
Abstract
Background
Women with breast cancer who have multiple affected relatives are more likely to have inherited genetic risk factors for the disease. All the currently known genetic risk factors for breast cancer account for less than half of the average familial risk. Furthermore, the genetic factor(s) underlying an increased cancer risk for many women from multiple-case families remain unknown. Rare genomic duplications and deletions, known as copy number variants (CNVs), cover more than 10% of a human genome, are often not assessed in studies of genetic predisposition, and could account for some of the so-called “missing heritability”.
Methods
We carried out a hypothesis-generating case-control study of breast cancer diagnosed before age 40 years (200 cases, 293 controls) using population-based cases from the Australian Breast Cancer Family Study. Genome-wide scanning for CNVs was performed using the Human610-Quad BeadChip and fine-mapping was conducted using PennCNV.
Results
We identified deletions overlapping two known cancer susceptibility genes, (BRCA1 and BLM), and a duplication overlapping SMARCB1, associated with risk. The number of deletions across the genome was 1.5-fold higher for cases than controls (P = 10-16), and 2-fold higher when only rare deletions overlapping genes (frequency <1%) were assessed (P = 5 × 10-4). Association tests of CNVs, followed by experimental validation of CNV calls, found deletions overlapping the OR4C11 and OR4P4 genes were associated with breast cancer (P = 0.02 and P = 0.03, respectively).
Conclusion
These results suggest rare CNVs might have a role in breast cancer susceptibility, at least for disease at a young age.
Similar content being viewed by others

Background
Breast cancer is the most common malignancy among women in the developed world, and is increasing rapidly in the developing world. A proportion of women with breast cancer have multiple affected relatives and are therefore more likely to have inherited genetic factors that increase their risk of developing the disease. All the currently known genetic risk factors for breast cancer currently only account for around 48% of average familial risk, and account for a lower proportion of the familial risk of disease at a young age, and the vast majority of women from multiple-case families do not have a known genetic explanation for their increased cancer risk [1, 2]. Thus, for a substantial fraction of women, including young affected women and those with a family history, the cause of their disease remains unexplained.
Copy number variants (CNVs) are estimated to cover 5–10% of the human genome [3] and, based on nucleotide coverage, are responsible for the majority of genetic variability in human populations. CNVs have been reported to disrupt genes known to be involved in breast cancer susceptibility, including BRCA1, BRCA2, TP53 and CHEK2 [4], and could similarly alter other genes involved in pathways related to breast cancer susceptibility. Furthermore, several array-based studies have reported candidate rare CNVs that overlap genes; variants in these might contribute to breast cancer susceptibility [5–7]. However, there has been a notable lack of consistency across these studies, probably because many women carry rare CNVs or because of false CNV calls caused by technical issues [8]. Evidence from some studies has suggested that the frequency and size of germline CNVs are increased in women with breast cancer [5, 7, 9], and that this might be strongest for CNVs that overlap gene regions [5, 7]. Thus, functional disruption of genes by CNVs across the genome might contribute to the genetic basis of breast cancer risk.
Only a few studies have examined the relationship between common germ-line CNVs and breast cancer risk. A large genome-wide association study (2000 breast cancer cases and 3000 controls) from the Wellcome Trust Case Control Consortium suggested that such CNVs were unlikely to have a major role in the genetic basis of breast cancer [10]. However, more recent genome-wide association studies of common CNVs (mean allele frequency (MAF) ≥5%) in Chinese and European women identified a deletion in the APOBEC3 gene cluster associated with up to 1.3-fold and up to 2.3-fold increase in risk of breast cancer associated with hemizygous and homozygous deletions, respectively [11, 12].
To better understand the role of CNVs in breast cancer risk we have conducted a hypothesis-generating study of breast cancer at a young age (diagnosed before the age of 40 years). Our study aimed to assess whether CNVs across the genome are more frequent in such breast cancer cases when compared with unaffected controls, and whether cancer susceptibility genes are disrupted by rare CNVs.
Methods
Subjects
We conducted a hypothesis-generating study of 258 women who were diagnosed with breast cancer before the age of 40 years from the population-based Australian Breast Cancer Family Study [13–15]. These cases had been previously screened for germline mutations in BRCA1, ATM, CHEK2, PALB2, TP53, BRCA2, CDH1, and FANCM [13, 14, 16–25]. A total of 348 women unaffected with breast cancer (controls) were selected from participants in the Australian Mammographic Density Twins and Sisters Study, a cross-sectional study of twins and their sisters [15, 26]. All study participants provided written informed consent.
Genotyping and identification of CNVs
All DNA samples were genotyped with the Human610-Quad BeadChip (Illumina, Inc, San Diego, CA, USA) with approximately 610,000 markers (including approximately 20,000 non-polymorphic markers) for single nucleotide polymorphism (SNP) and CNV analysis. Samples were processed using Illumina’s recommended protocol for Infinium HD assays. Data for each array were normalised using GenomeStudio 2011.1 software (Illumina). Probe information, including genomic location, signal intensity (Norm R), allele frequency (Norm theta), log R ratios (LRRs), and B allele frequencies (BAF), for each sample was calculated and exported from GenomeStudio. All samples had a call rate >95%. The CNV calls were generated using the PennCNV program (version 27 Aug. 2009), using the default program parameters, library files, and genomic wave adjustment.
Quality control procedures were performed to remove poor-quality array data (Additional file 1: Figure S1). Samples were excluded if they met the following criteria: log R ratio standard deviation >0.28; B allele frequency drift >0.01; waviness factor deviating from 0 by >0.04, or with the number of CNV calls exceeding 70. To reduce false positives, CNV calls were excluded if they contained <5 probes, and/or were ≥1000 kb in size. A total of 200 cases and 293 controls passed quality control steps and were assessed in the study. CNV data used in the study are shown in Additional file 2: Table S1.
Identification of genes overlapping CNVs and defining rare CNVs
To avoid examining multiple isoforms from genes, we annotated 39,544 UCSC RefSeq (NCBI36/Hg18) transcripts using the SOURCE database [27] and defined the genomic intervals for a total of 18,791 unique genes. Thus, each gene interval encompassed the start and end of all associated RefSeq transcripts (Additional file 1: Figure S2). CNVs and gene regions that were estimated to overlap by at least one base pair were identified in a genome-wide scan using Intersect and Join tools from the Galaxy web server [28–30].
Because putative CNV calls do not typically conform to discrete genomic regions in different women, we used the genome coordinates of 18,791 RefSeq gene (NCBI36/Hg18) boundaries to define a CNV region (Additional file 1: Figure S1). Each of these regions therefore represented a cluster of one or more CNVs overlapping a well-characterised gene in the human genome and was used to measure the frequency of CNVs in our study. Rare CNVs were defined as those with a frequency <1% in the total sample.
CNV validation by quantitative PCR
DNA samples were used to experimentally validate putative CNVs at 12 genomic regions using Human TaqMan® Copy Number Reference Assays (Thermofisher Scientific Inc). Primer and probe sequences are presented in Additional file 3: Table S2. RNaseP was used as an endogenous reference gene. All assays were carried out in triplicate.
Statistical analysis of CNV load
Welch’s t test was conducted to establish the level of significance associated with the difference in CNV carrier frequencies between the cases and controls. This test is an adaptation of Student’s t test designed to cope with datasets that have unequal variances. The statistical package R version R 2.14.2 was used to perform statistical analyses. P values <0.05 were considered significant.
Genome-wide CNV association analysis
Genes overlapping CNVs identified in cases or controls were assigned as having DNA loss (copy number states zero or one) or DNA gain (copy number states three or four). Perl 5.14.2 (ActiveState, Canada) was used to produce counts of the CNVs based on copy number state and the gene region by which they were defined. The CNV state with the most CNVs for each region was identified. A corresponding table of incidence of the most numerous CNV state in that region was analysed between cases and controls using Fisher’s exact test. This was used to calculate odds ratios, 95% confidence intervals and P values for the association between each CNV and cancer status. P values were adjusted for multiple testing with a false discover rate of 5%, using the method of Benjamini and Yekutieli (2001). Tests with a corrected P value <0.05 were considered statistically significant. The analysis was performed based on CNVs defined by their location within a gene region.
Results
CNV discovery in cases of early-onset breast cancer and in controls
A total of 58 cases and 55 controls were removed from the study after quality control criteria were applied, leaving 200 cases and 293 controls for downstream analyses (Additional file 1: Figure S1). Using PennCNV software, a total of 5109 and 6133 CNV calls were generated for cases and controls, respectively, ranging from 0.6 to 998 kb. The average number of CNVs observed in the two study groups was larger in cases than controls (25.6 vs. 20.9; P = 2 × 10-10) (Table 1). When accounting for copy number type, the average number of deletions was 1.5-fold greater in cases compared with controls (18.1 vs. 12.4; P = 2 × 10-16), whereas the average number of copy number gains in cases was slightly lower than that of controls (7.4 vs. 8.5; P = 0.01). These results suggest that women with early-onset breast cancer carried a greater CNV load across the genome compared with controls and that this feature was due to the inheritance or de novo formation of genomic deletions.
To assess the potential disruption of CNVs to functional regions across the genome, CNVs overlapping 18,791 reference sequence (RefSeq) genes (NCBI36/Hg18) were identified in cases and controls. Thirty-four percent of CNVs were predicted to overlap genes in both study groups (Table 1). Deletions affecting gene regions were shown to be 1.6-fold more frequent in cases compared to controls (5.2 vs. 3.3; P = 4 × 10-10). Concordantly, the average number of RefSeq genes predicted to be disrupted by genomic deletions was also 1.6-fold higher in cases compared with controls (7.3 vs. 4.7; P = 2 × 10-8). By comparison, there was no significant difference observed between cases and controls when the number of copy number gains overlapping genes (P = 0.1), and the number of genes overlapping copy number gains (P = 0.5) were measured.
To determine whether CNV size contributed to genomic burden we calculated the genomic distance between the start and end probes of each PennCNV call. These data showed that there was a slight decrease in the average size of CNVs in cases compared with controls (59.4 kb vs. 65.7 kb; P = 0.001); however, there was no statistically significant difference in the average size of the CNVs between the groups when considering copy number type (deletion or duplication) (Table 2). Thus, the enrichment of genes disrupted by genomic deletions in cases of early breast cancer is the result of increased frequency rather than the size of these variants.
Rare CNVs in cases of early-onset breast cancer and in controls
To examine the prevalence of rare CNVs, RefSeq gene regions containing five or more CNVs (>1% frequency in the study cohort) were excluded from downstream analyses. By this process, all remaining rare CNVs assessed in this study overlapped RefSeq gene regions. The number of rare deletions in the cases were twofold greater than in controls (1.6 vs. 0.8; P = 5 × 10-4), but there was no statistically significant difference in copy number gains between the two groups (P = 0.6) (Table 3). The average number of RefSeq genes predicted to be disrupted by genomic deletions was 1.8-fold higher in cases compared with controls (2.0 vs. 1.2; P = 5 × 10-3). The ratio of genes disrupted per rare deletion event in each sample was 1.2 in cases and 1.4 in controls (data not shown). There was no significant difference in ratios between the two groups (P = 0.1). Together these results suggest that in most samples rare deletions are disrupting a single gene, and such events in cases of early-onset breast cancer were twice as common as in controls.
Rare CNVs disrupting known cancer susceptibility genes
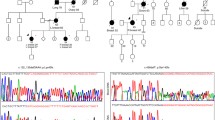
Annotation of CNVs in cases and controls against genes known to be involved in cancer-associated syndromes (Additional file 3: Table S3) revealed four cases with deletions in APC (n = 2), BRCA1 (n = 1) and BLM (n = 1), and one case with a duplication overlapping SMARCB1 (n = 1). One control was also found to harbour a putative deletion overlapping APC (Fig. 1). Subsequent evaluation with qPCR confirmed deletions overlapping BLM and BRCA1, the duplication overlapping SMARCB1, but none of the deletions overlapping the APC locus (two cases and one control) (Additional file 3: Table S4). Each validated CNV was shown to overlap one or more of the coding exons within the respective cancer susceptibility gene, suggesting a deleterious effect on the encoded proteins. CNVs potentially associated with risk of breast cancer did not overlap any of the genes that are commonly included in breast cancer predisposition gene panel tests (Additional file 4: Table S5). The BLM deletion was genotyped in relatives of the affected proband and identified two additional carriers of deletion but none were affected with breast cancer and an (obligate) non-carrier of this deletion had had breast cancer diagnosed at age 42 years (Additional file 1: Figure S3).

Putative copy number variation (CNV) calls overlapping known cancer susceptibility genes in five cases of early-onset breast cancer. UCSC Genome Browser screenshots (NCBI36/hg18) show the location of the CNVs (solid red rectangles) in relation to each gene. RefSeq reference sequence
Genome-wide CNV association study
To identify other new genomic loci contributing to breast cancer, we applied a gene-centric-based approach that defines copy number events by their location within a RefSeq gene region and compares the frequency of these events in cases and controls. Thus, genes overlapping CNVs identified in case and control cohorts were subsequently assigned status as DNA loss or DNA gain. The odds ratio estimates for the association between CNV status and risk of breast cancer are presented in Table 4. Deletions in three genes were associated with increased risk of breast cancer (DOCK5, P = 3 × 10-3; OR4C11, P = 2 × 10-2; and OR4P4, P = 3 × 10-2 ). qPCR did not confirm the small 646-base-pair deletion at the DOCK5 locus (Table 4). The CNV was predicted by six probes mapping to a 1.4 kb simple tandem repeat region (chr8:25,073,452-25,074,806; GRCh37/hg19). In contrast, deletions were overlapping the ORC4C11 and OR4P4 loci were verified by qPCR (Table 4; Additional file 3: Table S4).
Discussion
In comparison with the large amount of single nucleotide variant data available from breast cancer studies, the contribution of inherited copy number variation to breast cancer risk remains relatively understudied. To our knowledge, this is the largest genome-wide CNV analysis of early-onset (<40 years of age) breast cancer in a population-based study. Our results suggest that CNV frequency (or CNV load) may be associated with breast cancer risk, which is consistent with non-statistically significant data from a previous study of cases of familial and early-onset (<40 years of age) breast cancer [5]. Moreover, our study showed that on average, women with early-onset breast cancer carried one extra deletion within their genome that overlapped a coding gene.
Consistent with our data, a recent analysis of the Exome Aggregation Consortium (ExAC) database showed that the average human genome contains 2.1 rare (<0.5%) CNVs (0.8 deletions, 1.3 duplications) that intersect at least one protein-coding gene [31]. These results are similar to those from controls analysed in this study in which there were 2.6 rare (<1%) CNVs (1.2 deletions, 1.4 duplications) that intersect at least one protein-coding gene. The slightly larger values seen in our study might be explained by the less stringent criterion we used for defining a rare CNV. A possible reason for the larger number of deletions seen in the cases may be related to chemotherapeutic and/or radiological treatment effects on patients. However, in contrast to deletions we observed a reduced total number of duplications in cases compared with controls, and no significant difference between groups when assessing rare CNVs. Such a trend is difficult to explain if the genomic rearrangements are a result of treatment alone. Our results were not directly comparable with results from three other breast-cancer-related studies [5–7] that used a different and more stringent approach to define rare CNVs, that is those showing no overlap or minimal overlap with CNVs listed in the Database of Genomic Variants.
Our study identified a deletion overlapping the known breast cancer susceptibility gene, BRCA1, in a woman diagnosed with infiltrating ductal carcinoma of the breast (grade 3) at the age of 39 years, who did not have a family history of the disease and did not have any relatives participating in our study. A rare deletion overlapping the Bloom syndrome RecQ-like helicase gene, BLM, was also detected in a patient and some of her family members. Although data were limited for segregation analysis, our results are consistent with the previous finding that BLM mutations are associated with a perhaps more moderate increased risk of breast cancer [32]. These data are consistent with this rare allele being associated with a low-to-moderate risk of breast cancer but our study did not have the capacity to formally address or measure a possible link to breast cancer risk. No other women in this study had CNV disruption to cancer susceptibility gene(s). Thus, it remains unclear whether the additional genes disrupted by rare deletions in cases have a causal role in breast cancer risk.
A genome-wide association study of the cases and controls identified deletions overlapping three gene regions (DOCK5, 6.9-fold, P = 0.003; OR4C11, 2.6-fold, P = 0.02, and OR4P4, 2.4-fold, P = 0.03) that were associated with an increased risk of breast cancer after accounting for multiple testing (Additional file 4: Table S5). Assessment of these regions in a subset of samples using orthogonal technology verified the deletions overlapping the olfactory receptor genes, OR4C11 and OR4P4, but not DOCK5. OR4C11 and OR4P4 are located at 11q11 and have been previously found to overlap a common bi-allelic deletion [33, 34]. Of note, deletions overlapping OR4C6 and OR4S2 that were not associated with breast cancer risk after multiple testing (Table 4) are located adjacent to the OR4C11/OR4P4 gene locus. To our knowledge, variants overlapping the OR4C11/OR4P4 gene locus have not previously been shown to be associated with breast cancer risk. It is unclear how this locus would have a causative role in breast cancer development, although 11q deletions are commonly found in breast tumours, particularly those classified as having a high histological grade [35]. A review of two breast tumour datasets from Pereira et al. (n = 1980) and The Cancer Genome Atlas (n = 960) showed that expression of OR4C11 and OR4P4 did not correlate with copy number loss (data not shown) [36, 37], although this locus may harbour regulatory element(s) that control key genes from long range.
CNVs overlapping more than 100 genes have been found exclusively or at a greater frequency in cases of familial and/or early-onset breast cancer; however, none of these loci have been identified in more than one study [8]. Large-scale studies of women with early-onset breast cancer are now required to better understand the contribution of germline CNVs to breast cancer risk. Such CNV-based studies are now possible by utilising available SNP genotyping data generated by massive genome-wide association studies that include cases of early-onset breast cancer [38, 39].
Conclusions
We report that the frequency of rare CNVs may be associated with breast cancer risk, and that compared with controls, patients with early-onset breast cancer carried one extra deletion within their genome that overlapped a coding gene. A genome-wide analysis of CNVs identified deletions at the OR4C11/OR4P4 locus that were also associated with breast cancer risk. Larger studies are required to further investigate these possible associations to understand the role of CNVs in the development of breast cancer.
Abbreviations
- BAF:
-
B allele frequency
- CI:
-
Confidence interval
- CNV:
-
Copy number variation
- Kb:
-
kilobase
- LRR:
-
Log R ratio
- MAF:
-
Minor allele frequency
- OR:
-
Odds ratio
- qPCR:
-
Quantitative polymerase chain reaction
- RefSeq:
-
reference sequence
- SNP:
-
single nucleotide polymorphism
References
Sakoda LC, Jorgenson E, Witte JS. Turning of COGS moves forward findings for hormonally mediated cancers. Nat Genet. 2013;45(4):345–8.
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45(4):353–61. 361e1-2.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–83.
Kuiper RP, Ligtenberg MJ, Hoogerbrugge N, Geurts van Kessel A. Germline copy number variation and cancer risk. Curr Opin Genet Dev. 2010;20(3):282–9.
Pylkas K, Vuorela M, Otsukka M, Kallioniemi A, Jukkola-Vuorinen A, Winqvist R. Rare copy number variants observed in hereditary breast cancer cases disrupt genes in estrogen signaling and TP53 tumor suppression network. PLoS Genet. 2012;8(6):e1002734.
Krepischi AC, Pearson PL, Rosenberg C. Germline copy number variations and cancer predisposition. Future Oncol. 2012;8(4):441–50.
Kuusisto KM, Akinrinade O, Vihinen M, Kankuri-Tammilehto M, Laasanen SL, Schleutker J. Copy number variation analysis in familial BRCA1/2-negative Finnish breast and ovarian cancer. PLoS One. 2013;8(8):e71802.
Walker LC, Wiggins GAR, Pearson JF. The role of constitutional copy number variants in breast cancer. Microarrays. 2015;4(3):17.
Krepischi AC, Achatz MI, Santos EM, Costa SS, Lisboa BC, Brentani H, Santos TM, Goncalves A, Nobrega AF, Pearson PL, et al. Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res. 2012;14(1):R24.
Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713–20.
Xuan D, Li G, Cai Q, Deming-Halverson S, Shrubsole MJ, Shu XO, Kelley MC, Zheng W, Long J. APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry. Carcinogenesis. 2013;34(10):2240–3.
Long J, Delahanty RJ, Li G, Gao YT, Lu W, Cai Q, Xiang YB, Li C, Ji BT, Zheng Y, et al. A common deletion in the APOBEC3 genes and breast cancer risk. J Natl Cancer Inst. 2013;105(8):573–9.
Hopper JL, Southey MC, Dite GS, Jolley DJ, Giles GG, McCredie MR, Easton DF, Venter DJ. Population-based estimate of the average age-specific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Australian Breast Cancer Family Study. Cancer Epidemiol Biomarkers Prev. 1999;8(9):741–7.
Dite GS, Jenkins MA, Southey MC, Hocking JS, Giles GG, McCredie MR, Venter DJ, Hopper JL. Familial risks, early-onset breast cancer, and BRCA1 and BRCA2 germline mutations. J Natl Cancer Inst. 2003;95(6):448–57.
John EM, Hopper JL, Beck JC, Knight JA, Neuhausen SL, Senie RT, Ziogas A, Andrulis IL, Anton-Culver H, Boyd N, et al. The Breast Cancer Family Registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6(4):R375–89.
Apicella C, Dowty JG, Dite GS, Jenkins MA, Senie RT, Daly MB, Andrulis IL, John EM, Buys SS, Li FP, et al. Validation study of the LAMBDA model for predicting the BRCA1 or BRCA2 mutation carrier status of North American Ashkenazi Jewish women. Clin Genet. 2007;72(2):87–97.
Leong T, Whitty J, Keilar M, Mifsud S, Ramsay J, Birrell G, Venter D, Southey M, McKay M. Mutation analysis of BRCA1 and BRCA2 cancer predisposition genes in radiation hypersensitive cancer patients. Int J Radiat Oncol Biol Phys. 2000;48(4):959–65.
Andrulis IL, Anton-Culver H, Beck J, Bove B, Boyd J, Buys S, Godwin AK, Hopper JL, Li F, Neuhausen SL, et al. Comparison of DNA- and RNA-based methods for detection of truncating BRCA1 mutations. Hum Mutat. 2002;20(1):65–73.
Southey MC, Tesoriero AA, Andersen CR, Jennings KM, Brown SM, Dite GS, Jenkins MA, Osborne RH, Maskiell JA, Porter L, et al. BRCA1 mutations and other sequence variants in a population-based sample of Australian women with breast cancer. Br J Cancer. 1999;79(1):34–9.
Smith LD, Tesoriero AA, Ramus SJ, Dite G, Royce SG, Giles GG, McCredie MR, Hopper JL, Southey MC. BRCA1 promoter deletions in young women with breast cancer and a strong family history: a population-based study. Eur J Cancer. 2007;43(5):823–7.
Neuhausen SL, Ozcelik H, Southey MC, John EM, Godwin AK, Chung W, Iriondo-Perez J, Miron A, Santella RM, Whittemore A, et al. BRCA1 and BRCA2 mutation carriers in the Breast Cancer Family Registry: an open resource for collaborative research. Breast Cancer Res Treat. 2009;116(2):379–86.
Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, Sabbaghian N, Apicella C, Byrnes GB, Winship I, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010;12(6):R109.
Goldgar DE, Healey S, Dowty JG, Da Silva L, Chen X, Spurdle AB, Terry MB, Daly MJ, Buys SM, Southey MC, et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. 2011;13(4):R73.
Le Calvez-Kelm F, Lesueur F, Damiola F, Vallee M, Voegele C, Babikyan D, Durand G, Forey N, McKay-Chopin S, Robinot N, et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. 2011;13(1):R6.
Mouchawar J, Korch C, Byers T, Pitts TM, Li E, McCredie MR, Giles GG, Hopper JL, Southey MC. Population-based estimate of the contribution of TP53 mutations to subgroups of early-onset breast cancer: Australian Breast Cancer Family Study. Cancer Res. 2010;70(12):4795–800.
Odefrey F, Stone J, Gurrin LC, Byrnes GB, Apicella C, Dite GS, Cawson JN, Giles GG, Treloar SA, English DR, et al. Common genetic variants associated with breast cancer and mammographic density measures that predict disease. Cancer Res. 2010;70(4):1449–58.
Diehn M, Sherlock G, Binkley G, Jin H, Matese JC, Hernandez-Boussard T, Rees CA, Cherry JM, Botstein D, Brown PO, et al. SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res. 2003;31(1):219–23.
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010; Chapter 19: Unit 19. 10. 1–21
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–5.
Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86.
Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Exome Aggregation Consortium, Daly MJ, MacArthur DG, Fromer M et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet. 2016;48(10):1107-11.
Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, Thorne H, kConFab, Barnes DR, Li J, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8(9):e1002894.
Waszak SM, Hasin Y, Zichner T, Olender T, Keydar I, Khen M, Stutz AM, Schlattl A, Lancet D, Korbel JO. Systematic inference of copy-number genotypes from personal genome sequencing data reveals extensive olfactory receptor gene content diversity. PLoS Comput Biol. 2010;6(11):e1000988.
Young JM, Endicott RM, Parghi SS, Walker M, Kidd JM, Trask BJ. Extensive copy-number variation of the human olfactory receptor gene family. Am J Hum Genet. 2008;83(2):228–42.
Roylance R, Gorman P, Harris W, Liebmann R, Barnes D, Hanby A, Sheer D. Comparative genomic hybridization of breast tumors stratified by histological grade reveals new insights into the biological progression of breast cancer. Cancer Res. 1999;59(7):1433–6.
Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):l1.
Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, Casey G, Hunter DJ, Sellers TA, Gruber SB, et al. The OncoArray Consortium: A network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev. 2017;26(1):126–35.
Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Maranian MJ, Bolla MK, Wang Q, Shah M, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015;47(4):373–80.
Acknowledgements
LCW was supported by the Health Research Council of New Zealand Sir Charles Hercus Health Research Fellowship, and the Royal Society of New Zealand Rutherford Discovery Fellowship. JLH is a Senior Principal Research Fellow and MCS is a Senior Research Fellow of the NHMRC. We wish to thank participants and staff of the Australian Breast Cancer Family Study.
Funding
The Australia site of Breast Cancer Family Registry was supported by the National Health and Medical Research Council of Australia (NHMRC), the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and grant UM1 CA164920 from the USA National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The AMDTSS was funded by the NHMRC, Cancer Australia and the National Breast Cancer Foundation. Genotyping was funded by NHMRC as part of an Australia Fellowship awarded to JLH.
Availability of data and materials
Please contact the corresponding author for additional information on how to obtain the study data.
Authors’ contributions
LCW and MCS conceived of, designed and coordinated the study. JFP and LCW performed the bioinformatic and statistical analyses. GARW carried out the molecular assays. MCS, GGG and JLH provided the resources from the ABCFS and AMDTSS, including the Human610-Quad BeadChip data for CNV calling. LCW drafted the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Written informed consent was obtained from each participant of the Australian Breast Cancer Family Study and the Australian Mammographic Density Twins and Sisters Study. These studies are approved by the Human Research Ethics Committee of the University of Melbourne. The bioinformatic, statistical and laboratory component of this research was approved by the University of Otago Ethics Committee (H14/131).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Study design for CNV discovery, quality control and analysis. Figure S2. The protocol use to define RefSeq gene boundaries. Figure S3. BLM deletions identified in a familial breast cancer pedigree. Male and female individuals are represented by squares and circles, respectively. The index case patient, who underwent genome-wide CNV profiling, is indicated by an arrow. Individuals with breast cancer are represented by closed circles. Other cancers are indicated by black shading in the lower left quadrant. Age at death, last known diagnosis or cancer diagnosis is indicated where known. Copy number genotype at the BLM locus is noted as BLM deletion (BLM del) or wildtype (wt) copy number. (DOCX 30 kb)
Additional file 2: Table S1.
CNV data for cases and controls. Chromosome coordinates given according to Hg18. (XLSX 819 kb)
Additional file 3: Table S2.
Taqman assays used for CNV assessment at 7 gene loci. Table S3. Cancer-predisposing genes disrupted by rare CNVs. Table S4. Results of Taqman assays carried across 7 gene loci. (DOCX 18 kb)
Additional file 4: Table S5.
Results from a genome-wide association analysis of CNVs overlapping gene loci. (XLSX 205 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Walker, L.C., Pearson, J.F., Wiggins, G.A.R. et al. Increased genomic burden of germline copy number variants is associated with early onset breast cancer: Australian breast cancer family registry. Breast Cancer Res 19, 30 (2017). https://doi.org/10.1186/s13058-017-0825-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-017-0825-6