific genomic loci. One striking feature of chromatin is the large biochemical diversity in the modifications and their substrates [4, 11]. For example, with histone modifications, a variety of residues displayed on histone tails act as substrates for a range of post-translational modifications (PTMs), including methylation, acetylation, phosphorylation, and ubiquitination. A leading hypothesis to explain this biochemical diversity is that the marks (individual and/or in combination) comprise a code that is read by modular reader domains in order to drive specific transcriptional and remodeling functions [12]. This form of regulation has the potential for vast combinatorial power. From the standpoint of designing epigenome editors, this diversity requires that the biochemical specificities (both the type of chemical modification and the target residue) are defined carefully. The location within the genome at which these modifications are made is another important consideration, because different genomic loci exhibit distinct chromatin modifications depending on developmental and cell states. Thus, another key factor in the design of editors is genome site or locus specificity.
Rapid advances in targeted epigenome editors
Cells use a system of chromatin effectors and associated histone and DNA modifications to modulate and establish gene-expression states. A central goal has been to try to link these modifications to specific functional roles, such as transcriptional activation and repression [2, 3, 13]. To date, our knowledge of chromatin-effector functions has largely derived from the pharmacological inhibition or genetic knockout of histone-modifying enzymes. More recently, precise and comprehensive genome-wide maps of chromatin modifications have been generated, mapped to transcriptomes, and used to provide further correlative evidence for chromatin functions [14]. Nevertheless, these two approaches—genome-wide perturbations and mapping analyses—neither account for potential pleiotropic effects nor directly demonstrate causal relationships between chromatin and functional states. Therefore, in order to complement these studies and to acquire causal and functional connections between chromatin modifications and their putative functions systematically, we need approaches that can selectively perturb chromatin biochemistry at specific genomic loci.
The advent of programmable DNA-targeting technologies, including ZFs [15], TALEs [16–18], and the CRISPR/Cas systems [19–21], has begun to make this possible. These technologies have been used, with tremendous success and excitement, to create programmable nucleases for genome editing in a wide range of cells and organisms [15, 16, 22–24]. The ability to target specific DNA sequences in eukaryotic genomes is now being harnessed to explore whether the epigenome can be similarly edited in a site-specific manner. The basic design of an epigenome editor is a fusion of a DNA-targeting module to one or more chromatin regulators (CRs; Fig. 1b). To date, efforts have largely focused on creating programmable writers (fusions to enzymes that catalyze chemical modifications of DNA or histone residue(s)) and erasers (fusions to enzymes that remove chemical modifications) (Table 1).
Early examples of epigenome editors include programmable DNA methyltransferases [25–27] and demethylases [28–31], histone methyltransferases and demethylases [32–34], and histone acetyltransferases and deacetylases [33]. In addition, the use of transcriptional activators or repressors that have been reprogrammed to target specific loci can initiate chromatin-mediated changes. For example, ZF fusions to the Krüppel-associated box (KRAB) repressor domain of the transcription factor Kox1 have been shown to suppress the expression of endogenous target genes, such as Sox2, in breast cancer cells through chromatin modifications [35]. The KRAB domain recruits co-repressor KAP1 (KRAB-associated protein 1), which in turn assembles a repressive state by nucleosome-remodeling and deacetylation (NuRD), de-acetylation of histones, incorporation of H3K9me3 (SETDB1), and ultimately heterochromatin formation [36, 37]. Other approaches have used the chromoshadow domain of heterochromatin protein 1 (HP1) to induce heterochromatin formation when targeted to a defined locus by ZFs [38] or LacI [39]. Similarly, fusions to the p65 domain of the mammalian transcription factor NFkB have been used to activate a variety of endogenous genes (and transgenes), principally by promoting histone acetylation via recruitment of p300/CBP [40].
Genomic specificity
Ideally, the activity of an engineered epigenome editor is localized to a specific genomic location. One key way of controlling this is through the DNA-targeting module. Indeed, the targeting specificity of the DNA-binding module is likely to be important in defining the overall activity of an editor, specifically by directing CR activity to a specific genomic locus and thereby minimizing opportunities for off-target effects. Studies that directly compare the activity of an editor across the different classes of DNA-binding modules are lacking, but different patterns of off-target activity have been detected, for example, for KRAB fusions to ZFs and nuclease-null dCas9 [41–43].
The genome-wide specificities of programmable DNA-binding modules, and strategies for improving them, have been the subject of considerable recent study [15, 44], which will not be discussed here. Epigenome editing will certainly benefit from these strategies, which include directed evolution [45], reducing non-specific DNA-binding energy [46, 47], truncating guide RNAs (gRNAs) in CRISPR systems [48], and structure-guided rational protein engineering [49, 50].
The genomic specificity of an editor can also, in some cases, be enhanced by altering the activity of the CR by changing its catalytic activity or its intrinsic interactions with binding partners, such as other regulatory proteins or DNA [41]. For example, for ZF fusions of DNA methyltransferases, mutants that had reduced catalytic activity gave rise to methylation that was more specific to targeted sites than that in the wild type [51, 52], presumably because the catalytic activity of the editors was more dependent on DNA binding.
Biochemical specificity
The use of full-length CRs and potent transcriptional activators or repressors, such as KRAB and p65, can be effective in inducing chromatin-mediated transcriptional changes. However, these components are known to recruit multiple chromatin-modifying activities and to induce broad chromatin changes, which confound our ability to link specific modifications to specific functional roles. Addressing this issue requires epigenetic editors that have precise control over the desired chromatin-modifying activities. It also requires quantifying the biochemical specificity of an epigenetic editor, that is, quantifying the full array of modifications made to a locus that has been targeted by an editor. These modifications are inherently more challenging to quantify than genomic specificity: a comprehensive panel of DNA histone modifications must be assessed using techniques such as chromatin immunoprecipitation (ChIP) with many different antibodies.
Strategies to create epigenetic editors that have improved functional or biochemical specificity have been explored. One key strategy is to truncate chromatin-modifying enzymes to their catalytic core domains. A notable recent example involved the human co-activator protein p300, which functions as a histone acetyltransferase and mediates interactions with multiple transcription factors to regulate many genes in tissues throughout the body. By fusing the catalytic core of the p300 acetyltransferase to dCas9, Hilton et al. [53] created a programmable histone acetyltransferase. They showed that this minimal fusion protein was able to catalyze the acetylation of H3K27 at target promoter sites, which led to robust transcriptional activation of target genes. This elegant study provides strong support for histone acetylation as a causal mechanism for transcriptional activation, but it also highlights the challenges associated with functionally annotating specific chromatin modifications. In this particular study, it remained unclear whether H3K27 acetylation causes the observed transcriptional effects or whether another histone lysine at the site (or perhaps even a lysine residue on a completely different protein) causes these effects. These efforts would benefit from new and improved methods for quantifying biochemical specificity in the context of epigenome-editing experiments.
A related strategy for improving the functional specificity of epigenetic editors is to remove non-catalytic domains or components from CRs in order to minimize the potential for non-specific interactions. For example, site-specifically recruiting the minimal catalytic domain of the histone methyltransferase SUV39H1 with a ZF array efficiently repressed the VEGF-A promoter, whereas full-length SUV39H1 did not cause repression [54]. Presumably this was because the intact HP1 interaction domain present in full-length SUV39H1 functioned to titrate the protein away from the VEGF-A gene. Related examples include coupling of the catalytic domains of chromatin-modifying enzymes to dCas9 [53], ZFs [25, 40, 53–59], TALEs [33, 53, 60, 61], or use of the Gal4 DNA-binding domain [26] to repress or silence endogenous genes.
Collectively, these studies have used fusions to minimal catalytic domains to develop epigenetic editors that have improved functional specificity. Efforts to truly isolate and re-engineer the catalytic domains of CRs will be key to improving the functional specificity of epigenetic editors.
Ongoing challenges
In addition to improving biochemical and site specificities, several important challenges remain. Current efforts have been focused predominantly on constructing epigenome editors by fusing writer or eraser domains with DNA-targeting elements. Engineered readers remain largely underdeveloped (Table 2). Potential applications of epigenomic readers include in vivo reporting on aberrant or disease-related modifications. An in vivo ChIP approach could feedback to an epigenome effector for reconfiguration of a detected aberrant modification state. In one example, a synthetic transcription factor was engineered by fusing the VP64 activation domain to the Polycomb chromodomain (PCD) [62]. The PCD of this synthetic transcription factor recognizes H3K27me3 that is associated with silenced genes and reactivates these genes. Engineering readers remains challenging for two reasons. First, it may be difficult to engineer a single histone reader domain that is specific for a particular histone residue. Combining multiple different reader domains, which is a common mode of natural chromatin regulation, may solve this problem. Second, as all similarly modified nucleosomes will look alike to chromatin readers, the readers will bind modifications throughout the genome rather than at specific locations. A combination of DNA- and chromatin-binding modalities may provide a solution. Given the complexity of chromatin biochemistry, there are probably many other features that will be important for the design of future epigenome-modifying tools. For example, histone lysine residues can exist in mono-, di-, and trimethylated states. Being able to finely tune this feature of chromatin modification could reveal its functional role and potentially provide fine-tuned control of transcriptional activity.
Continued work on characterizing and discovering new catalytic domains will expand the list of available parts from which to select for improved properties such as substrate specificity [63–71]. Another interesting approach for improving the catalytic activity of epigenome editors is to fuse the catalytic core domains of multiple subunits or co-recruiting synergistic co-factors. For example, fusion of the catalytic C-terminal domains of DNA methyltransferase 3a (DNMT3a) and DNMT3L induced DNA methylation at the VEGF-A promoter with better efficiency than did the DNMT3a catalytic domain alone, by mimicking a stable Dnmt3a–Dnmt3L heterodimer [59]. DNMT3L, despite its lack of catalytic activity, directly interacts with and stimulates the catalytic activity of DNMT3a. Targeting chromatin modification by coupling multiple subdomains that have catalytic or structural functions may be a better reflection of the natural mode of chromatin regulation.
Combination and context
There exist a surprisingly large number of epigenome modifications. The combinatorial interactions between these modifications and other chromatin-bound proteins increase this complexity even further. In fact, most chromatin states that are associated with regions such as active promoters and enhancers are characterized by specific combinations of chromatin modifications [72]. Why did this combinatorial complexity evolve? One reason could be that single modifications alone are not sufficient to account for all the distinct states that need to be specified or marked. Perhaps a more intriguing possibility is that combinatorial interactions set the stage for context-dependent regulation and enhance locus-specific recruitment.
With context dependency, one modification could mask, modulate, or enhance the binding interaction of a reader of a second modification. This is seen in the association of HP1 with H3K9me3, which is abolished by the dynamic and transient phosphorylation of the adjacent Ser10 residue [73]. Similarly, association of the double chromodomains of CHD1 with H3K4me3 is reduced by demethylation of Arg2 (a two-fold reduction) or by phosphorylation of Thr3 (a 25-fold reduction). Trans-histone crosstalk can also occur, as found in COMPASS (Complex of Proteins Associated with Set1), the yeast homolog of the mammalian MLL complex [74]. A global functional proteomic screen revealed that monoubiquitination of histone H2B by Rad6 is required for H3K4 methylation by COMPASS and for H3K79 methylation by Dot1 [75]. The recruitment of Cps35, an essential subunit of COMPASS, to chromatin in the presence of H2B monoubiquitination facilitates the recruitment of COMPASS and Dot1. Thus, combinatorial modifications may act as gates, allowing events to occur only in a particular order.
Combinatorial modifications could also prime a gene to follow one of multiple possible paths. Certain domains of the embryonic stem (ES) cell genome possess both activating and repressive histone modifications, known as bivalent domains; these are typically enriched at developmentally important genes [76, 77]. It is proposed that genes that have bivalent domains are poised for either activation or repression, depending on the differentiation path that the cell ultimately follows.
Gene expression is precisely controlled in time and space by the integration of this diverse array of PTM signals and the actions of multiple chromatin-regulating factors operating in multifactorial ways [3, 78]. If we can design epigenome editors to control these complex states, we may be able to fully unveil the context dependency of chromatin regulation and thus understand whether the pre-established chromatin context will affect (cancel out, enhance, or synergize) the effectiveness of the following chromatin regulation. We might then be able to adopt the true combinatorial features of natural chromatin communication in a range of applications.
Combinatorial and high-throughput techniques reveal contextual and combinatorial principles
The interactions between chromatin proteins, chromatin modifications, and the surrounding DNA sequence and chromatin state determine local transcriptional outputs. This is key for the design of functional epigenome editors because behaviors that are observed at one specific locus may not hold at another locus where the presence of existing proteins may alter the activity of a recruited epigenome editor. Therefore, one important goal for epigenome engineers is to reveal the rules of chromatin context. Accessing and deciphering these rules will require high-throughput and combinatorial techniques.
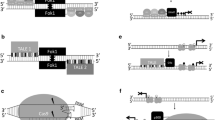
There have been several in vitro methods for the rapid assessment of combinatorial and contextual properties of epigenome editors [79], but the intracellular and intranuclear environments are likely to have significant effects. To overcome the technical hurdles of working in the cellular environment, library-based methods can functionally assay comprehensive sets of regulators in vivo. For example, Akhtar and colleagues [80] randomly integrated thousands of barcoded reporter transgenes into the genome using piggyback transposition (Fig. 2a). By assaying cells with integrated reporters (IRs), these authors could test whether the local chromatin compaction state prior to integration had predictive power for IR expression levels. Analysis of normalized transgene expression by high-throughput sequencing of the library revealed non-random patterns of IR expression, which was strongly dependent on local chromatin context.

Interrogating the contextual and combinatorial principles of epigenome regulation. a A method for the parallel monitoring of the transcriptional activities of thousands of randomly integrated, barcoded reporters was used to study chromatin position effects across the genome. b Synthetic chromatin regulators (synCRs), composed of fusions of programmable zinc fingers (ZFs) and subunit proteins derived from diverse chromatin-regulating complexes, were used to study and program transcriptional outputs produced by individual and combinations of CRs at integrated reporters. GFP green fluorescent protein. c CRISPR/dCas9 can be exploited for high-throughput functional assays of epigenetic regulators thanks to its experimental tractability for combinatorial and multiplexed recruitment. Scaffolding multiple RNA-hairpin motifs to a guide RNA (gRNA) allows multivalent recruitment of chromatin regulators (CRs). Scaffolding different RNA motifs to gRNA allows heterologous recruitment of CRs. The same CR can be simultaneously recruited to multiple loci by using different gRNAs specific to each gRNA locus
In our group, Keung et al. [81] fused a comprehensive set of 223 yeast CRs to programmable ZF proteins (Fig. 2b). We site-specifically co-recruited the CRs together with the commonly used transcriptional activator VP16 to diverse arrays of synthetic reporters. This revealed a range of transcriptional logic and behaviors, demonstrating the complexity of chromatin regulation. We divided this range of logic into six distinct classes of combinatorial regulation: dominant repressors, repressors, neutral factors, enhancers of VP16-mediated activation, additive activators, and synergistic activators.
Future work
The simplicity of programming the CRISPR-Cas9 system to target multiple endogenous genomic loci simultaneously [82–84] and/or to recruit multiple different protein domains [85] to a locus offers a powerful platform with which to decipher the combinatorial and contextual complexity of the epigenome (Fig. 2c). The experimental tractability of CRISPR/Cas9 genome editing tools for high-throughput approaches exceeds that of any other currently available DNA-targeting platform [86–90]. Creatively leveraging previous systems could also expand the parameter space that is explored. For example, the platform that Akhtar and colleagues [80] developed could be adapted to study additional contextual effects. With only minor modifications in the experimental design, DNA sequence elements could be added or other chromatin modifiers recruited in front of the reporter gene to ask how each component interacts with each endogenous state.
Memory and epigenetics
Among the myriad modifications being written and erased on chromatin, a subset is stably inherited through mitotic or meiotic cell divisions. These epigenetically inherited modifications are important for the maintenance of gene expression patterns throughout the differentiation and developmental processes of mammals and may result in disease or cancer when mis-regulated [8, 91]. Several important examples of behavioral and disease traits are inherited across generations in complex organisms, including mice [92]; here we focus on cellular studies because studies of the mechanistic roles of epigenome modifications are more feasible. Understanding and controlling epigenetic modifications could also have an impact on biotechnology and synthetic biology, where stable biological switches are highly desired.
A variety of different mechanisms underlie epigenetic properties but they all depend on some form of feedback. Broadly, feedback mechanisms can be trans- or cis-acting or a combination of both [93]. Trans-acting mechanisms typically involve positive feedback of a transcription factor in the regulation of its own gene. This mechanism is utilized both to establish and to self-sustain a specific transcriptional state of a gene, as demonstrated in the activation and maintenance of differentiated functions of nematode sensory neurons [94, 95] and widely in maintaining differentiated cell identity [96, 97]. Cis-acting mechanisms more often involve chromatin modifications directly. DNA methylation in mammals is a prime example [98]. DNA methylation is crucial in the establishment of epigenetic memory that is essential for normal development [99, 100]. Work in vertebrates has been focused mostly on the methylation of cytosine in the context of CpG di-nucleotides at transcription start sites (TSSs), which is believed to maintain genes in a locked-in off state. Recent advances in the genome-scale mapping of methylation has found additional context-dependent functions (in, for example, TSSs, gene bodies, and enhancers) that go beyond the repressive association of DNA methylation [101]. Epigenetic memory by DNA methylation is established through the DNA-strand to DNA-strand copying action of DNMT1 and through the recruitment of repressive regulatory proteins upon de novo methylation by DNMT3 [98]. However, this classic model for epigenetic memory, with the canonical distinction between the roles of DNMT3 and DNMT1, is being challenged by recent experimental evidence [102, 103].
Histone modifications are also involved in maintaining epigenetic regulation. For example, antagonizing groups of protein complexes, the Polycomb (PcG) and trithorax (trxG) groups, mediate the mitotic inheritance of repressive and active transcriptional states, respectively [104]. There is also evidence that some heterochromatic histone modifications crosstalk with and may derive their stability from DNA methylation [105, 106]. These examples point to the important role of chromatin in stably maintaining the transcriptional state of critical lineage-specifying genes. The exact mechanisms that underlie these epigenetic properties of chromatin modifications have been difficult to pin down given the time-dependent nature of gene-expression memory. Nevertheless, several temporally dynamic experimental approaches using epigenome editors have and will continue to shed light on the molecular feedback underlying memory in chromatin systems.
Synthetic systems can directly induce epigenetic chromatin states
In a landmark study, Hathaway et al. [38] developed a chemically inducible system to establish and erase heterochromatin in vivo at the Oct4 locus (Fig. 3a). The chromoshadow domain of HP1α was site-specifically directed to ZFHD1-binding sites via FKBP-Frb dimerization domains in the presence of rapamycin. Upon transient recruitment of HP1α, a >10-kb region of H3K9 methylation was established and maintained through multiple cell divisions (over at least several weeks), even after the release of HP1α. By measuring the kinetics and stability of chromatin modification establishment and turnover, Hathaway et al. [38] generated a computational model that incorporated a feedback mechanism between DNA methylation and H3K9 methylation.

Use of epigenome editing tools to study the dynamics and memory of epigenetic regulation. a The selective recruitment of HP1α to specific loci in live cells was used to establish H3K9me3-dependent gene silencing and to study the kinetics and extent of heterochromatin. b In another study, doxycyline (DOX) was used to selectively recruit four repressive CRs that are associated with diverse chromatin modifications (Krüppel-associated box (KRAB) (associated with H3K9 methylation), embryonic ectoderm development (EED) (associated with H3K27 methylation), DNA methyltransferase 3B (DNMT3B) (associated with DNA methylation), and histone deacetylase 4 (HDAC4) (associated with histone deacetylation)). By tracking transcriptional output of a reporter gene in individual cells, researchers discovered that cells stochastically transition between active and silent states. These dynamics were described by a simple three-state model, in which different CRs operate over different time scales to modulate the fraction of cells in a population that are in each state. YFP yellow fluorescent protein
The relationship between DNA methylation and H3K9 methylation, as well as other types of repressive modifications, was further investigated by Bintu et al. [107] in an elegant synthetic biology study. These authors developed a framework to quantitatively interrogate the kinetics and stability of gene repression induced by four proteins that act through different types of chromatin modifications: (1) embryonic ectoderm development (EED) of Polycomb Repressive Complex 2 (PRC2) (H3K27 methylation), (2) KRAB (H3K9 methylation), (3) DNMT3B (DNA methylation), and (4) histone deacetylase 4 (HDAC4) (histone deacetylation) (Fig. 3b). Each protein was transiently recruited for different periods of time to a fluorescent reporter gene using the reverse Tet repressor (rTetR). Using single-cell time-lapse microscopy, Bintu et al. [107] observed that the reporter turned on and off in an all-or-none fashion for all of the chromatin modifiers studied. Yet the time it took for the reporter to turn off and the stability of repressed reporter differed depending on the modifier. In fact, each type of chromatin modification led to different kinetics and stabilities of gene repression, suggesting that the epigenome may encode different operational types of gene regulation.
The strong epigenetic properties of DNA methylation were confirmed in both studies. Nevertheless, studies are still attempting to confirm whether various histone modifications are truly epigenetic, that is, self-sustaining in the absence of the initial triggering signal or any necessary DNA sequence [95, 108, 109]. For example, the artificial recruitment of the PRC2 complex via a tetracycline-inducible GAL4–EED fusion protein induced H3K27me3, and this modification was maintained even after repression of GAL4–EED [110]. More recently, two studies have provided compelling evidence for the epigenetic inheritance of H3K9 methylation in the fission yeast Schizosaccharomyces pombe [111, 112]. A particularly important aspect of these findings was that the epigenetic inheritance of H3K9 methylation was decoupled from any DNA sequence and could be established at genomic loci that are normally devoid of H3K9 methylation and heterochromatin. In these two studies, the H3K9 methyltransferase Clr4 was recruited to the ade6+ gene [111, 112]. Transient recruitment of Clr4 was controlled by tetracycline-dependent release of TetR–Clr4. Interestingly, while the establishment of high levels of H3K9 methylation was subsequently lost upon release of the TetR-Clr4 initiator (within around ten cell divisions), deletion of the putative demethylase Epe1 resulted in H3K9-methylation-mediated silencing at the tethering site through many mitotic and meiotic divisions. These results suggest that the inheritance of H3K9 methylation is determined by the balance of a feedback loop between methylation by Clr4 through a reader–writer mechanism and active demethylation by Epe1. These studies demonstrate the synergy of forward-engineering approaches (such as those involving control of the genomic locus and of the timing of Clr4 recruitment) and chromatin biology techniques and genetics in demonstrating the factors required in the epigenetic maintenance of H3K9 methylation.
Future work
Many other histone modifications still remain to be tested for their epigenetic properties and many molecular details of epigenetic mechanisms remain to be discovered [27]. These ongoing studies may benefit from technical advances that will make it possible to dynamically recruit proteins and to interrogate large parameter spaces in high-throughput screens for minimal factors that are required for epigenetic maintenance. For example, to identify the minimal factors required for epigenetic chromatin states, CRISPR-Cas9 systems could be used either to knockout chromatin proteins and/or to recruit multiple factors to specific genomic loci [38, 111–113]. In addition, greater temporal control could provide more precise information on the stability and kinetics of epigenetic systems. This could be achieved through the use of light-activated protein systems. Konermann et al. [33] demonstrated that 32 repressive histone effector domains could be conditionally targeted to a genomic locus via the light sensitive cryptochrome 2 (CRY2) protein and its interacting partner CIB1 from Arabidopsis thaliana [33]. This particular study was not focused on identifying the epigenetic properties of the chromatin modifiers, but this technique holds potential as a toolkit that can provide high temporal resolution with which to study epigenetic mechanisms and identify epigenetic factors [114].
Many opportunities for exploiting the unique features of epigenetic regulation lie ahead. Researchers could work to harness any potential restricted or conditional epigenetic inheritance of histone modifications for developing “short-term” or “flexible” epigenetic memory circuitry [99], which could be intentionally designed to maintain the edited epigenome state for a short period of time. For example, there may be instances, in normal development or for transient therapeutic applications, that require that genes are regulated such that they are suppressed for a short period of time and subsequently reactivated. The repressive state of a gene could be induced with repressive histone methyltransferases and later (before one cell cycle is completed or within very few cell divisions) reversed by either demethylases or a passive histone dilution mechanism. By contrast, complete and permanent repression of genes could be achieved with the incorporation of DNA-methylation-mediated gene silencing [25, 56]. It is important to note that there is evidence to suggest that transiently induced DNA methylation is not maintained, highlighting the importance of multivalent deposition of functionally related epigenetic marks for truly stable reprogramming [57]. Either short-term or long-term epigenetic memory could be a valuable feature of many applications, including gene and cell therapy. Finally, while the epigenetic maintenance of chromatin and gene expression states has been demonstrated in several cellular systems, exciting but challenging work lies ahead in using epigenome editing tools to study the long-term heritability of chromatin modifications (such as DNA methylation [92, 98]) across generations of complex organisms such as mice.
Artificial perturbations of chromatin structure
Chromatin adds a unique spatial element to gene regulation at multiple scales [115, 116]. For example, certain histone modifications have been observed to demarcate and preserve chromatin domains such as silent heterochromatic and active euchromatic regions. These regions are hypothesized to be established and preserved by highly dynamic processes involving histone modifications; these include self-reinforcing mechanisms that spread modifications along adjacent nucleosomes [111, 112], so-called “reader-writer” mechanisms [117]. Chromatin’s three-dimensional conformation and positioning in the nucleus also orchestrate gene expression. For example, looping mediates long-range genomic interactions by juxtaposing distal regulatory elements such as enhancers with distant loci, to either coordinate their expression or co-localize regulatory factors. This type of spatial organization is observed in tissue-specific gene regulation, in which genomic elements cluster together at certain stages of development [118]. Tools that can replicate or perturb chromatin’s spatial properties will enhance our ability to study and potentially harness these complex mechanisms.
Several molecular approaches have already been used successfully to perturb chromatin structure and these studies suggest that continued work in this area could reveal important and potentially useful regulatory principles relating to chromatin shape. For example, an ectopic repressor assay using a drug-inducible ZF-KRAB fusion protein demonstrated that KRAB-mediated repression spans tens of kilobases and is established by the long-range propagation of H3K9me3 and HP1 β [119]. This and similar approaches [38, 81] provide us with the unique ability to regulate multiple genes in tandem using a single regulator. Furthermore, transcriptional activators and repressors that are recruited site-specifically to regions more than 1 kb downstream of promoters can activate [120] and repress [121] yeast genes, respectively, when they are placed near telomeres. This effect “at a distance” is mediated by a telomere-position effect in yeast, which is analogous to the position effect variegation (PEV) observed in Drosophila, wherein a normally active euchromatic gene is juxtaposed with heterochromatin by structural rearrangement and becomes silenced [122]. Modeling efforts along with site-specific recruitment approaches have also provided insights into how multiple regulators that have opposing functions (active or repressive) are coordinated to regulate genes in a way that is determined by the spatial distribution of nucleation sites along the chromosome [123, 124]. These studies can help to explain the expression pattern of adjacent genes in a certain positioning context and could potentially unveil the mechanisms of variegated gene expression.
Recent efforts have begun to directly manipulate chromatin looping and to change the three-dimensional contact profile of genes with other loci or nuclear structures (Fig. 1). Deng and colleagues [125, 126] employed ZFs to override a stringent developmental gene expression pattern by artificially forcing chromatin looping. Specifically, these researchers forced chromatin looping between the β-globin gene and its distal regulatory region, the locus control region (LCR) which is positioned 40 kb away. This looping was induced by synthetically tethering Ldb1, a protein present at the LCR, to the β-globin promoter, which led to Ldb1–Ldb1-mediated chromatin looping. Deng and colleagues demonstrated that forced chromatin looping was sufficient for activation of the β-globin gene [125, 126]. They then showed that forced chromatin looping that was achieved by tethering Ldb1 to a developmentally silenced embryonic globin gene was sufficient to trigger the gene’s reactivation. These studies demonstrate a novel approach to control the three-dimensional structure of the epigenome.
There are other ways to induce structural perturbations in chromatin. Even a change in the direction of a small fragment (~20 bp) of DNA sequence can control transcriptional activity by reconfiguring the topology of chromatin loops [127]. CCCTC-binding factor (CTCF) insulators and the associated cohesion complex play important roles in higher-order chromatin organization in mammalian genomes. By reversing the relative orientation of CTCF-binding sites by CRISPR/Cas9-based genome editing, changes in the directionality of DNA looping and gene expression can be made [127]. Such efforts will be key to elucidating the relationship between DNA sequence elements and the three-dimensional structure of chromatin.
Structural- or spatial-factor-dependent regulation of gene expression can also be mediated by spatially positioning genes in the nucleus. The randomly integrated reporter platform of Akhtar and colleagues [80], for example, revealed spatial positioning effects that correlated with gene expression. Lamina-associated domains (LADs), late-replicating domains, and regions marked by the histone modification H3K9me2 often coincide with one another and harbor mostly inactive endogenous genes [128]. In addition, the integrated reporters, much of whose fold change was unaccounted for by local chromatin compaction, were more actively expressed when integrated near active genes. Akhtar and colleagues proposed that these effects are a result of the collective actions of enhancers and transcription units in creating transcription-promoting regions, again highlighting the functional importance of how genes are spaced along a chromosome.
Concluding remarks
In this review, we have discussed important features that must be considered when designing functional epigenome engineering tools and current challenges that need to be addressed. The impact of recent advances in epigenome engineering has been remarkable in terms of both understanding the underlying mechanisms of epigenome regulation and designing new ways to regulate genes for future biomedical and biotechnological applications. Forward-engineering approaches allow researchers to directly interrogate the relationships between the epigenome and transcriptional function. These approaches are highly complementary to other cell biology methods and are particularly useful for systematically exploring large parameter spaces [9]. In addition, epigenome editing technologies hold considerable promise for engineering applications. The application of engineering principles toward the construction of novel biological systems (i.e., synthetic biology) could take advantage of this additional class of chromatin-based regulation. The many features of epigenome regulation present interesting properties or functional connections that could be exploited in assembling synthetic biological networks [10]. Ultimately, epigenome editing may emerge in new forms of gene therapy by modifying/correcting diseased epigenome states without making permanent and potentially deleterious genetic changes in cells [8, 26, 129].
Perhaps one of the most exciting prospects in developing new epigenome editing tools is how they may alter our perspective of the function and nature of the epigenome’s complexity. Several current models depict chromatin modifications as an additional layer of regulatory nodes that act in concert with genetic networks to coordinate cellular programs [130]. With our increasing ability to interface, perturb, and establish these regulatory nodes, we can begin to think of the epigenome as a powerful set of operations that can be performed on signals from and between various levels of cellular regulation. Given the widespread use of the epigenome in nature, there is good reason to believe that epigenome editing, and the predictable manipulation of chromatin modifications, will serve as a powerful new paradigm for synthetic biology and bioengineering. No longer will the epigenome be a complex problem to decipher, but rather a powerful platform to harness.
Abbreviations
- ChIP:
-
Chromatin immunoprecipitation
- COMPASS:
-
Complex of Proteins Associated with Set1
- CR:
-
Chromatin regulator
- CTCF:
-
CCCTC-binding factor
- DNMT3a:
-
DNA methyltransferase 3a
- EED:
-
Ectoderm development
- gRNA:
-
Guide RNA
- HDAC4:
-
Histone deacetylase 4
- HP1:
-
Heterochromatin protein 1
- IR:
-
Integrated reporter
- KRAB:
-
Krüppel-associated box
- LCR:
-
Locus control region
- PCD:
-
Polycomb chromodomain
- PRC2:
-
Polycomb Repressive Complex 2
- PTM:
-
Post-translational modification
- TALE:
-
Transcription activator-like effector
- TSS:
-
Transcription start site
- ZF:
-
Zinc finger
References
Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27:351–7.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80.
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59.
Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53.
Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–40.
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63.
Bashor CJ, Horwitz AA, Peisajovich SG, Lim WA. Rewiring cells: synthetic biology as a tool to interrogate the organizational principles of living systems. Annu Rev Biophys. 2010;39:515–37.
Keung AJ, Joung JK, Khalil AS, Collins JJ. Chromatin regulation at the frontier of synthetic biology. Nat Rev Genet. 2015;16:159–71.
Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–81.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5.
Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27:389–96.
Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18.
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–46.
Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55.
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12.
Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–6.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Jinek M, Chylinski K, Fonfara I, Hauer M. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–22.
Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–8.
Esvelt KM, Wang HH. Genome-scale engineering for systems and synthetic biology. Mol Syst Biol. 2013;9:641.
Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78.
Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405.
Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, et al. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics. 2012;7:350–60.
Li F, Papworth M, Minczuk M, Rohde C, Zhang Y, Ragozin S, Jeltsch A. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. 2007;35:100–12.
Minczuk M, Papworth MA, Kolasinska P, Murphy MP, Klug A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc Natl Acad Sci U S A. 2006;103:19689–94.
Chen H, Kazemier HG, De Groote ML, Ruiters MHJ, Xu GL, Rots MG. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014;42:1563–74.
Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137–42.
Gregory DJ, Mikhaylova L, Fedulov AV. Selective DNA demethylation by fusion of TDG with a sequence-specific DNA-binding domain. Epigenetics. 2012;7:344–9.
Gregory DJ, Zhang Y, Kobzik L, Fedulov AV. Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics. 2013;8:1205–12.
Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013;31:1133–6.
Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–6.
Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. Functional annotation of native enhancers with a Cas9–histone demethylase fusion. Nat Methods. 2015;12:401–3.
Stolzenburg S, Rots MG, Beltran AS, Rivenbark AG, Yuan X, Qian H, et al. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012;40:6725–40.
Urrutia R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003;4:231.
Sripathy SP, Stevens J, Schultz DC. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol Cell Biol. 2006;26:8623–38.
Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149:1447–60.
Brink MC, van der Velden Y, de Leeuw W, Mateos-Langerak J, Belmont AS, van Driel R, Verschure PJ. Truncated HP1 lacking a functional chromodomain induces heterochromatinization upon in vivo targeting. Histochem Cell Biol. 2006;125:53–61.
Heller EA, Cates HM, Peña CJ, Sun H, Shao N, Feng J, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17:1720–7.
Grimmer MR, Stolzenburg S, Ford E, Lister R, Blancafort P, Farnham PJ. Analysis of an artificial zinc finger epigenetic modulator: widespread binding but limited regulation. Nucleic Acids Res. 2014;42:10856–68.
O’Geen H, Henry IM, Bhakta MS, Meckler JF, Segal DJ. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015;43:3389–404.
Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143–9.
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–43.
Hubbard BP, Badran AH, Zuris JA, Guilinger JP, Davis KM, Chen L, et al. Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat Methods. 2015;12:939–42.
Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK, Liu DR. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. 2014;11:429–35.
Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–70.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Keith JJ. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5.
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2015;351:84–8.
Smith AE, Ford KG. Specific targeting of cytosine methylation to DNA sequences in vivo. Nucleic Acids Res. 2007;35:740–54.
McNamara AR. Characterisation of site-biased DNA methyltransferases: specificity, affinity and subsite relationships. Nucleic Acids Res. 2002;30:3818–30.
Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7.
Snowden AW, Gregory PD, Case CC, Pabo CO. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol. 2002;12:2159–66.
Falahi F, Huisman C, Kazemier HG, van der Vlies P, Kok K, Hospers GAP, Rots MG. Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol Cancer Res. 2013;11:1029–39.
Stolzenburg S, Beltran AS, Swift-Scanlan T, Rivenbark AG, Rashwan R, Blancafort P. Stable oncogenic silencing in vivo by programmable and targeted de novo DNA methylation in breast cancer. Oncogene. 2015;34:5427–35.
Kungulovski G, Nunna S, Thomas M, Zanger UM, Reinhardt R, Jeltsch A. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenetics Chromatin. 2015;8:12.
Nunna S, Reinhardt R, Ragozin S, Jeltsch A. Targeted methylation of the epithelial cell adhesion molecule (EpCAM) promoter to silence its expression in ovarian cancer cells. PLoS One. 2014;9:e87703.
Siddique AN, Nunna S, Rajavelu A, Zhang Y, Jurkowska RZ, Reinhardt R, et al. Targeted methylation and gene silencing of VEGF-A in human cells by using a designed Dnmt3a-Dnmt3L single-chain fusion protein with increased DNA methylation activity. J Mol Biol. 2013;425:479–91.
Bernstein DL, Le Lay JE, Ruano EG, Kaestner KH. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J Clin Invest. 2015;125:1998–2006.
Cho H-S, Kang JG, Lee J-H, Lee J-J, Jeon SK, Ko J-H, et al. Direct regulation of E-cadherin by targeted histone methylation of TALE-SET fusion protein in cancer cells. Oncotarget. 2015;6:23837–44.
Haynes KA, Silver PA. Synthetic reversal of epigenetic silencing. J Biol Chem. 2011;286:27176–82.
Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, et al. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U S A. 2001;98:12902–7.
Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–32.
Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 2005;19:1444–54.
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4.
Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–81.
Messmer S, Franke A, Paro R. Analysis of the functional role of the Polycomb chromo domain in Drosophila melanogaster. Genes Dev. 1992;6:1241–54.
Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20.
Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–9.
Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30.
Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14:211–24.
Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–22.
Sun Z-W, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–8.
Lee J-S, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, et al. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–96.
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26.
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9.
Suganuma T, Workman JL. Crosstalk among histone modifications. Cell. 2008;135:604–7.
Nguyen UTT, Bittova L, Müller MM, Fierz B, David Y, Houck-Loomis B, et al. Accelerated chromatin biochemistry using DNA-barcoded nucleosome libraries. Nat Methods. 2014;11:834–40.
Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 2013;154:914–27.
Keung AJ, Bashor CJ, Kiriakov S, Collins JJ, Khalil AS. Using targeted chromatin regulators to engineer combinatorial and spatial transcriptional regulation. Cell. 2014;158:110–20.
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8.
Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell. 2014;54:698–710.
Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol. 2013;31:684–6.
Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2014;160:339–50.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–91.
Koike-Yusa H, Li Y, Tan E-P, Velasco-Herrera MDC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–73.
Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–4.
Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8.
Whitelaw E, Morgan HD, Sutherland HGE, Martin DIK. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–8.
Moazed D. Mechanisms for the inheritance of chromatin states. Cell. 2011;146:510–8.
Hobert O. Maintaining a memory by transcriptional autoregulation. Curr Biol. 2011;21:R146–7.
Ptashne M. Epigenetics: core misconcept. Proc Natl Acad Sci U S A. 2013;110:7101–3.
Holmberg J, Perlmann T. Maintaining differentiated cellular identity. Nat Rev Genet. 2012;13:429–39.
Lassar AB, Buskin JN, Lockshon D, Davis RL, Apone S, Hauschka SD, Weintraub H. MyoD is a sequence-specific DNA binding protein requiring a region of myc homology to bind to the muscle creatine kinase enhancer. Cell. 1989;58:823–31.
Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21.
Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32.
Bestor TH. The DNA, methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–402.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–11.
Jeltsch A, Jurkowska RZ. New concepts in DNA methylation. Trends Biochem Sci. 2014;39:310–8.
Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–43.
Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16:519–32.
Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304.
Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, et al. Dynamics of epigenetic regulation at the single-cell level. Science. 2016;351:720–4.
Sengupta AK, Kuhrs A, Müller J. General transcriptional silencing by a Polycomb response element in Drosophila. Development. 2004;131:1959–65.
Cheng TH, Gartenberg MR. Yeast heterochromatin is a dynamic structure that requires silencers continuously. Genes Dev. 2000;14:452–63.
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–300.
Ragunathan K, Jih G, Moazed D. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science. 2015;348:1258699.
Audergon PNCB, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, Allshire RC. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science. 2015;348:132–5.
Ayyanathan K, Ayyanathan K, Lechner MS, Lechner MS, Bell P, Bell P, et al. Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 2003;17:1855–69.
Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–5.
Van Bortle K, Corces VG. Nuclear organization and genome function. Annu Rev Cell Dev Biol. 2012;28:163–87.
Talbert PB, Henikoff S. Spreading of silent chromatin: inaction at a distance. Nat Rev Genet. 2006;7:793–803.
Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–8.
Cope NF, Fraser P, Eskiw CH. The yin and yang of chromatin spatial organization. Genome Biol. 2010;11:204.
Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Dénervaud N, et al. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010;6:e1000869.
de Bruin D, Zaman Z, Liberatore RA, Ptashne M. Telomere looping permits gene activation by a downstream UAS in yeast. Nature. 2001;409:109–13.
Zaman Z, Heid C, Ptashne M. Telomere looping permits repression ‘at a distance’ in yeast. Curr Biol. 2002;12:930–3.
Wakimoto BT. Beyond the nucleosome: epigenetic aspects of position–effect variegation in Drosophila. Cell. 1998;93:321–4.
Ratna P, Scherrer S, Fleischli C, Becskei A. Synergy of repression and silencing gradients along the chromosome. J Mol Biol. 2009;387:826–39.
Kelemen JZ, Ratna P, Scherrer S, Becskei A. Spatial epigenetic control of mono- and bistable gene expression. PLoS Biol. 2010;8:e1000332.
Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–44.
Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849–60.
Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162:900–10.
Bickmore WA, van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270–84.
Falahi F, Sgro A, Blancafort P. Epigenome engineering in cancer: fairytale or a realistic path to the clinic? Front Oncol. 2015;5:1–11.
Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NACH, Margaritis T, et al. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell. 2011;42:536–49.
Nowling T, Bernadt C, Johnson L, Desler M, Rizzino A. The co-activator p300 associates physically with and can mediate the action of the distal enhancer of the FGF-4 gene. J Biol Chem. 2003;278:13696–705.
Kwaks THJ, Sewalt RGAB, van Blokland R, Siersma TJ, Kasiem M, Kelder A, Otte AP. Targeting of a histone acetyltransferase domain to a promoter enhances protein expression levels in mammalian cells. J Biotechnol. 2005;115:35–46.
Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105.
Beisel C, Imhof A, Greene J, Kremmer E, Sauer F. Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature. 2002;419:857–62.
Strahl BD, Grant PA, Briggs SD, Sun Z-W, Bone JR, Caldwell JA, et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298–306.
Osipovich O, Milley R, Meade A, Tachibana M, Shinkai Y, Krangel MS, Oltz EM. Targeted inhibition of V(D)J recombination by a histone methyltransferase. Nat Immunol. 2004;5:309–16.
Firestein R, Cui X, Huie P, Cleary ML. Set domain-dependent regulation of transcriptional silencing and growth control by SUV39H1, a mammalian ortholog of Drosophila Su(var)3-9. Mol Cell Biol. 2000;20:4900–9.
Mohan M, Herz H-M, Shilatifard A. SnapShot: histone lysine methylase complexes. Cell. 2012;149:498.
Peterson CL, Laniel M-A. Histones and histone modifications. Curr Biol. 2004;14:R546–51.
Kutateladze TG. SnapShot: histone readers. Cell. 2011;146:842.
Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–94.
Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nat Chem Biol. 2012;8:417–27.
Acknowledgements
We would like to thank members of the Khalil laboratory for helpful discussions and comments and K. Flynn for help with artwork.
Funding
This work was supported by a NIH-NIGMS Ruth L. Kirschstein Postdoctoral Fellowship (1F32GM105272-01A1 to A.J.K.), a National Science Foundation CAREER Award (MCB-1350949 to A.S.K.), a National Science Foundation Expeditions in Computing grant (CCF-1522074 to A.S.K.), and a Defense Advanced Research Projects Agency grant (W911NF-11-2-0056 to A.S.K.).
Authors’ contributions
MP, AJK, and ASK conceived and wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Park, M., Keung, A.J. & Khalil, A.S. The epigenome: the next substrate for engineering. Genome Biol 17, 183 (2016). https://doi.org/10.1186/s13059-016-1046-5
Published:
DOI: https://doi.org/10.1186/s13059-016-1046-5