
Abstract
Background
Maternal and paternal high-fat (HF) diet intake before and/or during pregnancy increases mammary cancer risk in several preclinical models. We studied if maternal consumption of a HF diet that began at a time when the fetal primordial germ cells travel to the genital ridge and start differentiating into germ cells would result in a transgenerational inheritance of increased mammary cancer risk.
Methods
Pregnant C57BL/6NTac mouse dams were fed either a control AIN93G or isocaloric HF diet composed of corn oil high in n-6 polyunsaturated fatty acids between gestational days 10 and 20. Offspring in subsequent F1–F3 generations were fed only the control diet.
Results
Mammary tumor incidence induced by 7,12-dimethylbenz[a]anthracene was significantly higher in F1 (p < 0.016) and F3 generation offspring of HF diet-fed dams (p < 0.040) than in the control offspring. Further, tumor latency was significantly shorter (p < 0.028) and burden higher (p < 0.027) in F1 generation HF offspring, and similar trends were seen in F3 generation HF offspring. RNA sequencing was done on normal mammary glands to identify signaling differences that may predispose to increased breast cancer risk by maternal HF intake. Analysis revealed 1587 and 4423 differentially expressed genes between HF and control offspring in F1 and F3 generations, respectively, of which 48 genes were similarly altered in both generations. Quantitative real-time polymerase chain reaction analysis validated 13 chosen up- and downregulated genes in F3 HF offspring, but only downregulated genes in F1 HF offspring. Ingenuity Pathway Analysis identified upregulation of Notch signaling as a key alteration in HF offspring. Further, knowledge-fused differential dependency network analysis identified ten node genes that in the HF offspring were uniquely connected to genes linked to increased cancer risk (ANKEF1, IGFBP6, SEMA5B), increased resistance to cancer treatments (SLC26A3), poor prognosis (ID4, JAM3, TBX2), and impaired anticancer immunity (EGR3, ZBP1).
Conclusions
We conclude that maternal HF diet intake during pregnancy induces a transgenerational increase in offspring mammary cancer risk in mice. The mechanisms of inheritance in the F3 generation may be different from the F1 generation because significantly more changes were seen in the transcriptome.
Similar content being viewed by others


Background
With 1.7 million new cases in 2012, breast cancer is the most common cancer in women worldwide, and the incidence is projected to continue growing [1]. However, only 5–10% of breast cancers are attributed to an inherited genetic cause [2, 3], and this leaves over 90% of breast cancers to be caused by some other factors. It is believed that environmental and lifestyle factors, such as diet, play a critical role in affecting breast cancer risk [4], and their effects on cancer susceptibility are likely to be mediated by epigenetic modifications [5].
Studies show that a Western dietary pattern, one that is higher in dietary fats, has been increasing around the world [6, 7] and may be linked to increased breast cancer risk [4]. Intake of dietary fats, especially saturated fatty acids and n-6 polyunsaturated fatty acids (n-6 PUFAs), has been climbing over the years [8], with intake of n-3 PUFAs declining [6]. A high-fat (HF) diet, especially one where n-6 PUFAs are high, increases estrogenic activities [9, 10] and is inflammatory [11, 12], which can stimulate breast cancer growth. Our laboratory and others have shown in animal models that maternal exposure to a HF diet during pregnancy increases female offspring’s mammary cancer risk [9, 13,14,15,16]. In addition, we found that maternal intake of an n-6 PUFA HF diet consumed before and during pregnancy increased mammary cancer risk of daughters (F1) and granddaughters (F2), but not of great-granddaughters (F3) [13], indicating multigenerational but not transgenerational inheritance [17]. Multigenerational inheritance of a trait caused by maternal exposures during pregnancy, such as an increased susceptibility to breast cancer, is seen in F1 and/or F2 but not in F3 generation offspring. If the trait is also seen in F3 generation, inheritance is transgenerational [18].
The lack of transgenerational inheritance in the offspring of HF diet-fed dams was puzzling because previously we and others discovered that several different exposures during pregnancy led to a transgenerational increase in mammary cancer risk and other adverse health effects in F1, F2, and F3 generations. Specifically, transgenerational inheritance of adult-onset diseases and abnormalities, such as mammary cancer [13], prostate disease [19], obesity [20], and autism-like phenotype [21], has been found in offspring of dams that were exposed to compounds such as ethinylestradiol [13], vinclozolin [19], dichlorodiphenyltrichloroethane (DDT) [20], and valproic acid [21] during pregnancy. Common to all these studies was that the maternal exposure took place after fetal implantation, whereas a HF diet that resulted in a multigenerational increase in mammary cancer risk was started before conception [13].
Mammals undergo two rounds of global DNA demethylation and remethylation during the embryonic and fetal periods [22]. It is possible that in our previous study, the reason why we did not see true transgenerational inheritance of increased breast cancer risk was due to introducing the HF exposure throughout the first round of DNA methylation erasure in the zygote and the subsequent remethylation, as well as during the second round of methylation erasure, which took place in primordial germ cells (PGCs), and the second remethylation, which reestablished epigenetic marks on mature germ cells [23]. Because PGCs originate from the posterior endoderm of the blastocyst, the second round of epigenetic programming may have been affected by the presence of the HF diet during the first round. To test a possibility that maternal HF intake induces a transgenerational inheritance of mammary cancer risk, we timed the HF exposure to start on gestational day (GD) 10. Pregnant women consume more dietary fats than nonpregnant women, and the increase takes place between the first and second trimesters [24, 25].
Our study shows that maternal intake of a HF diet between GDs 10 and 20 caused a transgenerational increase in mammary cancer risk tumors without any other intervening dietary exposures. Results from RNA sequencing (RNA-seq) analysis suggested that there was a difference in how mammary cancer risk was affected in F1 and F3 generation offspring by maternal HF intake (i.e., whether the exposure directly affected fetal somatic cells or the germline).
Methods
Breeding and dietary exposure
Male and female C57BL/6NTac mice were obtained from Taconic Biosciences (Germantown, NY, USA) and housed in standard rodent housing at constant temperature and humidity and with a 12-h/12-h light/dark cycle at Georgetown University’s Department of Comparative Medicine, in accordance with all institutional and federal regulations.
To generate F1 offspring (Fig. 1a), 7-week-old mice were mated by housing two females together with one male per cage. Upon mating, mice were randomly divided into two groups and fed a control AIN93G diet until pregnancy was verified by the presence of a mucus copulatory plug in the vaginal opening (GD 0). From GDs 10 to 20, the pregnant dams (F0) were divided into two groups and fed either a HF diet containing 41.1% energy from fat, 38.9% kcal of corn oil (CO), and 2.2% kcal of soybean oil (SBO) (Additional file 1: Table S1), or they were continued on the control diet (16.0% energy from fat, 13.7% kcal of CO, 2.2% kcal of SBO) (Additional file 1: Table S1). Because the length of pregnancy in these mice is 19–21 days, the HF exposure took place during the second and third mouse trimesters. The HF diet was made isocaloric with the control diet by replacing some carbohydrates with non-energy-containing cellulose (fiber). CO contains high levels of n-6 PUFAs. Both diets contained 10 g/kg SBO to ensure that pregnant dams received sufficient levels of n-3 PUFAs. All subsequent generations (F1–F3) were fed only the control diet.

Transgenerational study design. a Pregnant C57BL/6NTac mice (F0) were fed either a high-fat (HF; n = 10) or control (CON; n = 10) diet. The HF diet was fed to dams from gestational day (GD) 10 to GD 20. All offspring were fed the CON diet after birth for the remainder of the study, including during pregnancies of F1 and F2 generation offspring. b All pups were cross-fostered at birth (postnatal day [PND] 1) to a CON mother to eliminate litter bias. Pups were weighed on PNDs 2 and 3 and weaned on PND 21. Tumorigenesis was initiated on PND 42 by priming female mice with medroxyprogesterone acetate (MPA; 15 mg/kg), followed by oral gavage of 7,12-dimethylbenz[a]anthracene (DMBA; 1 mg/dose/week) for 3 weeks. Tumorigenesis was monitored by palpation once per week starting 3 weeks after final DMBA administration up to 20 weeks post-DMBA. Mammary glands (MGs) and tumors were collected and processed from F1 and F3 offspring unexposed to DMBA on PND 50 for whole mounts and on PND 100 to perform RNA sequencing analysis
To obtain F3 generation offspring, female F1 generation offspring of dams fed a HF diet during pregnancy were mated to F1 HF diet-fed males, respectively. No sibling matings were performed. The control group was mated similarly. Pregnant F1 dams of both groups were fed the CON diet. These steps were repeated with F2 offspring to obtain the F3 generation. The F2 generation was used for breeding purposes only; that is, no experiments were performed with or tissues collected from them.
Mammary tumorigenesis
To induce mammary tumors (Fig. 1b), female C57BL6/NTac mice (n = 30 for F1 control, n = 30 for F1 HF; n = 20 for F3 control, and n = 25 for F3 HF offspring) were first primed with 15 mg/kg of medroxyprogesterone acetate (MPA; Greenstone, Peapack, NJ, USA) at postnatal day (PND) 42. One milligram of 7,12-dimethylbenz[a]anthracene (DMBA; Sigma-Aldrich, St. Louis, MO, USA) in 0.1 ml of CO was administered by oral gavage on PNDs 49, 56, and 63. After the last DMBA dose, tumor development was monitored by palpation once weekly for 20 weeks. If tumors were detected, their sizes were measured by calipers. The following endpoints were determined: incidence (number of mice with tumors), latency (time to first tumor), multiplicity (number of tumors per mouse), and burden (total tumor volume per mouse). The overall health of mice was monitored daily. A mouse was killed prior to the end of the tumor-monitoring period if it lost a significant amount of weight or if the tumor reached 10% of the mouse’s body weight. Mice killed for health reasons not pertaining to the study were excluded from the analysis. Tumor histopathology was assessed by a certified pathologist.
Kaplan-Meier survival curves were used to assess differences in tumor incidence between groups, followed by log-rank tests. Tumor latency and multiplicity differences were assessed by t test. Difference in tumor burden was assessed by repeated measures analysis of variance (ANOVA).
Tissue collection
Fourth mammary glands were obtained on PND 50 and PND 100 from offspring not exposed to the carcinogen to assess changes in mammary gland morphology and transcriptome, respectively (Fig. 1b). At the end of the tumor-monitoring period, offspring in F1 and F3 generations were killed, and whole blood was collected via cardiac puncture and placed into serum gel separator tubes. We then collected mammary tumors, resected a portion for formalin-fixed, paraffin embedding, and flash-froze the remaining tissues in cryotubes in liquid nitrogen.
Terminal end buds
Whole mounts were prepared according to an established protocol [26]. For that purpose, the left fourth abdominal mammary glands were stretched onto a slide, fixed, and stained with carmine aluminum solution. Slides were examined blindly under a microscope to determine the total number of terminal end buds (TEBs). These are the structures that give rise to malignant mammary tumors in mice and rats [27] and to similar structures in humans, called terminal ductal lobular units, which also are the sites of most breast cancers [28]. Differences in TEB numbers between the offspring of dams fed the HF or control diet were analyzed by t tests in both generations separately.
RNA sequencing
To identify differentially expressed genes (DEGs) between offspring of dams exposed to the HF or control diet during pregnancy, RNA-seq was performed. Total RNA was extracted from the right fourth mammary glands obtained on PND 100 from F1 and F3 generation offspring that were not treated with DMBA by using the RNeasy Lipid Tissue Mini Kit (QIAGEN Sciences, Germantown, MD, USA) with on-column DNase digestion (QIAGEN Sciences) per the manufacturer’s protocol. The concentration and purity of the extracted RNA were determined by using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The quality of the samples was assessed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) for RNA integrity number (>7.0) and concentration (minimum 70 ng/μl). RNA-seq was performed by GENEWIZ (South Plainfield, NJ, USA). The HiSeq 2500 platform (Illumina, San Diego, CA, USA) was used for RNA-seq in a 1 × 50-bp single-read configuration in rapid run mode, with a total of at least 120 million reads per lane over 5 lanes with a read depth of at least 10 million reads per sample. A total of five control and three HF offspring in the F1 generation and four control and five HF offspring in the F3 generation were used for RNA-seq analysis.
Rsem was used to quantify transcript abundance using the mouse genome Mus musculus as a reference. With a p value cutoff of 0.05, we obtained 5620 DEGs for further analysis. We then selected those 48 DEGs that were seen in both F1 and F3 generation offspring, with the direction (up- or downregulation) of differential expression being similar in the two generations, and performed Ingenuity Pathway Analysis (IPA; QIAGEN Bioinformatics, Redwood City, CA, USA) to assess function of the DEGs. Knowledge-fused differential dependency network (KDDN) analysis was also performed on the 48 DEGs to assess transcriptional gene interaction unique to either control or HF offspring [29].
Validation of RNA-seq data by quantitative real-time polymerase chain reaction
Of the 48 DEGs, we attempted to validate the expression of 13 genes that were selected on the basis of their reported association with breast cancer. RNA was extracted from the right fourth mammary glands of six control and six HF offspring of F1 generation and also of six control and six HF offspring of F3 generation using the RNeasy Lipid Tissue Mini Kit per the manufacturer’s protocol. These mice included those used for RNA-seq. The concentration, purity, and quality of RNA samples were assessed as described above. A quantity of 2 μg of RNA per sample was used to generate complementary DNA (cDNA) via reverse transcription using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) and was run on a PTC-100 thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA). Product cDNA was brought to a working concentration of 5 ng/μl and mixed with ABsolute QPCR, SYBR Green, ROX mix (Thermo Fisher Scientific, Waltham, MA, USA) and gene-specific forward and reverse primers. Primers used in quantitative polymerase chain reaction (qPCR) analysis were designed using IDT tool primer design (Integrated DNA Technologies, Coralville, IA, USA) (primer sequence provided in Additional file 2: Table S2). Real-time qPCR was carried out using a 7900HT Real-Time PCR system (Applied Biosystems). Expression of target genes was calculated by the relative standard curve method normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Statistical differences between the control and F1 HF and F3 HF groups were assessed by one-way ANOVA.
Results
Effect of maternal HF intake on offspring mammary tumorigenesis
Maternal exposure to a HF diet during pregnancy did not affect the distribution of benign versus malignant mammary tumors among the offspring. Malignant mammary tumors were those that were either papillary or tubular adenocarcinomas or mammary carcinomas. Among the control offspring, 58.3% and 64.3% were malignant in the F1 and F3 generations, respectively (Additional file 3: Figure S1a), compared with 66.0% and 77.3% in the F1 and F3 generation HF offspring (Additional file 3: Figure S1b).
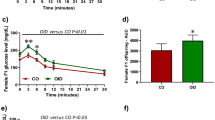
When we assessed differences in tumor incidence, we observed that only those with a malignant phenotype were included in the analysis. Female offspring exposed to the HF diet through a pregnant dam exhibited increased tumor incidence in F1 (Fig. 2a) (p < 0.016) and F3 (Fig. 2b) (p < 0.040) generations compared with control offspring. Mammary tumor burden was also increased in the F1 generation (Fig. 2c) (p < 0.027), but the increase failed to reach statistical significance in the F3 generation (Fig. 2d) (p < 0.242).

Transgenerational effect of maternal control (CON) or high-fat (HF) diet on offspring mammary tumorigenesis. Differences in mammary tumor incidence in (a) F1 (p < 0.016; CON, n = 30 mice; HF, n = 28 mice) and (b) F3 (p < 0.040; CON, n = 19 mice; HF, n = 24 mice) generation female offspring of dams fed either CON or HF diet during pregnancy. Differences in mammary tumor burden in (c) F1 (p < 0.027) and (d) F3 (p < 0.242) generation female offspring. Differences in mammary tumor latency in (e) F1 (p < 0.028) and (f) F3 (p < 0.110) generation female offspring. Mean ± SEM data are shown in c–f. TEB Terminal end bud
Maternal HF exposure during pregnancy induced earlier onset of mammary cancer in F1 generation (Fig. 2e) (p < 0.028) and had a similar trend in F3 generation offspring (Fig. 2f) (p < 0.110). Mammary tumor multiplicity was unaffected by maternal HF exposure (Additional file 4: Figure S2).
Offspring mammary gland morphology
Mammary gland morphology was assessed using whole mounts obtained from female offspring at PND 50. The number of TEBs (indicated by the arrows in Fig. 3a) was counted and found to be significantly higher in HF offspring for both F1 (Fig. 3b) (p < 0.035) and F3 generations (Fig. 3c) (p < 0.023).

Effect of maternal control (CON) or high-fat (HF) diet exposure on offspring mammary gland development. a The left fourth mammary glands were obtained on postnatal day 50 for whole mounts. Terminal end buds, structures in the enlarged image indicated by the arrows, were counted for (b) F1 (p < 0.035; n = 8 for HF and n = 6 for CON) and (c) F3 (p < 0.023; n = 5 for HF and n = 4 for CON). Mean ± SEM data are shown. DMBA 7,12-Dimethylbenz[a]anthracene
Differentially expressed genes in mammary glands of HF diet-exposed offspring
To elucidate the potential differences in mammary cancer risk in the F1 and F3 generation offspring of HF diet-fed dams, RNA-seq analysis was performed on normal mammary glands. In the F1 generation, 1587 DEGs were identified, and in the F3 generation, 4423 DEGs were seen (Fig. 4a). Of these, 390 were the same genes in both F1 and F3 HF offspring. However, only 48 of the DEGs were altered in the same direction (up- or downregulated) in both generations. Heat maps of these genes are shown in Fig. 4b and c and Additional file 5: Table S3.

Differentially expressed genes (DEGs) in mammary glands of F1 and F3 generation offspring of dams fed either control (CON) or high-fat (HF) diet during pregnancy. a RNA-sequencing analysis identified 1587 DEGs in F1 and 4423 DEGs in F3 generation mammary glands obtained on postnatal day 100 from mice not exposed to 7,12-dimethylbenz[a]anthracene (n = 5 CON and n = 3 HF offspring in F1 generation, and n = 4 CON and n = 5 HF offspring in F3 generation). A total of 390 common DEGs were found in the F1 and F3 generations, with 48 regulated in the same direction in both generations. Heat map of common 48 DEGs in (b) F1 mammary glands and (c) F3 mammary glands. Knowledge-fused differential dependency networks cluster map of nodes uniquely connected to different sets of genes in HF or CON offspring in (d) F1 or (e) F3 generation. Yellow ovals indicate nodes. Single-lined green connections indicate gene interactions in HF offspring. Double-lined red connections indicate gene interaction in CON offspring
IPA indicated that the top pathways that were different between HF and control offspring in both F1 and F3 generations were related to vitamin D receptor/retinoid X receptor (VDR/RXR) activation, phosphatase and tensin homolog (PTEN) signaling, farnesoid X receptor/RXR (FXR/RXR) activation, hereditary breast cancer signaling, and Notch signaling (Additional file 6: Table S4). Top upstream regulators of these pathways were interferon regulatory factors 3 and 7 (IRF3 and IRF7, respectively) linked to interferon and macrophage regulation [30], and delta like canonical ligand 3 (DLL3), Jagged 1 (JAG1), and mesogenin 1 (MSGN1), which all are linked to Notch signaling [31, 32] (Additional file 6: Table S4). Top diseases and biofunctions involved development, cellular functions, cancer and tumor morphologies, and inflammatory responses (Additional file 7: Table S5).
KDDN analysis (Fig. 4d and e, Table 1) performed to identify unique gene signaling interactions present in either the HF or control offspring highlighted the following ten genes as nodes: ALG6, DPF3, GRHL3, MAGIX, MT-TS2, and OPTC (full gene names provided in Abbreviations section below; upregulated in HF offspring), as well as AKR1C14, SLC5A3, SLC6A2, and ZBP1 (downregulated in HF offspring). These nodes had different signaling connections in HF and CON offspring. The node genes in the offspring of HF diet-fed dams were linked to changes in genes that are indicative of increased cancer risk (downregulation of ANKEF1, IGFBP6; upregulation of SEMA5B), increased resistance to cancer treatments (upregulation of EGR3, SLC26A3), poor cancer prognosis (upregulation of ID4, JAM3, TBX2), increased risk of metastasis (upregulation of GPCPD1) and impaired anticancer immune response (downregulation of ZBP1, upregulation of EGR3) (Additional file 8: reference table). In contrast, the ten node genes in the mammary glands of the offspring of dams fed the CON diet were linked to changes in the expression of genes indicative of reduced cancer risk (downregulation of DPF3, SNORA41) and improved immune functions (upregulation of ZBP1, ZFP683; downregulation of EGR3). These differences potentially play a regulatory role in causing transgenerational inheritance of increased mammary cancer risk in the offspring of dams fed the HF diet during pregnancy.
Verification of differential gene expression
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis indicated that all 13 genes differentially expressed in the RNA-seq dataset between offspring of HF and control diet-fed dams were validated in the F3 generation (Fig. 5). However, among the F1 generation, none of the eight upregulated genes were validated: AKT2, EGR3, HES1, ID4, JAM3, PCDHGA8, SLC26A10, and TBX2 (Fig. 5a–h). Of the five downregulated genes (IGFBP6, OAS3a, P21, SLFN1, and ZBP1) (Fig. 5i–m), four were significantly and one was nonsignificantly downregulated in both the F1 (OAS3a was not significant) and F3 (IGFBP6 was not significant) generations.

Verification of differential gene expression. Validation by quantitative real-time polymerase chain reaction of the following 13 differentially expressed genes identified in RNA-sequencing analysis: (a) Akt2, (b) Egr3, (c) Hes1, (d) Id4, (e) Jam3, (f) Pcdhga8, (g) Slc26a10, (h) Tbx2, (i) Igfbp6, (j) Oas3a, (k) p21, (l) Slfn1, and (m) Zbp1 (p < 0.05, a different from control diet [CON], b different from F1 high-fat (HF) diet, c different from F3 HF; p < 0.06, d marginally different from CON). We used fourth mammary glands obtained on postnatal day 100 from six CON and six HF offspring in F1 generation, as well as from six control and six HF offspring in F3 generation for the analysis. Mean ± SEM data are shown. Akt2 Serine/threonine kinase 2, CON Control diet, Egr3 Early growth response 3, Hes1 Hairy and enhancer of split-1, HF High fat, Id4 DNA-binding protein inhibitor ID-4, Igfbp6 Insulin-like growth factor binding protein 6, Jam3 Junctional adhesion molecule 3, Oas3 2′-5′-Oligoadenylate synthetase 3, Pcdhga8 Protocadherin gamma subfamily A, 8, Slc26a10 Solute carrier family 26 member 10, Slfn1 Schlafen 1, Zbp1, Z-DNA binding protein 1
Discussion
We found that maternal intake of a HF n-6 PUFA diet, starting on GD 10 during the second half of pregnancy and continuing until the end of pregnancy, increased estrogen receptor-positive (ER+) mammary cancer risk in F1 and F3 generation mouse offspring. Transgenerational inheritance likely requires that epigenetic changes induced by maternal exposures during pregnancy persist in germ cells. This possibility has been debatable because when F1 generation germ cells become fertilized, parental DNA methylation patterns are erased in the zygote [17, 22, 23]. However, a growing number of studies indicate that some genes in preimplantation zygotes can escape the complete loss of methylation marks that were established during reprogramming events of germ cells [32, 33]. Further, it has been shown that changes in histone marks in the preconception oocytes can be transmitted across generations [34]. Results of our previous studies, as well as results of multiple other studies, indicate that various maternal exposures after the first week of gestation, such as endocrine disruptors ethinylestradiol (EE2) [13], 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [35], vinclozolin [19], or DDT in rats [20], or bisphenol A [36], cause transgenerational alterations seen in F3 generation offspring.
Why, then, is the F3 generation not affected [13] if the exposure starts before conception and involves both the preimplantation period and the period when PGCs travel to the genital ridge [22, 23]? It is possible that although F1 generation PGCs are affected directly by the in utero HF environment in both our previous study and the present study, the changes can be transgenerationally inherited only if the somatic cells in the blastocyst giving rise to PGCs were not also affected. If true, maternal exposures that start before conception and continue through pregnancy can have multigenerational effects only involving the F1 and F2 generations, but not the F3 generation.
To determine if similar changes occur in gene transcription in F1 and F3 generations, when they are caused by a direct exposure in F1 generation or inherited in F3 generation through the germline, we performed RNA-seq analysis using normal mammary glands unexposed to the carcinogen DMBA, which were obtained from 100-day-old offspring of HF and control diet-fed dams. Surprisingly, over three times more DEGs were seen in the F3 than in the F1 generation mammary glands. Other groups have also reported greater gene transcription differences in F3 than F1 generation offspring. Ma et al. [35] found that maternal exposure to TCDD between GDs 8 and 14 resulted in a greater increase in both messenger RNA and protein expression of the imprinted gene insulin-like growth factor 2 (IGF2) in the rat liver of F3 than in F1 offspring. The increase was not caused by differences in DNA methylation patterns, which appeared similar in F1 and F3 generation offspring [35]. Consistent with these data, the differences in DNA methylation that we found earlier in F1 and F3 generations between control and maternal EE2 exposure groups were similar [13]. Thus, mechanisms other than changes in DNA methylation likely explain the increase in DEGs in the F3 generation offspring compared with F1 generation offspring.
From among all the DEGs found in our RNA-seq analysis, we selected the ones that were similarly altered in both F1 and F3 generation offspring of dams fed a HF diet during pregnancy compared with their controls for further analysis, because there possibly exists a connection between these 48 DEGs and the transgenerational inheritance of increased breast cancer risk. According to IPA, top biological functions of the DEGs were (1) development (cellular, embryonic, organ, organismal, and tissue), (2) cellular functions (cell cycle, proliferation, and morphology), (3) cancer and tumor morphology, and (4) inflammatory response (Additional file 7: Table S5). Considering that the main component of the HF diet was CO, a source of n-6 PUFAs, it is not surprising to find inflammatory response on this list of affected biofunctions. n-6 PUFAs and their arachidonic acid-derived eicosanoids are considered proinflammatory and associated with many inflammatory diseases, such as various types of cancer [11].
Five top upstream regulators of the 48 DEGs were DLL3 and JAG1 (Notch ligands) [37]; MSGN1 (transcriptional activator of a Notch signaling program) [32]; and IRF3 and IRF7, which are both key transcriptional regulators of interferons and macrophages [30]. Notch signaling may also regulate IRF activity [38]. These results suggest that Notch signaling may be altered in the offspring by maternal HF intake. Notch signaling was also among the five top pathways altered in the offspring of HF diet-fed dams, in addition to VDR/RXR and FXR/RXR activation, hereditary breast cancer signaling, and PTEN signaling. Researchers in earlier studies have found that maternal HF diet intake increases offspring Notch signaling in the hippocampus [39] and neural stem cells in mice [40]. Because the Notch pathway regulates stem cell maintenance, cell fate specification, differentiation, proliferation, motility, and survival during embryonic development and in cancers [41], our findings indicate that upregulation of Notch signaling in the normal mammary glands in F1 and F3 generation offspring of HF diet-fed dams may contribute to their increased mammary cancer risk.
To further characterize the functional roles of the DEGs, the networks hosted by these genes were constructed using KDDN analysis [29]. KDDN analysis identified differential connections among transcription factors that exist only in the mammary glands of offspring of HF diet-fed dams or only in the control offspring. In the HF group, genes identified as connected to the node genes were related to poor prognosis (SEMA5B, ID4, TBX2, GRHL3, DPF3), increased cellular proliferation and migration (GRHL3, PARP8, JAM3), and altered immune response (ZBP1, EGR3). As an example, the KDDN analysis indicated that upregulated grainyhead like transcription factor 3 (GRHL3) interacted with inhibitor of DNA binding 4 (ID4) and insulin-like growth factor binding protein 6 (IGFBP6) in the HF group, whereas its downregulation in the control group interacted with glycerophosphocholine phosphodiesterase 1 (GPCPD1) and small nucleolar RNA, H/ACA Box 41 (SNORA41). Upregulation of GRHL3 is strongly implicated in breast cancer [42], possibly by increasing the epithelial-mesenchymal transition [43]. ID4, upregulated in the offspring of HF diet-fed dams, is associated with poor prognosis of breast cancer, inhibits BRCA1 function in basal-like breast cancer [44], and promotes chemoresistance [45]. IGFBP6 was downregulated in HF offspring, and it acts as a tumor suppressor [46]. The interactions of downregulated GRHL3 in the control offspring with downregulated GPCPD1 and SNORA41 are indicative of good cancer prognosis [47, 48]. Changes in the expression of these genes and their interaction with each other suggest that maternal HF n-6 PUFA diet may not only lead to a transgenerational increase in breast cancer risk but also increase breast cancer mortality. This conclusion is consistent with our recent finding that maternal EE2 exposure during pregnancy increased resistance to antiestrogen therapy of ER+ mammary tumors in the offspring [49].
Conclusions
Our findings indicate that consuming a HF n-6 PUFA diet between GDs 10 and 20 during pregnancy causes a transgenerational increase in mammary cancer risk in mice. We also observed over three times more changes in the mammary gland transcriptome in F3 than in F1 generation offspring of HF diet-fed dams, suggesting that germline inheritance of increased mammary cancer risk may involve additional pathways to those altered in the adult mammary gland by a direct exposure of the fetal somatic cells.
Abbreviations
- AKR1C14:
-
Aldo-keto reductase family 1, member C14
- AKT2:
-
Serine/threonine kinase 2
- ALG6:
-
α-1,3-Glucosyltransferase
- ANKEF1:
-
Ankyrin repeat and EF-hand domain containing 1
- ANOVA:
-
Analysis of variance
- cDNA:
-
Complementary DNA
- CO:
-
Corn oil
- CON:
-
Control
- DDT:
-
Dichlorodiphenyltrichloroethane
- DEG:
-
Differentially expressed gene
- DLL3:
-
Delta like canonical Notch ligand 3
- DMBA:
-
7,12-Dimethylbenz[a]anthracene
- DPF3:
-
Double PHD fingers 3
- E2:
-
17-β-Estradiol
- EE2:
-
Ethinylestradiol
- EGR3:
-
Early growth response 3
- ER:
-
Estrogen receptor
- FXR/RXR:
-
Farnesoid X receptor/retinoid X receptor
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GD:
-
Gestational day
- GPCPD1:
-
Glycerophosphocholine phosphodiesterase 1
- GPCR:
-
G protein-coupled receptor
- GRHL3:
-
Grainyhead like transcription factor 3
- GWAS:
-
Genome-wide association study
- HES1:
-
Hairy and enhancer of split-1
- HF:
-
High fat
- ID4:
-
DNA-binding protein inhibitor ID-4
- IGF-1:
-
Insulin-like growth factor 1
- IGF2:
-
Insulin-like growth factor 2
- IGFBP6:
-
Insulin-like growth factor binding protein 6
- IPA:
-
Ingenuity Pathway Analysis
- IRF3:
-
Interferon regulatory factor 3
- IRF7:
-
Interferon regulatory factor 7
- JAG1:
-
Jagged 1
- JAM3:
-
Junctional adhesion molecule 3
- KDDN:
-
Knowledge-fused differential dependency network
- MAGIX:
-
MAGI family member, X-linked
- MPA:
-
Medroxyprogesterone acetate
- MSGN1:
-
Mesogenin 1
- MT-TS2:
-
Mitochondrially encoded transfer RNA serine 2 (AGU/C)
- OAS3:
-
2′-5′-Oligoadenylate synthetase 3
- OPTC:
-
Opticin
- P21:
-
Cyclin-dependent kinase inhibitor 1
- PCDHGA8:
-
Protocadherin gamma subfamily A, 8
- PGC:
-
Primordial germ cell
- PND:
-
Postnatal day
- PTEN:
-
Phosphatase and tensin homolog
- PUFA:
-
Polyunsaturated fatty acid
- qPCR:
-
Quantitative polymerase chain reaction
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- RNA-seq:
-
RNA sequencing
- SBO:
-
Soybean oil
- SEMA5B:
-
Semaphorin 5B
- SFA:
-
Saturated fatty acid
- SLC26A10:
-
Solute carrier family 26 member 10
- SLC26A3:
-
Solute carrier family 26 member 3
- SLC5A3:
-
Solute carrier family 5 member 3
- SLC6A2:
-
Solute carrier family 6 member 2
- SLFN1:
-
Schlafen 1
- SNORA41:
-
Small nucleolar RNA, H/ACA box 41
- SNP:
-
Single-nucleotide polymorphism
- STAT5:
-
Signal transducer and activator of transcription 5
- TBX2:
-
T-box 2
- TCDD:
-
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TEB:
-
Terminal end bud
- VDR/RXR:
-
Vitamin D receptor/retinoid X receptor
- ZBP1:
-
Z-DNA binding protein 1
- ZFP683:
-
Zinc finger protein 683
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Antoniou AC, Easton DF. Models of genetic susceptibility to breast cancer. Oncogene. 2006;25:5898–905.
Oldenburg RA, Meijers-Heijboer H, Cornelisse CJ, Devilee P. Genetic susceptibility for breast cancer: how many more genes to be found? Crit Rev Oncol Hematol. 2007;63:125–49.
Albuquerque RC, Baltar VT, Marchioni DM. Breast cancer and dietary patterns: a systematic review. Nutr Rev. 2014;72:1–17.
Walker CL, Ho SM. Developmental reprogramming of cancer susceptibility. Nat Rev Cancer. 2012;12:479–86.
Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93:950–62.
Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81:341–54.
Guyenet SJ, Carlson SE. Increase in adipose tissue linoleic acid of US adults in the last half century. Adv Nutr. 2015;6:660–4.
Hilakivi-Clarke L, Clarke R, Onojafe I, Raygada M, Cho E, Lippman ME. A maternal diet high in n-6 polyunsaturated fats alters mammary gland development, puberty onset, and breast cancer risk among female rat offspring. Proc Natl Acad Sci U S A. 1997;94:9372–7.
Hilakivi-Clarke L, Onojafe I, Raygada M, Cho E, Clarke R, Lippman M. Breast cancer risk in rats fed a diet high in n-6 polyunsaturated fatty acids during pregnancy. J Natl Cancer Inst. 1996;88:1821–7.
Marion-Letellier R, Savoye G, Ghosh S. Polyunsaturated fatty acids and inflammation. IUBMB Life. 2015;67:659–67.
Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14:461–9.
de Assis S, Warri A, Cruz MI, Laja O, Tian Y, Zhang B, et al. High-fat or ethinyl-oestradiol intake during pregnancy increases mammary cancer risk in several generations of offspring. Nat Commun. 2012;3:1053.
Stark AH, Kossoy G, Zusman I, Yarden G, Madar Z. Olive oil consumption during pregnancy and lactation in rats influences mammary cancer development in female offspring. Nutr Cancer. 2003;46:59–65.
Walker BE. Tumors in female offspring of control and diethylstilbestrol-exposed mice fed high-fat diets. J Nat Cancer Inst. 1990;82:50–4.
Luijten M, Thomsen AR, van den Berg JA, Wester PW, Verhoef A, Nagelkerke NJ, et al. Effects of soy-derived isoflavones and a high-fat diet on spontaneous mammary tumor development in Tg.NK (MMTV/c-neu) mice. Nutr Cancer. 2004;50:46–54.
Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109.
Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol. 2008;25:2–6.
Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology. 2006;147:5515–23.
Skinner MK, Manikkam M, Tracey R, Guerrero-Bosagna C, Haque M, Nilsson EE. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Med. 2013;11:228.
Choi CS, Gonzales EL, Kim KC, Yang SM, Kim JW, Mabunga DF, et al. The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci Rep. 2016;6:36250.
Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, et al. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–62.
Skinner MK, Haque CG, Nilsson E, Bhandari R, McCarrey JR. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. PLoS One. 2013;8:e66318.
Liu FL, Zhang YM, Pares GV, Reidy KC, Zhao WZ, Zhao A, et al. Nutrient intakes of pregnant women and their associated factors in eight cities of China: a cross-sectional study. Chin Med J (Engl). 2015;128:1778–86.
Bosaeus M, Hussain A, Karlsson T, Andersson L, Hulthen L, Svelander C, et al. A randomized longitudinal dietary intervention study during pregnancy: effects on fish intake, phospholipids, and body composition. Nutr J. 2015;14:1.
de Assis S, Warri A, Cruz MI, Hilakivi-Clarke L. Changes in mammary gland morphology and breast cancer risk in rats. J Vis Exp. 2010;44:e2260.
Russo IH, Russo J. Mammary gland neoplasia in long-term rodent studies. Environ Health Perspect. 1996;104:938–67.
Russo J, Hu YF, Yang X, Russo IH. Developmental, cellular, and molecular basis of human breast cancer. J Natl Cancer Inst Monogr. 2000;27:17–37.
Tian Y, Zhang B, Hoffman EP, Clarke R, Zhang Z, Shih I, et al. KDDN: an open-source Cytoscape app for constructing differential dependency networks with significant rewiring. Bioinformatics. 2015;31:287–9.
Gonzalez MJ, Ruiz-Garcia A, Monsalve EM, Sanchez-Prieto R, LaBorda J, Diaz-Guerra MJ, et al. DLK1 is a novel inflammatory inhibitor which interferes with NOTCH1 signaling in TLR-activated murine macrophages. Eur J Immunol. 2015;45:2615–27.
Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. 2012;23:450–7.
Chalamalasetty RB, Dunty Jr WC, Biris KK, Ajima R, Iacovino M, Beisaw A, et al. The Wnt3a/β-catenin target gene Mesogenin1 controls the segmentation clock by activating a Notch signalling program. Nat Commun. 2011;2:390.
Orozco LD, Rubbi L, Martin LJ, Fang F, Hormozdiari F, Che N, et al. Intergenerational genomic DNA methylation patterns in mouse hybrid strains. Genome Biol. 2014;15:R68.
Gaydos LJ, Wang W, Strome S. Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science. 2014;345:1515–8.
Ma J, Chen X, Liu Y, Xie Q, Sun Y, Chen J, et al. Ancestral TCDD exposure promotes epigenetic transgenerational inheritance of imprinted gene Igf2: Methylation status and DNMTs. Toxicol Appl Pharmacol. 2015;289:193–202.
Ziv-Gal A, Wang W, Zhou C, Flaws JA. The effects of in utero bisphenol A exposure on reproductive capacity in several generations of mice. Toxicol Appl Pharmacol. 2015;284:354–62.
Murata A, Yoshino M, Hikosaka M, Okuyama K, Zhou L, Sakano S, et al. An evolutionary-conserved function of mammalian notch family members as cell adhesion molecules. PLoS One. 2014;9:e108535.
Restivo G, Nguyen BC, Dziunycz P, Ristorcelli E, Ryan RJ, Özuysal ÖY, et al. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 2011;30:4571–85.
Mendes-da-Silva C, Lemes SF, Baliani TS, Versutti MD, Torsoni MA. Increased expression of Hes5 protein in Notch signaling pathway in the hippocampus of mice offspring of dams fed a high-fat diet during pregnancy and suckling. Int J Dev Neurosci. 2015;40:35–42.
Yu M, Jiang M, Yang C, Wu Y, Liu Y, Cui Y, et al. Maternal high-fat diet affects Msi/Notch/Hes signaling in neural stem cells of offspring mice. J Nutr Biochem. 2014;25:227–31.
Harrison H, Farnie G, Brennan KR, Clarke RB. Breast cancer stem cells: something out of notching? Cancer Res. 2010;70:8973–6.
Xu H, Liu C, Zhao Z, Gao N, Chen G, Wang Y, et al. Clinical implications of GRHL3 protein expression in breast cancer. Tumour Biol. 2014;35:1827–31.
Zhao P, Guo S, Tu Z, Di L, Zha X, Zhou H, et al. Grhl3 induces human epithelial tumor cell migration and invasion via downregulation of E-cadherin. Acta Biochim Biophys Sin Shanghai. 2016;48:266–74.
Baker LA, Holliday H, Swarbrick A. ID4 controls luminal lineage commitment in normal mammary epithelium and inhibits BRCA1 function in basal-like breast cancer. Endocr Relat Cancer. 2016;23:R381–92.
Qi K, Li Y, Li X, Lei X, Wang B, Zhang L, et al. Id4 promotes cisplatin resistance in lung cancer through the p38 MAPK pathway. Anticancer Drugs. 2016;27:970–8.
Kaulsay KK, Ng EH, Ji CY, Ho GH, Aw TC, Lee KO. Serum IGF-binding protein-6 and prostate specific antigen in breast cancer. Eur J Endocrinol. 1999;140:164–8.
Lesjak MS, Marchan R, Stewart JD, Rempel E, Rahnenfuhrer J, Hengstler JG. EDI3 links choline metabolism to integrin expression, cell adhesion and spreading. Cell Adh Migr. 2014;8:499–508.
Bao L, Zhang Y, Wang J, Wang H, Dong N, Su X, et al. Variations of chromosome 2 gene expressions among patients with lung cancer or non-cancer. Cell Biol Toxicol. 2016;32:419–35.
Hilakivi-Clarke L, Wärri AM, Bouker KB, Zhang X, Cook KL, Jin L, et al. Effects of in utero exposure to ethinyl estradiol on tamoxifen resistance and breast cancer recurrence in a preclinical model. J Nat Cancer Inst. 2016;109:djw188.
Acknowledgements
We thank Samuel Wopperer, Helen Jung and Catherine Sevigny for their excellent technical assistance.
Funding
This work was supported by National Institutes of Health grants U54 CA149147 and R01 CA164384 (to LHC) and P30 CA51008 (to the Lombardi Comprehensive Cancer Center; funding for shared resources).
Availability of data and materials
The datasets supporting the conclusions of this study are included within the article and its additional files.
Authors’ contributions
LHC is the principal investigator of the study and study supervisor. LHC, SdA, and NMN conceptualized and designed the transgenerational study. NMN and LHC wrote and reviewed the manuscript. NMN and LHC developed the methodology for the breeding and treatment of the animals. NMN, MIC, CB, XZ, BW, and CY were responsible for all aspects of the study concerning the animal model, including breeding, treatment, diet, collection of tissues, and tissue postprocessing. LJ, XW, and JX performed the RNA-seq analysis and assisted in the interpretation of the data along with NMN and LHC. FdOA performed the validation qRT-PCR of the RNA-seq. MM performed the analysis of the mammary gland morphology. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval
All animal procedures were approved by the Georgetown University Animal Care and Use Committee (GUACUC).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Table S1. Nutritional content of control (modified AIN93-G) and high-fat n-6 PUFA diets fed to pregnant mouse dams. (DOCX 15 kb)
Additional file 2:
Table S2. Primer sequences used in this study. (DOCX 77 kb)
Additional file 3:
Figure S1. Tumor histopathology of all tumors collected from F1 and F3 generation offspring of dams exposed to either control (CON) or high-fat (HF) diet during pregnancy. a Tumor status of CON offspring. b Tumor status of HF offspring. (PDF 129 kb)
Additional file 4:
Figure S2. Mammary tumor multiplicity was not altered between (a) F1 control (CON; n = 30 mice) and high-fat (HF; n = 29 mice) offspring or (b) F3 CON (n = 19 mice) and HF (n = 24 mice) generation offspring. (PDF 54 kb)
Additional file 5:
Table S3. Common differentially expressed genes in the mammary glands of F1 and F3 generation offspring of dams fed high-fat n-6 PUFA diet during pregnancy, compared with control mice. (DOCX 104 kb)
Additional file 6:
Table S4. Top differentially expressed pathways and predicted upstream regulators in F1 and F3 offspring of control or high-fat fed dams, identified in Ingenuity Pathway Analysis. (DOCX 47 kb)
Additional file 7:
Table S5. Top differentially expressed diseases and biofunctions between F1 and F3 offspring of dams fed control or high-fat diet during pregnancy, identified using Ingenuity Pathway Analysis. (DOCX 65 kb)
Additional file 8:
Supplementary reference list for KDDN. (DOCX 102 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nguyen, N.M., de Oliveira Andrade, F., Jin, L. et al. Maternal intake of high n-6 polyunsaturated fatty acid diet during pregnancy causes transgenerational increase in mammary cancer risk in mice. Breast Cancer Res 19, 77 (2017). https://doi.org/10.1186/s13058-017-0866-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-017-0866-x