
Abstract
Background
Hepatocellular carcinoma (HCC) is a heterogeneous disease with high mortality rate. Recent genomic studies have identified TP53, AXIN1, and CTNNB1 as the most frequently mutated genes. Lower frequency mutations have been reported in ARID1A, ARID2 and JAK1. In addition, hepatitis B virus (HBV) integrations into the human genome have been associated with HCC.
Results
Here, we deep-sequence 42 HCC patients with a combination of whole genome, exome and transcriptome sequencing to identify the mutational landscape of HCC using a reasonably large discovery cohort. We find frequent mutations in TP53, CTNNB1 and AXIN1, and rare but likely functional mutations in BAP1 and IDH1. Besides frequent hepatitis B virus integrations at TERT, we identify translocations at the boundaries of TERT. A novel deletion is identified in CTNNB1 in a region that is heavily mutated in multiple cancers. We also find multiple high-allelic frequency mutations in the extracellular matrix protein LAMA2. Lower expression levels of LAMA2 correlate with a proliferative signature, and predict poor survival and higher chance of cancer recurrence in HCC patients, suggesting an important role of the extracellular matrix and cell adhesion in tumor progression of a subgroup of HCC patients.
Conclusions
The heterogeneous disease of HCC features diverse modes of genomic alteration. In addition to common point mutations, structural variations and methylation changes, there are several virus-associated changes, including gene disruption or activation, formation of chimeric viral-human transcripts, and DNA copy number changes. Such a multitude of genomic events likely contributes to the heterogeneous nature of HCC.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death, with a poor 5-year survival rate of less than 10% [1]. While more than 600,000 new cases are diagnosed annually, there is no effective targeted therapy. HCC is highly heterogeneous and associated with various etiological factors, including hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, alcohol consumption and exposure to aflatoxin and possibly vinyl chloride [2]. Several emerging themes were revealed by recent genomic studies [3]-[6], including recurrent mutations in TP53, Wnt-signaling components CTNNB1 and AXIN1, and chromatin regulators like ARID1A and ARID2, as well as HBV integration near TERT, CCNE1 and MLL4. The frequently altered genes discovered by these studies have differed, however, possibly due to small discovery panels (up to 25 patients) and the inherent heterogeneity of HCC due to several associated etiological factors. For example, activating mutations in CTNNB1 are mutually exclusive with HBV infection [6] and hence would not be prominent in a cohort enriched for HBV-infected patients. Therefore, it might not be surprising that different studies have identified different genes that are mutated in the population at low frequency, like ARID2, ARID1A and JAK1. Another difficulty in constructing the complete mutational landscape in HCC is the focused nature of most previous studies, since it is not common for a single study to comprehensively examine multiple types of genomic changes that include point mutations, deletions, structural variations, and virus-mediated mutations. We have previously shown that HBV DNA frequently integrates into the human genome, causing diverse changes such as DNA copy number variation, chimeric viral-human transcript fusions, and transcriptional activation [3]. Given the disruptive nature of HBV integration, it is pertinent to study all modes of genomic changes in the same context.
In this study, we analyzed a panel of 42 HCC patients with a combination of whole-genome, exome and transcriptome sequencing. We identified multiple high-allelic frequency mutations in LAMA2. LAMA2 encodes the α subunit of laminin, the major component of basal laminae. Besides being a structural component of the extracellular matrix, basal laminae can influence cell proliferation and differentiation. Defective anchoring to laminins is widespread in cancer [7]. The high incidence of mutations in an extracellular matrix component like LAMA2 adds a new dimension of underlying genetic components to this rather complex disease. We also found two patients with mutations in IDH1 at the R132 hotspot found in other cancers [8],[9], and one patient with truncated IDH2. The tumor suppressor BAP1 was mutated in two patients. We previously showed that HBV randomly integrates into the human genome and results in several genomic and genetic alterations [3]. Here we report HBV integrations in eight HBV-infected patients, including integration in the vicinity of three previously reported genes, TERT, CCNE1 and MLL4. Aside from individual genes mutated in HCC, we also examine the diverse modes of genomic alteration in this heterogeneous disease, delineating both conventional mutations and virus-associated changes that contribute to liver oncogenesis.
Results
The mutational landscape of hepatocellular carcinoma
We analyzed 42 HCC tumor-normal pairs to identify frequent and high-allelic frequency mutations. We sequenced whole genomes and transcriptomes of 12 patients, and exomes of 30 additional patients. We identified 49 non-silent mutations per patient (median value; Table S1 in Additional file 1; Figure S1A in Additional file 2) from the 12 whole genomes, and 54 non-silent somatic mutations per patient (median value; Table S2 in Additional file 1; Figure S2 in Additional file 2) from the 30 exomes. One of the patients had an unusually high mutation rate (Figure S2 in Additional file 2), with >99% of the point mutations of the C > T type (Table S2 in Additional file 1). The mechanism of such a high mutation rate in this particular patient is not clear, but such a C > T mutation pattern is consistent with APOBEC3B-mediated mutagenesis observed in other types of cancers [10],[11].
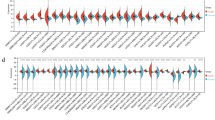
We found frequent non-silent mutations in TP53, CTNNB1, AXIN1, LAMA2, ZFPM2 and TAF1L (Figure 1A). We focused on mutations identified at high allelic frequencies (Figure 1B), as they are likely to be functional. Frequent mutations in TP53, CTNNB1 and AXIN1 have been reported previously in HCC, and these occur in 13 out of 42 patients (Figure 1A). Additionally, we found mutations in IDH1 at codon 132 (2/42 cases; Figure 2A), a hotspot for mutations in glioblastoma and intrahepatic cholangiocarcinoma [9], thus expanding the indications for IDH1 mutations. Mutations at codon 132 in IDH1 have been shown to dominantly inhibit the catalytic activity of IDH1, which normally functions to catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate, and contribute to tumorigenesis by inducing the HIF-1 pathway [12]. Interestingly, another patient carried a nonsense mutation in IDH2, which is also frequently mutated in cancer and has a similar effect to IDH1. A third IDH family member, IDH3A, which encodes the alpha-subunit of IDH3, carried a non-synonymous mutation (Table S2 in Additional file 1), although its significance is not clear. Further, we found mutations in the tumor suppressor BAP1[13] in two patients (Figure 2B). BAP1 is a deubiquitylase associated with protein complexes regulating key cellular pathways, including the cell cycle, cellular differentiation, cell death, gluconeogenesis and the DNA damage response [14]. One patient carried a deletion leading to a frame-shift, along with another non-synonymous mutation. The second patient had a D184V mutation, a position important for catalytic activity, based on sequence similarity to the residue D176 from UCHL1 [15]. While frequent somatic BAP1 mutations have been found in mesothelioma, uveal melanoma and cutaneous melanoma response [14], we report the first finding of BAP1 mutation in HCC. Another notable mutation was a E545K mutation in PIK3CA (Table S2 in Additional file 1), which is a hotspot mutation in the helical domain leading to constitutive activation [16],[17]. Several other mutations previously reported in cancer were found as singleton cases (Table S2 in Additional file 1).

Summary of genomic aberrations in hepatocellular carcinoma. (A) Select genes that have multiple point mutations at high allelic frequency or structural variations are shown. Select cancer gene census genes that showed mutations at high allelic frequency and highly clonal viral integrations are also shown. HBV infection status was known for the 12 whole-genome sequencing samples. For these samples, HBV infection status is shown as a plus sign if infected, and a minus sign if no infection was detected. (B) Allelic frequency profiles of frequently mutated genes across whole genomes and exomes. For each mutant allele found in the study, the percentile of its allelic frequency (calculated for each sample separately) is shown on the y-axis. The red bars indicate the median value of the percentile allelic frequency. Blue dots represent chosen mutations, where the median percentile value was more than 0.5, while the grey dots show examples of genes that had median values below 0.5 and were rejected.

Mutations in IDH1 , BAP1 and CTNNB1 . (A) IDH1 is mutated in two patients at the hotspot R132. (B) BAP1 mutation in two patients. Patient 3885 has a D184V mutation, a site that is important for the peptidase enzymatic activity (by homology). The other patient (9113) carries two mutations, including a point mutation, R237C, and a frameshift deletion at position 354. (C) An in-frame deletion in CTNNB1 in patient 30147. The deletion spans 121 amino acids near the amino-terminal. This region is involved in the degradation of β-catenin and is frequently mutated in cancer, as shown by clustering of mutations in this region in the COSMIC database. Each green box is a mutation instance in COSMIC. Overlapping, unique mutations have been stacked.
Aberrations in the members of the Wnt signaling pathway are reported to be frequent in HCC [6]. Besides point mutations in CTNNB1 and AXIN1, we also found partial deletions in these genes. The CTNNB1 deletion spans exons 3 and 4 (Figure S3A,B in Additional file 2; Table S5 in Additional file 1). This in-frame deletion removes the amino terminus of β-catenin, a region heavily mutated in multiple cancers (Figure 2C). Since the amino terminus is involved in degradation of β-catenin, the deletion likely results in its stabilization. We previously reported an AXIN1 deletion [3] that comprises the last three exons of the gene and results in a fusion with LUC7L (Table S6 in Additional file 1), likely leading to functional loss.
Hepatitis B virus integration into the human genome
HBV integration is another mechanism for influencing gene expression and function in HCC. HBV can randomly integrate into the genome of infected hepatocytes [3],[4],[18],[19]. Recurrent HBV integrations near cancer-related genes like TERT[4] indicate that HBV can play a causal role in HCC. From the whole genomes of HBV-infected patients we identified 146 HBV integration sites (Table S3 in Additional file 1), and from their transcriptomes we identified 545 chimeric transcripts (Table S4 in Additional file 1). The number of integration sites per patient ranged from 2 to 28 sites in the tumor samples and 0 to 19 sites in the tumor-adjacent samples. Clonal expansion of HBV-containing hepatocytes was specific to tumors (Figure 3A), as the tumors showed high amounts of human-viral chimeric DNA compared with the tumor-adjacent samples. We found integrations near three known recurrent integration targets [4]: two in the promoter of TERT, one upstream of CCNE1 (Figure S4 in Additional file 2) and another in the third exon of MLL4[3]. These integrations occur at high clonal frequency (Table S3 in Additional file 1). The HBV integration near CCNE1 correlated with extensive genomic instability at this region, featuring at least three inversions and one inter-chromosomal translocation to chromosome 7 (Figure S4 in Additional file 2). Intriguingly, none of the breakpoints disrupted the CCNE1 coding sequence. Other highly clonal integration sites were mapped near TNFSF4 and AGPAT6, in addition to ANGPT1 and CASP12 as previously reported [3].

Hepatitis B virus integration and chimeric transcription in hepatocellular carcinoma. (A) Viral integrations in 12 whole-genome sequenced samples were determined based on human-viral chimeric reads. Overlapping or nearby (within 500 bp) chimeric reads were clustered together, and the closest gene to the human-viral junction of each cluster was determined. In the plot, each point represents a chimeric read cluster, and the y-axis represents the total number of chimeric reads per cluster per gigabase of mapped human bases for that sample. The human junction is shown on the x-axis. For each sample, the cluster with the highest number of chimeric reads is labeled with the closest gene. (B) Two hotspots for viral breakpoints in viral-human chimeric transcripts. Human-viral chimeric RNA reads were clustered based on close vicinity (up to 500 bp). The scatter plot shows the mapping of the clusters on the viral genome (y-axis) versus the linearized human genome (x-axis). The size of the points is proportional to the number of reads belonging to the cluster. The histogram on the y-axis represents the frequency of chimera observed along the viral genome. Two hotspots are observable. The most frequent viral junction is the region at the 3' end of the X gene. A second, less frequent hotspot is seen at the S gene. (C) Chimeric transcripts show enrichment for viral promoter origin and human exon exclusion. Histograms representing chimeric RNA-Seq read counts are shown. Chimeric reads were classified as downstream, when the viral positive strand was 3' of the human sequence, or upstream, when the viral positive strand was 5' of the human sequence (consistent with viral promoter-driven transcription). They were further classified for inclusion or exclusion of human exon sequence. Specific enrichment of the chimeric transcripts can be seen when the viral sequence is upstream of the breakpoint, and human exons are excluded.
Hepatitis B virus-mediated transcription from viral promoters
Besides HBV viral integration at the DNA level, we also performed comprehensive analysis of available RNA-Seq data to identify fusion transcripts between HBV and human sequences. A large number of such chimeric transcripts were identified (Table S4 in Additional file 1). Chimeric transcripts were preferentially fused to two breakpoints in the viral genome: one near the 3' end of the X gene and the other within the S gene (Figure 3B; Figure S5A,B in Additional file 2). Human-viral chimeric transcripts may include the viral sequence upstream or downstream of the breakpoint. The former, which is consistent with transcription initiation from a viral promoter, is enriched in the chimera (Figure 3C). If chimeric transcription initiated from a human promoter, we would expect to see enrichment for inclusion of human exons in the chimera; however, that was not the case (Figure 3C). The larger prominence of the breakpoint at the 3' end of the X gene was likely due to linearization (and consequent integration) of the viral DNA at the DR1 site, which was previously identified as a fusion hotspot for human-HBV chimeric RNA [3],[18]. Full length HBx has been shown to have oncogenic potential [20]. Interestingly, truncation of HBsAg can confer advantage over immune surveillance [21] and pre-S deletions are associated with development of HCC [22].
Multiple modes of activation of TERT
The telomerase gene TERT is upregulated in multiple cancers [23]. Here, we observed two modes of perturbation of TERT: viral integrations into the promoter of TERT in two patients (Figure 4, patients H166 and H219), and translocations in two other patients (Figure 4, patients 30996 and 30374). In all four cases, the breakpoints did not disrupt the exons and TERT was expressed in tumor (Figure 4) but not in the matched normal tissue (data not shown). In patient H219, chimeric reads showing fusion between the 5' UTR of TERT mRNA and viral sequence were evident (Table S4 in Additional file 1), suggesting a direct role of HBV in activating TERT expression. However, we note that there was bidirectional transcription at this insertion, and transcription in the direction of the TERT gene was the least prevalent of the two transcripts resulting from the insertion. In patient H166, evidence for fusion between viral sequence and a region upstream of the TERT transcription start site was found, but no direct evidence of a fusion between TERT mRNA and viral mRNA was found. In patient 30996, TERT was translocated to a region on chromosome 5 that in turn showed extensive rearrangements in a chromothripsis-like fashion (Figure S6 in Additional file 2). Lastly, in patient 30374, the promoter region of TERT was involved in an interchromosomal translocation to a region upstream of RXRA on chromosome 9 (Table S5 in Additional file 1), likely resulting in misregulation of TERT. TERT was not expressed in the non-tumor samples in three out of these four patients, while RNA-Seq data were not available for the fourth patient (30996).

Multiple modes of TERT activation. Viral integration and structural variations at the TERT locus in tumor samples from four patients. Patient 30996 has an intra-chromosomal inversion supported by 132 whole-genome sequencing reads (red dashed line). Patient 30374 has an inter-chromosomal translocation to chromosome 9 supported by 27 reads (purple block). Viral integration junctions are shown as red triangles and vertical grey lines (patients H166 and H219). RNASeq.T: RNA-Seq coverage in the tumor sample. Coverage is on a linear scale in patients 30996 and 30374, and on a log2 scale in patients H166 and H219. CNV: copy number ratio of the tumor sample relative to the non-tumor sample (log2 scale).
LAMA2 is frequently mutated in hepatocellular carcinoma
We found that approximately 14% of the samples analyzed (6/42) had non-silent mutations in LAMA2 (Figure 1A; Tables S1 and S2 in Additional file 1), a member of the laminin gene family. The relatively high allelic frequencies of these mutations suggest early occurrence during tumor development (Figure 1B). Although the co-occurrence of LAMA2 mutations and CTNNB1/AXIN1 mutations is low, the apparent exclusivity is not significant (Fisher exact P-value = 0.3848). To understand the prevalence of LAMA2 mutations, we examined data from other studies, and found that approximately 6% (5/88) of the samples in the Asia Cancer Research Group study [4], and approximately 5% (5/104) of the samples in the Riken liver cancer cohort from the International Cancer Genome Consortium also had LAMA2 mutations. The mutation prevalence will likely vary between different patient cohorts, partly due to extensive heterogeneity observed in HCC. We also found that LAMA2 is frequently mutated in other cancers, including lung adenocarcinoma (11%), lung squamous cell carcinoma (13%), uterine corpus endometrioid carcinoma (13%), and head and neck squamous cell carcinoma (10%) (data source: The Cancer Genome Atlas).
The non-focal nature of LAMA2 mutations in liver cancer (Figure S7 in Additional file 2) suggests it plays a tumor suppressor role. Accordingly, downregulation of LAMA2 expression was connected to tumor progression in other tumor types like laryngeal squamous cell carcinoma [24] and breast cancer [25]. We examined a comprehensive panel of tumor tissues for expression profiling, and found downregulation of LAMA2 across multiple cancer types, most notably in ovarian, lung and colorectal cancer (Figure 5A). In addition, we examined multiple cancer cell lines and found that a decrease in LAMA2 expression was accompanied by an increase in DNA methylation near the transcription start site (Figures S8 and S9 in Additional file 2). Furthermore, we observed significant downregulation of LAMA2 expression in a large cohort of un-treated HCC patients with clinical data from the University of Hong Kong [26] (Figure 5B). Promoter DNA methylation, downregulation across multiple cancer types, and mutations in a number of cancer indications support a tumor suppressor role for this gene.

LAMA2 status is associated with clinical outcome in hepatocellular carcinoma. (A) Microarray-based expression profile of LAMA2 across a panel of cancer and normal human tissues (Gene Logic, Gaithersburg, MD, USA). Numbers at the top are -log10 P-values for a two-sided t-test comparing the expression values between the normal (green) and cancer (red) samples of the same tissue. (B) Microarray-based expression profile of LAMA2 in 300 HCC patients from University of Hong Kong (data from Gene Expression Omnibus series GSE25097). The number at the top is the -log10 P-value for a two-sided t-test comparing the expression values between the non-tumor (blue) and cancer (red) samples. (C) Kaplan-Meier curves for survival of LAMA2-low versus LAMA2-high HCC patients from a cohort of 228 HCC patients from the University of Hong Kong. Patients with low LAMA2 expression show poorer overall survival (left panel) and disease-free survival (right panel) by log-rank test (P-values of 0.024 and 0.026, respectively). HR: hazard ratio between LAMA-high and LAMA-low samples.
To test whether laminin deficiency could impact the clinical outcome of HCC patients, we examined the patient survival data from the University of Hong Kong cohort of HCC patients (228 HCC samples with survival data). We found that patients with lower LAMA2 expression level showed significantly worse survival outcome (Figure 5C; P-value = 0.024, log-rank test). The effect size was greater upon stratifying the patients into upper and lower quartiles instead of median-based stratification (Figure S10 in Additional file 2). Among the 5 LAMA family members, only LAMA2 showed a significant relation with clinical outcome. Moreover, patients with low LAMA2 expression were 30% more likely to have tumor recurrence (odds ratio = 1.7, P = 0.034, Chi-Square test). Thus, LAMA2 low expressing tumors appear to represent a more aggressive subtype of HCC. Interestingly, compared with tumors with wild-type LAMA2, tumors with LAMA2 mutations showed histopathological features of poorly differentiated tumors, with substantial cellular and nuclear atypia and moderate to abundant stroma interspersed between cells (Figure 6). To understand the underlying molecular mechanism of such differences in clinical outcome, we compared the LAMA2-low with the LAMA2-high tumors and found striking enrichment for upregulation of cell cycle genes (Figure S11 in Additional file 2). A similar trend was also observed in breast, colorectal and lung cancers (Figure S11 in Additional file 2). Therefore, the LAMA2-deficient samples represent a subgroup of highly recurrent and proliferative hepatocellular carcinomas, and LAMA2-based stratification appears to apply to other cancer types as well.

Morphology of hepatocellular carcinomas with LAMA2 mutations. Hematoxylin and eosin (H&E)-stained sections of HCC with LAMA2 mutation (upper panels, 9193, 3206A7 009) show poorly differentiated neoplastic hepatocytes with cellular and nuclear pleomorphism, mitotic figures (arrow) and moderate to abundant stroma (asterisks). H&E-stained sections of HCC with wild-type LAMA2 generally have more uniform, well-differentiated neoplastic cell morphology (lower panels). Scale bar is 20 μm.
Discussion
Multiple efforts using deep sequencing of HCC are now revealing important players in this heterogeneous disease. Previous genomic studies have implicated the role of tumor suppressor TP53, Wnt-signaling components CTNNB1 and AXIN1, telomerase TERT, cell cycle regulator CCNE1, and chromatin regulators like MLL4, ARID1A and ARID2, although different studies have identified different subsets of these players. Here we report low frequency mutations in tumor suppressor BAP1 and metabolic genes IDH1 and IDH2 in HCC. Although mutations in these genes have been reported in other cancer types such as glioblastoma and mesothelioma [8],[14], BAP1, IDH1 and IDH2 have not been previously linked to HCC. Since mutant IDH1 has been pursued as an anticancer target in glioma [27], it is conceivable that HCC patients carrying the R132 mutation can also benefit from such IDH1 inhibitors. Similarly, HCC patients with PIK3CA E545K mutation can benefit from inhibitors effective against the kinase activity of this mutant [28]. It is conceivable that other PIK3CA mutations may also be present in HCC.
Interestingly, we observed frequent mutations in the extracellular matrix gene LAMA2 in liver cancer patients. Specific upregulation of LAMA2 expression in cirrhotic hepatocytes (Figure 5B) suggests that basal laminae may be required for controlled regeneration following liver injury. Low expression of LAMA2 is tied to poor survival outcome, high recurrence of HCC, and upregulation of cell cycle genes. We compared the LAMA2 expression profile, using the transcriptome data from 12 tumors, with those of poor-prognosis stemness markers like EPCAM, PROM1 (CD133), THY1 (CD90), NCAM and KRT9 (CK19), but did not find any conclusive evidence of correlation with these markers, although there was a weak correlation with CD90 (Pearson correlation coefficient = 0.65). It is conceivable that functional LAMA2 in the extracellular matrix may keep the proliferation of regenerating hepatocytes in check, and defective or lack of LAMA2 facilitates tumor progression. This is also supported by the fact that the loss of cell surface anchoring to basal laminae has been found to promote tumor growth and cell proliferation [7]. Moreover, soluble laminin in culture can suppress cell proliferation in mammary epithelial cells [29]. DNA methylation at the LAMA2 promoter region found by us and others [30] suggests that epigenetic mechanisms may target LAMA2 in multiple cancers. These findings suggest the importance of the extracellular matrix during HCC development and perhaps other types of cancers as well. This has potential implications on stratification of HCC patients and on decisions about therapeutic options for such categories of patients.
While the expression of LAMA2 seems to be related to DNA methylation, the TERT gene appears to be influenced by other modes of genomic alteration. In two of our HCC samples, the HBV viral DNA is integrated into the promoter of TERT, leading to the activation of TERT expression. In two other HCC patients, viral-independent translocation was observed, juxtaposing the TERT promoter on other active genomic regions. All four such patients exhibit much elevated TERT gene expression compared with adjacent liver tissues based on our RNA-Seq data (Figure 4). More recently, point mutations have been found in the promoter region of TERT in multiple types of cancer, leading to higher TERT mRNA expression [31],[32]. Clearly, TERT activation is common in human cancers and is likely oncogenic, but the mechanism of gene activation may differ between cancer types. While we did not find any point mutations in the TERT promoter in HCC, other HBV-dependent and HBV-independent genomic events seem to be involved in alternative mechanisms of TERT activation.
Overall, there is a constellation of genomic alterations in HCC (Figure 7). Among conventional mutations, point mutations are clearly the most studied type of changes in HCC due to mature technologies for detecting such mutations. Large coding region deletions, such as the CTNNB1 deletion we report here (Figure 2C), are usually understudied but their functional importance should not be ignored. Perhaps the most notable genomic alterations in the HBV-positive patients are various changes caused by viral DNA integration into the human genome. Based on our work and that of others [3]-[5], patterns of HBV-based mutagenesis are starting to emerge. In some patients, integrated HBV viral promoter would activate nearby genes such as TERT and ANGPT1. In others, HBV integration sites occur in the middle of human genes such as MLL4, causing gene disruption and/or viral-human chimera transcripts. HBV integration also leads to local genomic instability, further causing DNA copy number changes [3]. Such diverse modes of genomic alteration add genetic complexity to HCC (Figure 7), likely leading to its highly heterogeneous nature. Deeper understanding of these complex patterns of mutations could also provide better understanding of the etiology of HCC and possibly lead to development of novel anti-HCC therapeutics. For example, the HBx-MLL4 chimera fusions potentially produce cancer-specific proteins that can be specifically targeted therapeutically, and the TERT gene activation modes may suggest different diagnostic markers for a subset of HCC patients. With more comprehensive genomic characterization of liver cancer patients, more detailed and reproducible cancer subtypes will emerge that can eventually guide clinical diagnostics and treatment management.

Diverse modes of genetic alterations in hepatocellular carcinoma. Conventional mutations are the changes that can also be seen in other cancers, while the virus-associated changes are unique to HBV-infected HCC tumors. Examples of genes associated with each genetic alteration are shown, compiled from multiple studies.
Conclusion
We found diverse modes of genomic alteration in HCC that affect important players in this disease. Point mutations and structural variations affected both AXIN1 and CTNNB1. We found HBV integrations in the TERT promoter, and virus-independent TERT translocations, likely leading to activation of TERT expression. HBV integrations occurred in MLL4, CCNE1 and TERT, leading to increased expression of CCNE1 and TERT, or HBx-MLL4 fusion. TP53, AXIN1 and CTNNB1 were the most frequently mutated genes in our cohort. LAMA2 showed high allelic frequency mutations, and we also found point mutations in BAP1, IDH1, IDH2 and PIK3CA.
Materials and methods
Samples and sequencing
Frozen tissue and peripheral blood mononuclear cell (PBMC) samples were obtained from commercial sources (Seracare LifeSciences, Milford, MA, USA; ProteoGenex, Culver City, CA, USA and Indivumed, Baltimore, MD, USA). Appropriate informed consent and institutional review board approval was obtained based on a representation and warranty from the vendors. Four-micron thick frozen sections were obtained from both primary HCC and the matched non-neoplastic liver tissue for histopathological evaluation by standard hematoxylin and eosin stain. Two distinct sets of samples were used for whole-genome sequencing and exome-sequencing. For whole-genome sequencing, tumor and tumor-adjacent samples from 12 patients were sequenced at 78× to 132× coverage using the Complete Genomics (Mountain View, CA, USA) platform. Of these 12 patients, 4 were reported previously [3]. Eight patients were confirmed to be HBV-positive by PCR. Thirty sets of tumor and tumor-adjacent samples were exome-sequenced using Illumina Hi-Seq. Exome sequencing was performed at a median read count of 79.9 million reads (range was 44.2 to 250 million). Uniquely mapped reads (35 to 213 million; 67.5 million median value) were used for variant calling.
Variant calling
High-throughput reads from whole-genome sequencing were mapped by Complete Genomics to the human genome (NCBI build 37). The mean mate gap post-mapping was approximately 311 bp. Single nucleotide variation, short indels and substitutions, large-scale structural variation, and copy number variation data were provided by Complete Genomics (standard pipeline version 2.0.1.6). Somatic mutations were called using open-source cgatools v.1.5 [33]. Mutations with a somatic score greater than -5 were retained. Further filtering of somatic mutations was done by removing mutations that were common with dbSNP build 132, a set of 69 normal genomes from Complete Genomics [34], the 1000 Genomes Project, and 2,440 exomes from NHLBI [35]. Any mutations that were also present in COSMIC v.62 were retained. The resulting set of mutations constitutes high-confidence mutations used in all the analyses in this study. For determining viral reads, whole-genome sequencing reads where at least one arm was not mappable to the human genome were mapped to a consensus HBV genome sequence using mapping tools from Real Time Genomics® (Hamilton, New Zealand).
Exome sequencing reads from the Illumina platform were mapped to UCSC human genome (GRCh37/hg19) using the default parameters of the BWA software [36]. Duplicate read removal and local realignment were performed as described previously [37]. Variant calling was performed on the tumor and normal BAM files using the Strelka algorithm [38] with the BWA default configuration file and a minimum variant quality of 30. Known germline variations represented in dbSNP build 131 [39] or 6,515 previously published normal exomes [40], but not represented in COSMIC v62 [41], were excluded. In addition, predicted somatic variations were additionally filtered to have a minimum variant allele frequency of 5% and not exceeding that number in the matched normal sample.
Somatic mutation calls from the whole genomes and exomes were pooled and the number of tumor samples carrying a mutation was tallied for each gene. For genes mutated in three or more samples, we selected those that were preferentially mutated at high allele frequencies across multiple patients. This eliminated some genes that were apparently mutated in a large number of tumors, but showed low allelic frequencies across these tumors (Figure 1B). Among the genes that were mutated in two samples, we focused on cancer gene census genes. Among these cases, IDH1 was recurrently mutated at position R132, and BAP1 carried three high allelic frequency mutations, two of them in the same tumor. Other notable mutations included a nonsense mutation in IDH2 at amino acid 63, and a E545K mutation in PIK3CA, which is a hotspot mutation in several cancers [16].
Mapping to the hepatitis B virus genome
Complete Genomics reads were mapped to the HBV consensus genome using the Real Time Genomics® software [42]. The subset of reads with minimum one mate pair mapping to the HBV genome were mapped to a `hybrid genome' - a combination of the human genome (hg19) and the HBV virus genome. The hybrid genome was created by merging the FASTA files of the human genome (hg19) and the HBV consensus genome, so that the viral genome is presented as an additional `pseudo' chromosome of the human genome. Based on a Phred-like quality score cutoff of minimum 20, aligned reads were further filtered for reads with one mate pair mapping uniquely to the human genome and the other mate pair mapping uniquely to the HBV genome.
Determination of viral integration sites and viral-human fusion transcripts
For both whole-genome sequencing and transcriptome sequencing, human-viral chimeric reads were grouped into chimeric clusters. If a boundary of a read was within 500 bp of the boundary of another read on the human as well as the viral arm, the two reads were assigned to the same cluster. Each cluster represents a human-viral junction, and two such junctions are expected per viral insertion site in the human genome. However, two junctions were not always detectable, likely due to lack of coverage or difficulty in mapping at one of the junctions. For whole-genome sequencing, we identified 146 such clusters (Table S3 in Additional file 1), and 612 clusters for RNA-Seq (Table S4 in Additional file 1), with at least two reads per cluster. Since two nearby chimeric clusters on the human genome may represent the same viral integration event, for the purpose of estimating the clonality of each integration (Figure 3A), we reduced this redundancy by retaining only the cluster with the higher number of chimeric reads, if the boundaries of two clusters are within 2 kb of each other on the human genome.
Chimeric transcripts showed two hotspots for breakpoints on the viral genome (Figure 3B). To examine these in more detail, we classified the human-viral breakpoints from whole genome and transcriptome data into two types: one where the fused viral sequence is upstream of the breakpoint in the viral genome, and the other where the viral sequence is downstream of the breakpoint (Figure 3C). The enrichment observed was especially striking for chimeric transcripts with upstream viral breakpoints, indicating that viral promoters (for X and S genes) are the likely driving factors for these fusion transcripts.
LAMA2 expression in multiple tissues
Expression data were obtained from a panel of 37 tissues from Gene Logic (Gaithersburg, MD, USA; Affymetrix HG-U133 platform, representing 3,600 normal and 1,701 neoplastic samples from different human tissues). Tissues that showed significant expression change in LAMA2 (P ≤ 0.001, two-sided t-test) are shown (Figure 5A). Expression was significantly decreased in breast, cervix, colorectal, endometrium, kidney, lung, lymphoid, ovary, skin, and thyroid tumor tissues.
DNA methylation analysis
DNA methylation was measured using Illumina Infinium 450 K BeadChip and preprocessed using the Bioconductor lumi package [43] with default settings (within-sample quantile color bias adjustment plus across-sample quantile normalization of pooled probe intensities). The methylation plot was produced using Bioconductor methyAnalysis package.
Relation between clinical data and LAMA2 expression
A cohort of 228 primary HCC samples from the University of Hong Kong was used for evaluating the effect of LAMA2 expression on survival. Statistical analysis was performed using IBM SPSS version 16.0 (Armonk, NY, USA) for Windows. For clinical correlation analysis with survival rates, continuous variables were modeled as categorical variables. Univariate analysis of LAMA2 expression on overall survival and disease-free survival rates was performed using the Kaplan-Meier method. The subjects were equally divided into high- and low-expression arms using the median value as cutoff (Figure 5C). To achieve more stringent stratification, upper and lower quartiles were used instead of median-based separation. The resulting Kaplan-Meier plots show a higher level of separation (Figure S10 in Additional file 2).
Detection of structural variation and potential gene fusion events
We utilized the paired-end nature of the sequencing reads to detect structural variations (Table S5 in Additional file 1) and potential gene fusion events (Table S6 in Additional file 1). The method used to detect structural variation was as previously described [3]. We further filtered somatic structural variants for matched or unrelated normal samples. For these putative somatic structural variants, we further searched for structural variation events that potentially lead to fusion gene events by the following criteria: 1) the somatic structural variant breakpoints defined by DNA-Seq overlap with a pair of distinct human genes (refGene); 2) the RNA-Seq data support the fusion occurred between these two fusion partners. For detecting fusion at the RNA level, we applied both ChimeraScan [44] and gStruct (Thomas Wu, unpublished). The somatic structural variants that lead to putative fusion events are reported in Table S5 in Additional file 1.
Data availability
Sequence data have been deposited at the European Genome-phenome Archive [45], which is hosted by the EBI, under accession number EGAS00001000824.
ICGC mutation data were accessed from the ICGC data portal [46]. The raw data from this study can be obtained from the European Genome-phenome Archive, study accession EGAS00001000678. Expression data for LAMA2 from the university of Hong Kong are available from the Gene Expression Omnibus repository, with accession GSE25097. LAMA2 mutation data from Asia Cancer Research Group study can be accessed at [47]. The Cancer Genome Atlas data were obtained from dbGaP (study accession phs000178.v8.p7).
Authors' contributions
SJ and ZJ conducted the primary analyses. SJ, ZJ and ZZ designed the study and wrote the manuscript. EWS conducted mutation detection on the exome samples. FG performed viral-human chimeric read detection from whole genome sequencing data. JL performed the RNA-Seq data analysis. OM developed methods for filtering mutations. PD conducted the DNA methylation analysis. JD and SBK performed experimental validation on the HBV infection status. SJ and ZM managed sample handling and acquisition. KFW performed data analysis on the cohort from the University of Hong Kong. ZG and YL conducted LAMA2 mutation analysis on the ACRG study. TDW implemented the computational method for detection of fusions from RNA-Seq data. DMF conducted histopathological examination of tissue sections. JML supervised the study on the cohort from the University of Hong Kong. SS led the RNA and exome sequencing efforts. ZZ coordinated the study. All authors read and approved the final manuscript.
Additional files
Abbreviations
- bp:
-
base pair
- HBV:
-
hepatitis B virus
- HCC:
-
hepatocellular carcinoma
- PCR:
-
polymerase chain reaction
- UTR:
-
untranslated region
References
Altekruse SF, McGlynn KA, Reichman ME: Hepatocellular Carcinoma Incidence, Mortality, and Survival Trends in the United States From 1975 to 2005. JCO. 2009, 27: 1485-1491. 10.1200/JCO.2008.20.7753.
Zhang Z: Genomic landscape of liver cancer. Nat Genet. 2012, 44: 1075-1077. 10.1038/ng.2412.
Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, Diao J, Yeung S, Jubb A, Ye W, Wu TD, Kapadia SB, Sauvage FJD, Gentleman RC, Stern HM, Seshagiri S, Pant KP, Modrusan Z, Ballinger DG, Zhang Z: The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22: 593-601. 10.1101/gr.133926.111.
Sung W-K, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, Poon RT, Fan ST, Chan KL, Gong Z, Hu Y, Lin Z, Wang G, Zhang Q, Barber TD, Chou W-C, Aggarwal A, Hao K, Zhou W, Zhang C, Hardwick J, Buser C, Xu J, et al: Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012, 44: 765-769. 10.1038/ng.2295.
Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, et al: Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012, 44: 760-764. 10.1038/ng.2291.
Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clément B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouzé E, Calvo F, Zucman-Rossi J: Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012, 44: 694-698. 10.1038/ng.2256.
Akhavan A, Griffith OL, Soroceanu L, Leonoudakis D, Luciani-Torres MG, Daemen A, Gray JW, Muschler JL: Loss of cell-surface laminin anchoring promotes tumor growth and is associated with poor clinical outcomes. Cancer Res. 2012, 72: 2578-2588. 10.1158/0008-5472.CAN-11-3732.
Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu I-M, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Hartigan J, Smith DR, Strausberg RL, Marie SKN, Shinjo SMO, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, et al: An integrated genomic analysis of human glioblastoma multiforme. Science. 2008, 321: 1807-1812. 10.1126/science.1164382.
Wang P, Dong Q, Zhang C, Kuan P-F, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan T-X, Auman JT, Hoskins JM, Misher AD, Moser CD, Yourstone SM, Kim JW, Cibulskis K, Getz G, Hunt HV, Thorgeirsson SS, Roberts LR, Ye D, Guan K-L, Xiong Y, Qin L-X, Chiang DY: Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013, 32: 3091-3100. 10.1038/onc.2012.315.
Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS: APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013, 494: 366-370. 10.1038/nature11881.
Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS: DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013, 2: e00534-10.7554/eLife.00534.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan K-L, Xiong Y: Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science. 2009, 324: 261-265. 10.1126/science.1170944.
Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, Wu J, Phu L, Katavolos P, LaFave LM, Abdel-Wahab O, Modrusan Z, Seshagiri S, Dong K, Lin Z, Balazs M, Suriben R, Newton K, Hymowitz S, Garcia-Manero G, Martin F, Levine RL, Dixit VM: Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012, 337: 1541-1546. 10.1126/science.1221711.
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G: BAP1 and cancer. Nat Rev Cancer. 2013, 13: 153-159. 10.1038/nrc3459.
Larsen CN, Price JS, Wilkinson KD: Substrate binding and catalysis by ubiquitin C-terminal hydrolases: identification of two active site residues. Biochemistry. 1996, 35: 6735-6744. 10.1021/bi960099f.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE: High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004, 304: 554-554. 10.1126/science.1096502.
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM: The oncogenic properties of mutant p110α and p110β phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005, 102: 18443-18448. 10.1073/pnas.0508988102.
Nagaya T, Nakamura T, Tokino T, Tsurimoto T, Imai M, Mayumi T, Kamino K, Yamamura K, Matsubara K: The mode of hepatitis B virus DNA integration in chromosomes of human hepatocellular carcinoma. Genes Dev. 1987, 1: 773-782. 10.1101/gad.1.8.773.
Brechot C, Pourcel C, Louise A, Rain B, Tiollais P: Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980, 286: 533-535. 10.1038/286533a0.
Kim C-M, Koike K, Saito I, Miyamura T, Jay G: HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991, 351: 317-320. 10.1038/351317a0.
Sunyach C, Chassot S, Jamard C, Kay A, Trepo C, Cova L: In VivoSelection of duck hepatitis B virus pre-S variants which escape from neutralization. Virology. 1997, 234: 291-299. 10.1006/viro.1997.8665.
Yeung P, Wong DK-H, Lai C-L, Fung J, Seto W-K, Yuen M-F: Association of hepatitis B virus pre-S deletions with the development of hepatocellular carcinoma in chronic hepatitis B. J Infect Dis. 2011, 203: 646-654. 10.1093/infdis/jiq096.
Buseman CM, Wright WE, Shay JW: Is telomerase a viable target in cancer?. Mutat Res. 2012, 730: 90-97. 10.1016/j.mrfmmm.2011.07.006.
Ni R, Shen X, Qian X, Yu C, Haiyan WU, Xia GAO: Detection of differentially expressed genes and association with clinicopathological features in laryngeal squamous cell carcinoma. Oncol Lett. 2012, 4: 1354-1360.
Mefford D, Mefford J: Stromal genes add prognostic information to proliferation and histoclinical markers: a basis for the next generation of breast cancer gene signatures. PLoS One. 2012, 7: e37646-10.1371/journal.pone.0037646.
Tung EK-K, Mak CK-M, Fatima S, Lo RC-L, Zhao H, Zhang C, Dai H, Poon RT-P, Yuen M-F, Lai C-L, Li J, Luk JM-C, Ng IO-L: Clinicopathological and prognostic significance of serum and tissue Dickkopf-1 levels in human hepatocellular carcinoma. Liver Int. 2011, 31: 1494-1504. 10.1111/j.1478-3231.2011.02597.x.
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK: An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013, 340: 626-630. 10.1126/science.1236062.
Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O, Aoki Y: The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011, 17: 3272-3281. 10.1158/1078-0432.CCR-10-2882.
Spencer VA, Costes S, Inman JL, Xu R, Chen J, Hendzel MJ, Bissell MJ: Depletion of nuclear actin is a key mediator of quiescence in epithelial cells. J Cell Sci. 2011, 124: 123-132. 10.1242/jcs.073197.
Lee S, Oh T, Chung H, Rha S, Kim C, Moon Y, Hoehn BD, Jeong D, Lee S, Kim N, Park C, Yoo M, An S: Identification of GABRA1 and LAMA2 as new DNA methylation markers in colorectal cancer. Int J Oncol. 2011, 40: 889-898.
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA: Highly recurrent TERT promoter mutations in human melanoma. Science. 2013, 339: 957-959. 10.1126/science.1229259.
Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, Castro P, Castro L, Pardal F, Lopes JM, Santos LL, Reis RM, Cameselle-Teijeiro J, Sobrinho-Simões M, Lima J, Máximo V, Soares P: Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013, 4: 1-6. 10.1038/ncomms3185.
Cgatools. [], [http://cgatools.sourceforge.net]
69 Genomes Data. [], [http://www.completegenomics.com/public-data/69-Genomes/]
Exome Variant Server. [], [http://evs.gs.washington.edu/EVS]
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011, 43: 491-498. 10.1038/ng.806.
Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK: Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics. 2012, 28: 1811-1817. 10.1093/bioinformatics/bts271.
Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K: dbSNP: the NCBI database of genetic variation. Nucl Acids Res. 2001, 29: 308-311. 10.1093/nar/29.1.308.
Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Altshuler D, Shendure J, Nickerson DA, Bamshad MJ, Project NES, Akey JM: Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013, 493: 216-220. 10.1038/nature11690.
Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, Teague JW, Stratton MR, Futreal PA: COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010, 38 (Database issue): D652-D657. 10.1093/nar/gkp995.
Reumers J, De Rijk P, Zhao H, Liekens A, Smeets D, Cleary J, Van Loo P, Van Den Bossche M, Catthoor K, Sabbe B, Despierre E, Vergote I, Hilbush B, Lambrechts D, Del-Favero J: Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol. 2012, 30: 61-68. 10.1038/nbt.2053.
Du P, Kibbe WA, Lin SM: lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008, 24: 1547-1548. 10.1093/bioinformatics/btn224.
Iyer MK, Chinnaiyan AM, Maher CA: ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011, 27: 2903-2904. 10.1093/bioinformatics/btr467.
European Genome-phenome Archive. [], [http://www.ebi.ac.uk/ega/]
Liver cancer - Riken, JP. [], [https://dcc.icgc.org/projects/LIRI-JP]
Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, Hauptschein R, Rejto PA, Fernandez J, Wang G, Zhang Q, Wang B, Chen R, Wang J, Lee NP, Zhou W, Lin Z, Peng Z, Yi K, Chen S, Li L, Fan X, Yang J, Ye R, Ju J, Wang K, et al: Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. 2013, [], [http://www.ingenuity.com/acrg2012]
Acknowledgments
We would like to acknowledge Robert Gentleman, Gerard Manning, Fred de Sauvage and Christiaan Klijn for support and fruitful discussions, and May Whittake, Sara Verbeke, Jeremy Stinson and Joseph Guillory for mutation validations. We also thank Brian Hilbush and Richard Littin for support for the Real Time Genomics® software for mapping of Complete Genomics® raw reads.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
Suchit Jhunjhunwala, Zhaoshi Jiang, Eric W Stawiski, Florian Gnad, Jinfeng Liu, Oleg Mayba, Pan Du, Jingyu Diao, Stephanie Johnson, Thomas D Wu, Sharookh B Kapadia, Zora Modrusan, Dorothy M French, Somasekar Seshagiri and Zemin Zhang are all employees of Genentech, Inc.
Electronic supplementary material
13059_2014_436_MOESM1_ESM.xlsx
Additional file 1: Table S1.: High confidence mutations in 12 HCC tumors from whole-genome sequencing. Table S2. High confidence mutations in 30 HCC samples from exome sequencing. Table S3. Viral integrations identified from whole-genome sequencing of eight HCC patients carrying HBV infection. Table S4. Summary of human-viral chimeric RNA reads identified from RNA sequencing of eight HCC patients carrying HBV infection. Table S5. Somatic structural variation breakpoints identified computationally from whole-genome sequencing of 12 HCC patients. Table S6. Somatic fusion transcripts identified computationally from RNA sequencing and whole genome sequencing of 12 HCC patients. (XLSX 837 KB)
13059_2014_436_MOESM2_ESM.pdf
Additional file 2: Figure S1.: Somatic mutation frequency and mutation signature from whole genome sequencing of 12 HCC patients. Figure S2. Exome-based somatic mutation frequency in 30 HCC patients. Figure S3. CTNNB1 structural variant in patient 30147. Figure S4. Structural variation and viral insertion near CCNE1. Figure S5. Frequencies of human-viral chimeric reads based on whole genome and transcriptome. Figure S6. Translocation of TERT in patient 30996. Figure S7. Non-silent LAMA2 mutations. Figure S8. DNA methylation and expression of LAMA2 in breast cancer cell lines. Figure S9. DNA methylation and expression of LAMA2 in lung cancer cell lines. Figure S10. Survival analysis for LAMA2-low and LAMA2-high patients. Figure S11. LAMA2 downregulation is associated with cell cycle regulation.. (PDF 720 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Jhunjhunwala, S., Jiang, Z., Stawiski, E.W. et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol 15, 436 (2014). https://doi.org/10.1186/s13059-014-0436-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-014-0436-9