
Abstract
Genome editing is the process of precisely modifying the nucleotide sequence of the genome. It has provided a powerful approach to research questions but, with the development of a new set of tools, it is now possible to achieve frequencies of genome editing that are high enough to be useful therapeutically. Genome editing is being developed to treat not only monogenic diseases but also infectious diseases and diseases that have both a genetic and an environmental component.
Similar content being viewed by others

The potential therapeutic importance of genome editing
In 1901, Sir Archibald Garrod identified alkaptonuria as the first known human genetic disease. Today, we recognize that there are at least 8000 human diseases that are caused by mutations in single genes (monogenic diseases); the number increases almost every day [1, 2]. While all of these diseases are classified as ‘rare’ in the US because they affect fewer than 200,000 people, they may affect over 400 million people worldwide. Some, such as sickle cell disease, affect tens of millions of people around the world and are only ‘rare’ in certain parts of the world, including the US, Europe and far east Asia. For a tiny subset of patients, allogeneic hematopoietic stem cell transplantation (allo-HSCT) or solid organ transplantation can be used to cure their genetic disease, but for the vast majority of patients there is no cure and at best they are treated by management of symptoms.
Therapeutic genome editing was born out of the idea that the ideal therapy for monogenic diseases would be to develop a method that can correct the disease-causing mutations directly; but as genome editing has developed in concert with continuing improvements in our understanding of the genetic contribution to non-monogenic diseases, the principle of genome editing is being developed not only to cure monogenic diseases but also to cure more common diseases that have multifactorial origins. The use of genome editing to cure monogenic disease is conceptually simple (genome editing can be used to correct the underlying genomic typographical errors), but the power of genome editing is that it provides a mechanism that can do more than simply modify single nucleotides. It is a method that can make more sophisticated and nuanced genomic changes, which can be used to cure more common diseases or to modify their course.
The exact nature of the therapeutic edit has to be driven by a solid understanding of the interplay between the underlying genetics and the specific pathophysiology of the disease. That is, one editing strategy might be appropriate for one disease but not applicable to another. This review will describe the basic strategies of genome editing and the tools that are now available both to correct typographical errors and to make more sophisticated changes to the genome. I will then discuss how genome editing is being developed to treat genetic, infectious, and acquired diseases. Finally, I end with a brief discussion of issues surrounding the use of genome editing in situations that might cause the engineered genetic change to be passed from one generation to the next.
The development of genome editing and the contemporary toolbox
Genome editing, also previously known as gene targeting, has been a powerful research tool for scientists. In particular, the ease of gene targeting in yeast was one factor that made yeast such an important model organism in studies of the pathophysiology of human disease [3, 4]. The importance of gene targeting as a research tool was further highlighted by the awarding of the Nobel Prize in Physiology or Medicine in 2007 to Drs Oliver Smithies and Mario Capecchi for their development of gene targeting in mouse embryonic stem cells and for their subsequent precise genetic engineering of mice—a transformational advance in understanding human pathophysiology [5, 6]. Even in the earliest days of gene therapy, it was recognized that genome editing might be the ideal approach for curing genetic diseases, but the earliest studies were stymied by the low absolute frequency of gene correction by homologous recombination in human somatic cells (10−6) [7–9]. A critical breakthrough was the discovery that by creating a site-specific DNA double-stranded break (DSB) in the target gene it is possible to stimulate genome editing by homologous recombination by 2–5 orders of magnitude, providing overall frequencies of 5 % or more [10–13]. In addition to stimulating gene targeting by homologous recombination by five orders of magnitude, a site-specific DSB could stimulate mutations such as small insertions/deletions at the site of the DSB by nine orders of magnitude. Thus, the DSB became a key principle in the development of genome editing.
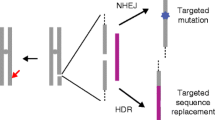
The basic process of nuclease-based genome editing is to create a specific DSB in the genome and then allow the cell’s own endogenous repair machinery to repair the break (Fig. 1). The cell can repair the break using one of two basic mechanisms: nonhomologous end-joining (NHEJ) or homologous recombination (HR) (see Box 1; Fig. 1) [14–17]. When the editing of a single break occurs by NHEJ, insertions/deletions are created at the site of the break [17] (Fig. 1a). The size of deletions tends to be larger than that of insertions, except when extrachromosomal DNA is captured at the site of the break (a rare but measurable occurrence), in which case insertions of hundreds of basepairs (bp) can occur [18, 19]. When editing of a single break occurs by HR using a provided donor sequence, precise nucleotide changes in the genome range from a single base insertion to the introduction of large cassette of genes (Fig. 1c) [20, 21]. When the editing of two breaks occurs by NHEJ, chromosomal deletions, inversions or translocations can be created (Fig. 1b) [22]. These gross chromosomal rearrangements can be generated intentionally for therapeutic purposes, but they also must be evaluated because any nuclease platform has the potential to produce off-target effects.

Nuclease-based genome editing creates a specific double-stranded break (DSB) in the genome and then allows the cell’s own endogenous repair machinery to repair the break. a When the editing of a single break occurs by nonhomologous end-joining (NHEJ), insertions/deletions are created at the site of the break. b When the editing of two DSBs occurs by NHEJ, chromosomal deletions, inversions or translocations can be created. c When editing of a single DSB occurs by homologous recombination (HR) using a provided donor sequence, precise changes in the nucleotide sequence ranging from a single base insertion to the introduction of a large cassette of genes can occur. Using NHEJ and HR mediated editing, it is now possible to inactivate genetic elements, create defined deletions ranging from a few bases to thousands of bases, and precise nucleotide changes to the sequence of the genome. AAV Adeno-associated virus, bps Basepairs, IDLV Integrase-deficient lentivirus
In seminal studies of the importance of DSBs, researchers used an artificial system in which the target site for a natural homing endonuclease (also sometimes called a ‘meganuclease’), I-SceI, was engineered into the genome of somatic cells; the frequency of genome editing was measured at that engineered I-SceI site [9, 13, 23–25]. The barrier to high-frequency editing was that neither I-SceI nor the other meganucleases could be easily re-engineered to recognize natural target sites in the genome. The first solution to this problem was the development of zinc-finger nucleases (ZFNs) (originally called ‘chimeric restriction enzymes’, then called ‘chimeric nucleases’) [26–28]. ZFNs are artificial proteins in which a zinc-finger DNA-binding domain is fused to the nonspecific nuclease domain derived from the FokI Type II S restriction endonuclease. At first, engineered ZFNs were shown to be as efficient as I-SceI in stimulating gene targeting in human somatic cells. Then, because the zinc-finger DNA binding domain can be engineered to recognize new target sites, ZFN-based protocols became the first method used to stimulate genome editing in human somatic cells to frequencies that are therapeutically relevant [9, 29, 30]. This work in human somatic cells paralleled the important work by Dana Carroll and his colleagues, who showed that ZFNs could be used to edit the complex genome of eukaryotic Drosophila melanogaster, both by mutagenic NHEJ and by HR [17, 31]. For a number of years, the only engineered nucleases in the genome editing toolbox were ZFNs [32, 33] and re-engineered meganucleases [34]. In the past 5 years, however, the development of TAL effector nucleases (TALENs) [35–37], CRISPR/Cas9 nucleases [38–40], and hybrid nuclease platforms [41–43] have dramatically expanded the engineered nuclease toolbox.
There are four basic and two hybrid engineered nuclease platforms that include engineered meganucleases, ZFNs, TALENs, CRISPR/Cas9 nucleases, mega-TAL nucleases, and Cas9-FokI nucleases (Box 2; Table 1). There are subtle differences between each of these nuclease platforms—for example, the type of break that is created is different: meganucleases and mega-TALs generate breaks with 3′ overhangs; ZFNs create breaks with 5′ overhangs; TALENs create breaks that are variable in position that are usually (but not always) 5′ overhangs, as determined by the properties of the FokI nuclease (Fn); and CRISPR/Cas9 nucleases create blunt breaks. In general, however, each of these platforms mediates their editing effects through the creation of a DSB and thus they share a fundamental mechanism of action.
The only tool needed for NHEJ-mediated genome editing is an engineered nuclease, but HR-mediated genome editing also requires an engineered donor vector. Donor vectors can be designed to template single bp changes or to insert large multi-gene cassettes into the genome. The homology arms for nuclease-mediated genome editing can be much shorter than those required for HR-mediated gene targeting in murine embryonic stem cells: instead of having to be 10 kilobases or greater, they can be as short as 400 bp [18]. Shortening the homology arms to below 400 bp does, however, seem to decrease the overall editing efficiency. Single-stranded oligonucleotides (ssODNs) have also been used to template small nucleotide changes after the induction of a DSB [44]. The ease with which ssODNs can be synthesized makes this approach relatively accessible to the researcher, but the mechanism by which ssODNs create a targeted change in the genome does not rely on the classic HR pathway and is not well understood. Moreover, ssODNs induce a replication and cell cycle arrest even in cancer cell lines [45] and would probably be even more problematic in primary therapeutically relevant human cell types, as demonstrated in part by the work of Hoban et al. [46].
Issues of delivery and process development
A mantra in the field has been that the three most important issues in gene therapy are delivery, delivery, and delivery. As the toolbox for genome editing has expanded, that mantra now also applies in many respects to therapeutic genome editing: what is the optimal process to deliver highly active genome-editing reagents into the most clinically relevant cell type? The answer to this question is increasingly disease specific. An important consideration in determining an appropriate delivery strategy is that genome editing, in contrast to gene-augmentation strategies, is a hit and run approach. In fact, sustained expression of the nuclease not only is not needed but should be avoided: continued expression of a nuclease increases the probability of deleterious genomic instability and may either compromise the edited cell’s fitness or predispose the exposed cell to transformation.
For ex vivo manipulation of cells, standard nonviral delivery of the nuclease as RNA, or ribonucleoprotein (RNP) for the CRISPR/Cas9 system, seems to be the most promising approach. Delivering the nuclease component as RNA or RNP ensures that both the activation of the Type I interferon response and the duration of the expression are minimized. The RNA or RNP can be delivered into a cell by a variety of mechanisms that are determined by the specific cell type’s ability to be transfected with different complexes. A universal method of delivery that is effective across all cell types is electroporation, in which cells are mixed with the RNA or RNP and a brief electrical pulse is passed through the mixture, thereby creating membrane holes through which the RNP or RNA enters. Multiple different electroporation devices are now available and, amazingly, electroporation conditions can be found that create minimal cellular toxicity as long as DNA or other nucleic acids that activate the innate immune system are not included in the mixture. For applications that simply require the delivery of the nuclease, this seems to be a robust solution. For applications that require HR-mediated editing, a DNA molecule also needs to be delivered. Delivery of naked DNA into cancer cell lines has been an effective method to deliver the donor vector, but the delivery of naked DNA into primary cells, particularly T cells and hematopoietic stem and progenitor cells, activates a deleterious innate immune response that both decreases the frequency of genome editing and compromises the fitness of the edited cell. Using adeno-associated virus (AAV) to deliver the donor template into cells may be a solution to this problem because AAV, like many viruses, has evolved to escape recognition by the innate intracellular immune response [47–50].
For therapeutic applications that require in vivo editing of cells, the challenge is greater and a solution has not been determined. Again, the solution to the in vivo delivery problem may differ depending on which target cell type needs to be identified. The solution for editing hepatocytes, for example, will probably be different than that for editing muscle, which will be different again from that for editing cells in the central nervous system. Nonetheless, with the development of multiple different serotypes of AAV that preferentially transduce different cell types in vivo [51, 52], the development of new methods of delivering mRNAs to cells, and the increasing sophistication of nanoparticles (both lipid and nonlipid based) to deliver to specific tissues, there are likely to be solutions forthcoming soon. Developing a delivery method in which the nuclease is not expressed for a sustained period of time is important both from a genotoxic standpoint and from an immunologic standpoint. It should be anticipated, until proven otherwise, that all of the engineered nuclease platforms will be seen by the immune system as foreign and will elicit a robust immune response that will both eliminate the therapeutically edited cells and perhaps cause toxic organ damage.
As therapeutic genome editing gathers momentum, an increasing number of innovative approaches are being developed. These can be classified along three different axes: NHEJ- vs. HR-mediated genome editing; ex vivo vs. in vivo delivery; and suitability for genetic vs. infectious vs. nongenetic diseases. Examples of some of these various strategies are discussed below.
Potential therapeutic applications of nonhomologous end-joining mediated genome editing
Diseases that can be addressed using NHEJ-mediated genome editing are those in which mutating a genetic element, whether a coding region, a regulatory element, or some other genetic element, might result in clinical benefit. One example of this approach is to delete the erythroid enhancer for Bcl11A in hematopoietic stem/progenitor cells (HSPCs) in order to upregulate γ-globin to treat sickle cell disease and β-thalassemia [53–55]. Both sickle cell disease and β-thalassemia are monogenic diseases caused by mutations in the HBB gene. Both diseases could be cured if HBG, a gene closely related to HBB, could be upregulated such that it could either replace the missing γ-globin protein (in β-thalassemia) or counteract the dysfunctional γ-globin protein (in sickle cell disease). Studies of the globin switch have demonstrated that Bcl11-A is a transcriptional repressor of HBG and that repression of Bcl11A results in the de-repression of HBG [56]. Moreover, when used as a research tool, genome editing demonstrated that deletion of a specific regulatory element in the Bcl11A gene, the erythroid enhancer, could repress Bcl11A in the erythroid lineage but not in the B-cell lineage, thus validating the inactivation of this element by NHEJ-mediated genome editing in HSPCs as a therapeutic strategy [53].
A different strategy using NHEJ-mediated genome editing is being developed to treat Duchenne’s muscular dystrophy, a monogenic disease cause by mutations in the Dystrophin gene. In this in vivo strategy, a single nuclease might be delivered into muscle fibers to create an insertion/deletion that compensates for the original frameshift mutation (thus reverting the pathologic reading frame mutation). Alternatively, a pair of nucleases might be delivered into muscle fibers to delete a set of exons in order to delete pathologic mutations, thereby converting Duchenne’s muscular dystrophy to the less severe Becker’s muscular dystrophy. Proof-of-concept studies have been published for both of these strategies, but the challenge that remains is to achieve the desired editing in a fraction of muscle fibers, including the heart and diaphragm tissues, large enough to alter the clinical course of the disease significantly [57–59]. In addition, as a general principle, any disease that might be treated by RNA interference (RNAi)-mediated knockdown of a gene [60, 61] might be more definitively treated by genome editing. Editing would provide permanent knockdown of the gene and therefore would not require repeated dosing of knockdown RNAi reagent.
For infectious diseases, ex vivo NHEJ-mediated genome editing has already reached Phase II clinical trials as a method to generate a T-cell population that is resistant to HIV infection. These studies are based on the discovery that people with bi-allelic mutations in the CCR5 gene are near completely resistant to HIV infection, and on the cure of an HIV patient by allo-HSCT using a donor whose stem cells contained a bi-allelic mutation in the CCR5 gene [62]. Sangamo Biosciences and their collaborators have engineered ZFNs to target the CCR5 gene, and then used these ZFNs to mutate the CCR5 gene in primary T cells derived from patients already infected with HIV [63–65]. In Phase I trials, they demonstrated that this approach was both feasible and safe, and Phase II trials are now underway [65].
In vivo NHEJ-based genome-editing approaches are also being developed for infectious diseases. In multiple proof-of-concept studies, nucleases have been engineered to recognize key elements of viral genomes (including those of HIV and hepatitis B) in order to create mutations that will inactivate the virus [66–68]. These studies have demonstrated that such nucleases can be engineered and that they can alter viral kinetics in in-vitro models, but real challenges remain in how to apply this strategy in an in vivo setting where delivery to nearly all infected cells must be achieved and in a way that does not require constitutive expression of the nuclease.
Finally, NHEJ-mediated genome editing has been applied in a proof-of-concept study as a potential approach to treat high cholesterol. PCSK9 is a regulator of cholesterol and those who have the rare homozygous deficiency in PCSK9 are otherwise healthy but have extremely low cholesterol levels. In vivo nuclease-mediated genome editing has been used to mutate the PCSK9 gene in livers, with a resultant drop in cholesterol levels [69, 70]. Although there are multiple caveats to these experiments, they do show, in principle, how in vivo editing might be used to treat multifactorial diseases whose course could be modified by using genome editing to create a clinically useful genotype.
Potential therapeutic applications of homologous recombination mediated genome editing
Deep understanding of the pathophysiology of certain diseases can show how NHEJ-mediated genome editing could be used as therapy for those diseases. In general, however, the ability to harness HR-mediated genome editing both ex vivo and in vivo has the potential to impact on an even larger number of diseases.
There are numerous genetic diseases of HSPCs, such as sickle cell disease, β-thalassemia, severe combined immunodeficiency and chronic granulomatous disease, that can be cured by allo-HSCT. In allo-HSCT for these types, the hematopoietic system is replaced by cells containing at least one wild-type version of the gene and, for this reason, some have called it ‘allogeneic gene therapy’ [71]. Using HR-mediated genome editing, it would be possible to replace genetically correct allogeneic stem cells with genetically corrected autologous cells. This can be done either by directly correcting the defective gene [46] or by using HR-mediated genome editing to target the therapeutic transgene to a ‘safe harbor’ [72]—a genomic site in which the transgene would be expressed at the needed levels without causing dysfunction or transformation of the modified cell [73]. One potential issue with gene correction by HR is that many genetic diseases, sickle cell disease being an exception, are caused by mutations throughout the gene. The engineered nuclease toolbox is now such that one might consider designing nucleases for each individual mutation. An alternative approach, however, is to design the donor vector such that, after HR, the integrated transgene would functionally correct all (or most) of the disease-causing mutations [74, 75]. Using this strategy, a single set of reagents could be developed to treat all individuals with the genetic disease—a strategy that would significantly simplify the development and regulatory process.
Proof-of-concept studies for in vivo HR-mediated genome editing have been described in which either the underlying mutant gene was directly corrected or a transgene was integrated into a specific location such that it would be expressed at sufficient levels to rescue the underlying defect [76]. In the direct gene correction strategy, nucleases and donor vectors were delivered to fumarylacetoacetate hydrolase (FAH)-deficient mice. Normally, FAH deficiency causes hepatocyte death but, after delivery of the genome-editing machinery, a small number of hepatocytes were corrected. These corrected hepatocytes then re-populated the remaining liver and rescued the mouse from liver failure. In these experiments, the corrected cells had a tremendous selective advantage over uncorrected cells, and the principle of selective advantage is one that is regularly used by the gene therapy community. In the transgene-targeting strategy, nucleases were used to stimulate the targeted introduction of a therapeutic transgene (either Factor IX or lysosomal storage enzymes) into a locus that drove high levels of expression from hepatocytes [77–79]. In this way, a small number of modified hepatocytes were able to rescue an underlying genetic defect at a systemic level.
Ex vivo HR-mediated genome editing is also being developed as a method to create an HIV-resistant immune system [20]. One of the hallmarks of HIV is its ability to mutate and escape any inhibition, and thus it is possible that simply mutating the CCR5 co-receptor will not be sufficient to confer cellular resistance to HIV. Moreover, many HIV patients have already developed HIV variants that enter cells through the CXCR4 coreceptor and thus would escape any approach that only targets CCR5; but using HR-mediated editing, one can simultaneously inactivate CCR5 while inserting a cassette of antiHIV genes, thereby creating multiple genetic blocks to the HIV lifecycle and inhibiting variants that enter through the CXCR4 co-receptor.
Finally, ex vivo HR-mediated genome editing has been shown in proof-of-concept experiments to be therapeutic for an acquired disease. In these experiments, fibroblasts were engineered by HR to secrete a wound-healing growth factor [80]. When these engineered fibroblasts were implanted into mouse wounds, they hastened wound healing by stimulating vascularization. In principle, this demonstrates that cells can be engineered to secrete therapeutic proteins that rescue nongenetic diseases. This might be applied to wound healing in humans but one can also speculate, for example, that a similar approach might be used to engineer cells, either ex vivo or in vivo, to secrete neuroprotective factors to slow or halt neurodegeneration or to facilitate neurogenesis or nerve regeneration after trauma.
Safety and toxicology
One of the tremendous potential advantages of genome editing, when compared to other methods of permanently altering the genome of cells, is the specificity of the process. Nevertheless, the induction of a DSB through a site-specific engineered nuclease is a critical aspect of gene editing and it is well known that DSBs can generate genomic instability, including chromosomal translocations, chromosome loss and aneuploidy [81]. Thus, a key aspect in the clinical development of nuclease-mediated genome editing is to establish a series of assays that assess the potential safety of the process. Unfortunately, the field is too young for there to be any single assay or set of assays that has been validated as establishing whether an editing process will be safe in humans [82]. Instead, safety and toxicology analysis is assessed according to the following principles: 1) minimizing or eliminating off-target DSBs and the consequent insertions/deletions that may be generated; 2) assessing the functional behavior of edited cells using the best available models; and 3) putting the process of genome editing in the context of the natural genomic instability that occurs continuously in everybody. These fundamental criteria apply to whichever nuclease platform is utilized because each platform works through the creation of a DSB.
There are biased and unbiased approaches to assessing the specificity of a nuclease [83, 84]. Bioinformatic tools that are based on searching for sites that have sequences similar to the intended target site can help to predict which off-target sites should be examined. Once a set of sites is identified, deep sequencing can be used to interrogate those sites to determine whether there are nuclease-generated insertions/deletions at those sites. Given the current error rate of deep sequencing methodologies, the limit of detection for a given site is ~0.01 % (or in 1 in 10,000). Moreover, the bioinformatics algorithms are still in their early development and still do not reliably identify all potential off-target sites. To complement the biased approach using bioinformatics, there are newly developed unbiased tools, including break Guide-seq [85], HTGTS [86], BLESS [70], and Digenome-seq [87]. Other assays use extrachromosomal DNA, including AAV [88], naked DNA plasmids [19], and integration-defective lentiviral vectors [89], to capture breaks and to help assess the specificity of a nuclease. These tools have particular power because they can also identify gross chromosomal rearrangements (like translocations) that are not identified using biased approaches. As chromosomal translocations will be a rare but inevitable consequence of induced DSBs, it would be prudent to avoid targeting genes that are involved in cancer-associated chromosomal translocations at this stage in the development of therapeutic genome editing.
The challenge with these unbiased approaches is that they have been developed in specialized cancer cell lines, which do not have intact DNA-repair pathways, and they need to be adapted to primary clinically relevant cell types with intact DNA repair pathways. Nonetheless, these tools still provide useful information in optimizing the specificity of a nuclease. For example, if these assays reveal that the nuclease has multiple off-target sites, then they suggest that the nuclease should be redesigned to be more specific. For ZFNs, this might entail using the obligate heterodimer structure for the nuclease domain or exploring specific alterations in the amino acids that mediate recognition of the target sequence [90]. For TALENs, this might entail using the obligate heterodimer structure for the nuclease domain or using alternative repeat variable di-residues (RVDs) for the TAL effector recognition domain [91]. For CRISPR/Cas9 nucleases, improved design might entail testing a different guide sequence, or using a truncated guide sequence [92] or a paired-nickase approach [93, 94]. For all of the platforms, specificity is increased by limiting the duration of expression of the nuclease, the pharmacologic equivalent of decreasing the AUC (‘area under the curve’) of nuclease exposure [39, 95, 96].
An approach that complements attempts to identify the potential off-target sites of the nuclease directly is to evaluate the genome-editing process by a more classical functional pharmacology–toxicology approach. This strategy evaluates whether the genome-editing process creates cells that are unable to perform their normal function (for example, the ability of hematopoietic stem cells to reconstitute multilineage hematopoiesis), that transform into cancer cells, or that cause the population to become clonally skewed (a possible harbinger of the creation of cells that might transform over a time frame beyond that which can be measured using current assays) [82].
The biased, unbiased, and functional approaches need to be performed in the therapeutic cell type of interest using the clinical-grade genome-editing process planned because assessment in other cell types, particularly already-transformed cancer cell lines, may not be relevant.
An important principle to keep in mind is that dividing cells are undergoing genomic challenge continuously. It is estimated that every time a cell divides, it must repair 20–40 DNA DSBs, not to mention millions of other types of DNA lesions [97, 98]. The consequence of this natural genomic challenge is that a normal dividing stem cell acquires ~3–30 mutations for each cell division; it is estimated 1 million mutations occur every second in an individual. It has been suggested that gene correction by genome editing of patient-derived induced pluripotent cells, followed by whole-genome sequencing to determine if deleterious mutations have occurred, might be a safer approach. The mutational burden caused by ex vivo expansion from a single cell to a therapeutically relevant cell number (for hematopoietic diseases this is in the order of 50–800 million cells depending on the size of the patient) may, in reality, be more oncogenic than just modifying a large number of somatic cells without being able to sequence the genome of any one cell.
Challenges of germline genome editing
The development of a powerful genome-editing toolbox, combined with the use of this toolbox to create a wide variety of genetically modified species by zygote injection [16], has raised the possibility that someone might use genome editing in human zygotes to create human beings [99, 100]. This possibility was further highlighted by researchers in China who used the approach in tripronuclear human zygotes (human zygotes that are genetically incapable of developing into a human but are nearly identical in concept to diploid zygotes) [101]. The tripronuclear zygote injection experiments highlighted the inefficiency and unpredictability of the process—findings that would have been predicted by animal experiments using healthy diploid zygotes. These results clearly demonstrate that genome-editing technology applied as zygote injection, even if judged ethically permissible or desirable, is not ready for human application. Nonetheless, these specific experiments and the general concept have generated a large number of headlines in both high-profile journals and the lay press. Which direction the conversation turns remains to be determined, but there are several principles that I hope remain at the forefront. First, the ethical issue should not impede the use of powerful genome-editing tools in research to provide a better understanding of germ cells, germ cell development, and early embryonic development. Second, the discussion should be led by thought leaders from a variety of different fields and should include voices from a broad range of stakeholders, including those families who lives have been impacted over multiple generations by the transmission of devastating genetic diseases through their family tree. Third, there is no single ethical viewpoint that has predominance and an ongoing, iterative process in which new understandings and viewpoints can be incorporated is the desired outcome rather than a defined resolution at a single point in time. Finally, the issue of using genome editing that might result in the transmission of specific genotypes to future generations needs to be put into the context of activities already taking place that similarly affect the genotypic makeup of future generations. Two such examples are pre-implantation genetic diagnosis with selective zygote implantation and curing or helping patients with genetic diseases (an unequivocally good thing) such that they might not pass down their disease-causing mutation to their children.
Future perspectives
The precision of genome editing and the ability to correct disease-causing typographical errors in the DNA sequence have always made the field conceptually appealing. All of the strategies of genome editing could be achieved using engineered meganucleases and ZFNs, but the challenge of making highly active and specific versions of these tools has limited the number of investigators who were committed to the concept. With the development of TALENs and then CRISPR/Cas9 nucleases, the barrier to entry for investigators was decreased so dramatically that essentially any researcher who is interested can begin to explore their own innovative ideas. With this explosion in interest, the pace of progress has increased exponentially. In the decade since the first use of ZFNs in human cells there has been just one clinical trial, so it is exciting to predict that in the next decade there will be tens (if not more) of genome editing-based clinical trials that will be developed by academics, biotechnology start-ups and pharmaceutical companies.
Nonetheless, there remain important issues to be resolved. These include developing a regulatory framework that is tailored to the underlying technology rather than one that is based on a different therapeutic foundation (such as small molecules or antibody biologics). There is also a need to develop safe and effective mechanisms to deliver the genome-editing machinery to a wide variety of tissues in vivo, including the liver, eye, muscle, heart, and brain. Finally, a flexible and adaptive regulatory framework needs to be developed to take into account the ethical and scientific issues around the potential use of genome editing that might alter the genetics of future generations (‘altering heredity’). This framework needs to take into account the diverse group of stakeholders who are affected by the issue and must respect culturally different perspectives.
Abbreviations
- AAV:
-
Adeno-associated virus
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- bp:
-
Base pairs
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DSB:
-
Double-stranded break
- FAH:
-
Fumarylacetoacetate hydrolase
- Fn:
-
FokI nuclease
- HR:
-
Homologous recombination
- HSPC:
-
Hematopoietic stem/progenitor cell
- NHEJ:
-
Nonhomologous end-joining
- RNAi:
-
RNA interference
- RNP:
-
Ribonucleoprotein
- RVD:
-
Repeat variable di-residue
- sgRNA:
-
Single-guide RNA
- ssODN:
-
Single-stranded oligonucleotide
- TALEN:
-
TAL effector nuclease
- ZFN:
-
Zinc-finger nuclease
References
Rosenberg LE. Legacies of Garrod’s brilliance. One hundred years—and counting. J Inherit Metab Dis. 2008;31:574–9.
Johnston JJ, Lewis KL, Ng D, Singh LN, Wynter J, Brewer C, et al. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am J Hum Genet. 2015;96:913–25.
Petes TD. Molecular genetics of yeast. Annu Rev Biochem. 1980;49:845–76.
Orr-Weaver TL, Nicolas A, Szostak JW. Gene conversion adjacent to regions of double-strand break repair. Mol Cell Biol. 1988;8:5292–8.
Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, Thompson S, et al. Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature. 1987;330:576–8.
Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–12.
Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature. 1985;317:230–4.
Sedivy JM, Dutriaux A. Gene targeting and somatic cell genetics—a rebirth or a coming of age? Trends Genet. 1999;15:88–90.
Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763.
Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–8.
Smih F, Rouet P, Romanienko PJ, Jasin M. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995;23:5012–9.
Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–106.
Choulika A, Perrin A, Dujon B, Nicolas J-F. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccaromyces cerevisiae. Mol Cell Biol. 1995;15:1968–73.
West SC, Chappell C, Hanakahi LA, Masson JY, McIlwraith MJ, Van Dyck E. Double-strand break repair in human cells. Cold Spring Harb Symp Quant Biol. 2000;65:315–21.
Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–83.
Carroll D. Genome engineering with targetable nucleases. Annu Rev Biochem. 2014;83:409–39.
Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764.
Hendel A, Kildebeck EJ, Fine EJ, Clark JT, Punjya N, Sebastiano V, et al. Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep. 2014;7:293–305.
Lin Y, Waldman AS. Capture of DNA sequences at double-strand breaks in mammalian chromosomes. Genetics. 2001;158:1665–74.
Voit RA, McMahon MA, Sawyer SL, Porteus MH. Generation of an HIV resistant T-cell line by targeted “stacking” of restriction factors. Mol Ther. 2013;21:786–95.
Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, et al. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci U S A. 2007;104:3055–60.
Lee HJ, Kim E, Kim JS. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–9.
Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91:6064–8.
Sargent RG, Brenneman MA, Wilson JH. Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol Cell Biol. 1997;17:267–77.
Taghian DG, Nickoloff JA. Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol. 1997;17:6386–93.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–60.
Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000;28:3361–9.
Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005;33:5978–90.
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–51.
Porteus MH. Mammalian gene targeting with designed zinc finger nucleases. Mol Ther. 2006;13:438–46.
Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–75.
Porteus MH, Carroll D. Gene targeting using zinc finger nucleases. Nat Biotechnol. 2005;23:967–73.
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–46.
Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. 2011;11:11–27.
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–61.
Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–6.
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–8.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, Duchateau P, et al. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2014;42:2591–601.
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32:569–76.
Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577–82.
Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods. 2011;8:753–5.
Ferrara L, Engstrom JU, Schwartz T, Parekh-Olmedo H, Kmiec EB. Recovery of cell cycle delay following targeted gene repair by oligonucleotides. DNA Repair (Amst). 2007;6:1529–35.
Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–604.
Porteus MH, Cathomen T, Weitzman MD, Baltimore D. Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol Cell Biol. 2003;23:3558–65.
Miller DG, Petek LM, Russell DW. Human gene targeting by adeno-associated virus vectors is enhanced by DNA double-strand breaks. Mol Cell Biol. 2003;23:3550–7.
Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7:307ra156.
Wang J, Exline CM, DeClercq JJ, Llewellyn GN, Hayward SB, Li PW, et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015; doi: 10.1038/nbt.3408.
Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14:316–27.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–80.
Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–7.
Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–7.
Vierstra J, Reik A, Chang KH, Stehling-Sun S, Zhou Y, Hinkley SJ, et al. Functional footprinting of regulatory DNA. Nat Methods. 2015;12:927–30.
Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–42.
Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244.
Ousterout DG, Kabadi AM, Thakore PI, Perez-Pinera P, Brown MT, Majoros WH, et al. Correction of dystrophin expression in cells from duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther. 2015;23:523–32.
Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, Qin X, et al. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther. 2013;21:1718–26.
Haussecker D. Current issues of RNAi therapeutics delivery and development. J Control Release. 2014;195:49–54.
Haussecker D, Kay MA. RNA interference. Drugging RNAi. Science. 2015;347:1069–70.
Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16.
Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–47.
Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–10.
Cradick TJ, Keck K, Bradshaw S, Jamieson AC, McCaffrey AP. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18:947–54.
Schiffer JT, Aubert M, Weber ND, Mintzer E, Stone D, Jerome KR. Targeted DNA mutagenesis for the cure of chronic viral infections. J Virol. 2012;86:8920–36.
Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510.
Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–92.
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–91.
Gaziev J, Lucarelli G. Allogeneic cellular gene therapy for hemoglobinopathies. Hematol Oncol Clin North Am. 2010;24:1145–63.
Chang CJ, Bouhassira EE. Zinc-finger nuclease-mediated correction of alpha-thalassemia in iPS cells. Blood. 2012;120:3906–14.
Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2012;12:51–8.
Voit RA, Hendel A, Pruett-Miller SM, Porteus MH. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014;42:1365–78.
Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–40.
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32:551–3.
Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21.
Barzel A, Paulk NK, Shi Y, Huang Y, Chu K, Zhang F, et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–4.
Sharma R, Anguela XM, Doyon Y, Wechsler T, DeKelver RC, Sproul S, et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–84.
Barker JC, Barker AD, Bills J, Huang J, Wight-Carter M, Delgado I, et al. Genome editing of mouse fibroblasts by homologous recombination for sustained secretion of PDGF-B and augmentation of wound healing. Plast Reconstr Surg. 2014;134:389e–401e.
Weinstock DM, Richardson CA, Elliott B, Jasin M. Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst). 2006;5:1065–74.
Corrigan-Curay J, O'Reilly M, Kohn DB, Cannon PM, Bao G, Bushman FD, et al. Genome editing technologies: defining a path to clinic. Mol Ther. 2015;23:796–806.
Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol Ther Nucleic Acids. 2014;3:e214.
Hendel A, Fine EJ, Bao G, Porteus MH. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015;33:132–40.
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–97.
Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33:179–86.
Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12:237–43. 1 p following 243.
Miller DG, Petek LM, Russell DW. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat Genet. 2004;36:767–73.
Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–23.
Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8:74–9.
Miller JC, Zhang L, Xia DF, Campo JJ, Ankoudinova IV, Guschin DY, et al. Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat Methods. 2015;12:465–71.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84.
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–8.
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9.
Pruett-Miller SM, Reading DW, Porter SN, Porteus MH. Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet. 2009;5:e1000376.
Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–9.
Friedberg ED, Walker GC, Siede W. DNA repair and mutagenesis. Washington, DC: ASM Press; 1995.
Nickoloff JA, Hoekstra MF. DNA damage and repair. Totowa, NJ: Humana Press; 1998.
Baltimore D, Berg P, Botchan M, Carroll D, Charo RA, Church G, et al. Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science. 2015;348:36–8.
Porteus MH, Dann CT. Genome editing of the germline: broadening the discussion. Mol Ther. 2015;23:980–2.
Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6:363–72.
Kanaar R, Hoeijmakers JH, van Gent DC. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8:483–9.
Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404.
Mandal P, Ferreira L, Collins R, Meissner T, Boutwell C, Friesen M, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–52.
Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, et al. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell. 2004;14:611–23.
Breese EH, Buechele C, Dawson C, Cleary ML, Porteus MH. Use of genome engineering to create patient specific MLL translocations in primary human hematopoietic stem and progenitor cells. PLoS One. 2015;10:e0136644.
Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697–700.
Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, et al. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods. 2011;8:671–6.
Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29:3757–74.
Jarjour J, West-Foyle H, Certo MT, Hubert CG, Doyle L, Getz MM, et al. High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res. 2009;37:6871–80.
Isalan M, Klug A, Choo Y. Comprehensive DNA recognition through concerted interactions from adjacent zinc fingers. Biochemistry. 1998;1;37(35):12026–33.
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12.
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501.
Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096.
Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78.
Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–85.
Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–92.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32.
Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol. 2015;33:175–8.
Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–84.
Acknowledgements
I thank all of the past and present members of my lab, the multiple collaborators, and various mentors I have worked with over the years for their hard work and support in helping to develop genome editing as a human therapeutic. I also am grateful to be supported as the Laurie Kraus Lacob Faculty Scholar in Pediatric Translational Research and thank the Amon G. Carter Foundation for their sustained and consistent support of the research in my lab.
Funding
The work in my lab has been consistently supported by the NIH and I thank the NIH for current support through grants PN2EY018244, R01-AI097320 and R01-AI120766.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author consults for and serves on the Scientific Advisory Board for CRISPR Therapeutics.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Porteus, M.H. Towards a new era in medicine: therapeutic genome editing. Genome Biol 16, 286 (2015). https://doi.org/10.1186/s13059-015-0859-y
Published:
DOI: https://doi.org/10.1186/s13059-015-0859-y