
Abstract
Background
Baricitinib has shown efficacy in hospitalized patients with COVID-19, but no placebo-controlled trials have focused specifically on severe/critical COVID, including vaccinated participants.
Methods
Bari-SolidAct is a phase-3, multicentre, randomised, double-blind, placebo-controlled trial, enrolling participants from June 3, 2021 to March 7, 2022, stopped prematurely for external evidence. Patients with severe/critical COVID-19 were randomised to Baricitinib 4 mg once daily or placebo, added to standard of care. The primary endpoint was all-cause mortality within 60 days. Participants were remotely followed to day 90 for safety and patient related outcome measures.
Results
Two hundred ninety-nine patients were screened, 284 randomised, and 275 received study drug or placebo and were included in the modified intent-to-treat analyses (139 receiving baricitinib and 136 placebo). Median age was 60 (IQR 49–69) years, 77% were male and 35% had received at least one dose of SARS-CoV2 vaccine. There were 21 deaths at day 60 in each group, 15.1% in the baricitinib group and 15.4% in the placebo group (adjusted absolute difference and 95% CI − 0.1% [− 8·3 to 8·0]). In sensitivity analysis censoring observations after drug discontinuation or rescue therapy (tocilizumab/increased steroid dose), proportions of death were 5.8% versus 8.8% (− 3.2% [− 9.0 to 2.7]), respectively. There were 148 serious adverse events in 46 participants (33.1%) receiving baricitinib and 155 in 51 participants (37.5%) receiving placebo. In subgroup analyses, there was a potential interaction between vaccination status and treatment allocation on 60-day mortality. In a subsequent post hoc analysis there was a significant interaction between vaccination status and treatment allocation on the occurrence of serious adverse events, with more respiratory complications and severe infections in vaccinated participants treated with baricitinib. Vaccinated participants were on average 11 years older, with more comorbidities.
Conclusion
This clinical trial was prematurely stopped for external evidence and therefore underpowered to conclude on a potential survival benefit of baricitinib in severe/critical COVID-19. We observed a possible safety signal in vaccinated participants, who were older with more comorbidities. Although based on a post-hoc analysis, these findings warrant further investigation in other trials and real-world studies.
Trial registration Bari-SolidAct is registered at NCT04891133 (registered May 18, 2021) and EUClinicalTrials.eu (2022-500385-99-00).
Similar content being viewed by others


Background
Baricitinib is an oral Janus kinase (JAK) 1/2 inhibitor, approved by the European Medicines Agency (EMA) for several chronic autoimmune diseases [1]. Early in the pandemic, baricitinib was suggested as COVID-19 therapy through anti-inflammatory effects by inhibiting the JAK-pathway and antiviral properties by inhibiting receptor-mediated endocytosis [2].
Five randomised controlled trials (RCTs) of baricitinib in COVID-19 have been published with promising results, although study design and effect estimates have varied [3, 4]. The ACTT2-trial met the primary endpoint of reduction in time to recovery with baricitinib plus remdesivir compared with remdesivir alone, although only a minority of the participants received glucocorticoids [5]. The ACTT4-trial comparing remdesivir and baricitinib with remdesivir and dexamethasone found no difference in mechanical ventilation-free survival by day 29. However, dexamethasone was associated with more adverse, treatment-related, and severe/life-threatening adverse events [6].
The manufacturer-sponsored double-blind, placebo-controlled COV-BARRIER trial investigated baricitinib or placebo added to standard of care (SoC), with approximately 80% receiving systemic corticosteroids. The trial failed to show a difference in the primary endpoint, occurrence of disease progression to high-flow oxygen/non-invasive ventilation, invasive mechanical ventilation, or death by day 28. However, a significant decrease of 28-day mortality in baricitinib recipients was observed, particularly in severe disease [7]. A subsequent addendum that included 101 critically ill patients on invasive mechanical ventilation or extracorporeal membrane oxygenation (ECMO), showed a marked reduction in 28-day all-cause mortality from 58% in the placebo group to 39% in the baricitinib group [8]. In the open-label RECOVERY trial, a modest, yet significant effect on mortality was reported, with 28-day mortality reduced from 14% in the control group to 12% in the baricitinib group [9]. Finally in the pragmatic PANCOVID-trial, 28-day survival was not significantly different in participants treated with baricitinib added to SoC versus SoC alone, although its small sample size precluded a definitive conclusion [10].
EU-SolidAct is a pan-European multicentre, adaptive platform trial, with its first sub-study Bari-SolidAct investigating baricitinib in patients with severe/critical COVID-19. The primary objective was to evaluate the effect of baricitinib vs. placebo, given in addition to SoC, on the occurrence of death within 60 days.
Methods
Study design and participants
EU-SolidAct is an investigator-initiated, randomised adaptive platform trial for COVID-19 and emerging infectious diseases. Data capture is modular depending on epidemic waves and available resources. Bari-SolidAct is a double-blind, randomized placebo-controlled phase 3 trial investigating baricitinib for severe or critical COVID-19. The Master protocol (EU-SolidAct) and subprotocol (Bari-SolidAct) are available on euclinicaltrials.eu.
Participants
Eligible participants were adults (≥ 18 years), with SARS-CoV2 infection confirmed by a polymerase chain reaction (PCR) no more than 9 days old, admitted to hospital with severe/critical COVID-19, defined as one of the following: (1) SpO2 < 90% on room air, (2) SpO2 90–94% with a downwards trend and/or signs of respiratory distress, (3) need of oxygen by non-invasive ventilation (NIV)/continuous positive airway pressure (CPAP), high-flow oxygen or non-rebreather mask, or iv) need of mechanical ventilation or ECMO.
Key exclusion criteria were suspected serious infection besides COVID-19, recent or recurrent thromboembolism, neutropenia, severe lymphopenia, severe renal dysfunction, pregnancy, breastfeeding, known hypersensitivity to constituents of study drugs, and immunosuppressive drugs including JAK inhibitors, except up to 4 days treatment with corticosteroids for COVID-19.
During the trial, amendments of eligibility criteria included a stricter cut-off for excluding patients with renal dysfunction (from eGFR < 15 to eGFR < 30 mL/min/1.73 m2) for consistency with other baricitinib protocols. All participants had eGFR above 30 mL/min/1.73 m2 at inclusion. Further amendments in the protocol specific for inclusion of immunocompromized participants with signs of hyperinflammation are reported in the Additional file 1: Online Appendix, as this part of the trial is still open for inclusion. Immunocompromized patients included before the decision to stop inclusion of immunocompetent patients from March 7th 2022 are included in this report, while those enrolled after March 7 are not. This amendment also increased the maximum time from PCR-confirmed SARS-CoV2 test to trial inclusion from 9 to 14 days, and maximum days with COVID-19 symptoms from 14 to 21.
Ethical considerations
The trial was conducted in accordance with ICH E6 (R2) Good Clinical Practice and the ethical principles of the Declaration of Helsinki. Informed consent by the study participant or legally authorised representative was given prior to inclusion in the study. This is an international trial conducted in several European countries, with approval from ethics committees and national competent authority in each country. The trial has been transferred to CTIS and is now accepted under the Clinical Trial Regulation (CTR), euclinicaltrials.eu (EU CT number 2022-500385-99-00). EU-SolidAct/Bari-SolidAct is also registered at www.clinicaltrials.gov (NCT04891133).
Randomisation and masking
Eligible patients were randomly allocated to baricitinib or matching placebo in an equal ratio, stratified by study centre and disease severity at baseline. An independent unblinded statistician used a computer-generated randomisation list with a permuted block size of 6 to assign participants to treatment groups and was the only person who knew the block size. All treatment assignments were securely stored on a server with restricted access. Participants, care providers and study personnel were masked to treatment assignment. Baricitinib and matching placebo were provided by Eli Lilly and Company, and drugs were stored, labelled, and shipped to the sites by a pharmaceutical logistics company.
Procedures
The participants were randomly assigned to one of the following groups: (1) 4 mg baricitinib once daily, or (2) matching placebo. Baricitinib or placebo were administered up to 14 days, and permanently stopped if the patient was discharged from hospital. Baricitinib and placebo were administered orally or by feeding tube. During the study, SoC changed in many countries as a result of updated World Health Organisation (WHO) guidelines recommending tocilizumab for severe and critical COVID-19 [11]. Tocilizumab was prohibited at inclusion in the trial but was allowed as rescue therapy in case of clinical progression.
Participants were assessed for study data, including outcomes and adverse events on days 1 (day of inclusion), 3, 5, 8 and every 7 days if still hospitalised, thereafter until day 90, and at discharge or early discontinuation. Participants were remotely followed up after hospital discharge until day 90. At day 90, participants were asked to answer patient reported outcome measures (PROM) provided via an electronic link. Viral loads and SARS-CoV2 serology were measured centrally from samples of serum and naso/oropharyngeal swabs collected at eligible sites at pre-defined time points during hospitalisation, see Additional file 1: Online Appendix for methods.
Outcomes
The primary endpoint was occurrence of death within 60 days (measured on day 61 after inclusion). Secondary endpoints were: (1) disease progression on the WHO progression scale within 28 days, (2) time from randomisation to sustained recovery defined as being home and without new complications within 14 days after discharge, (3) time from randomisation to first hospital discharge within 90 days, (4) modified WHO score (mild, moderate, severe or critical disease) on day 14 and 28, (5) occurrence of serious adverse events leading to study treatment discontinuation or death, (6) viral clearance assessed by SARS-CoV-2 PCR in naso/oropharyngeal specimens, (7) markers of systemic inflammation (CRP, ferritin, LDH, D-dimer, procalcitonin) during hospitalisation, and (8) PROM by Oslo COVID-19 QLQ-PW80 sub-scale scores (consisting of 80 items with recall timeframe the last 7 days) at day 90 [12].
Statistical analysis
Based on mortality rates in the DisCoVeRy [13] and NOR-Solidarity [14] trials, in addition to publicly available statistics from France, we assumed a 60-day mortality probability of 15% in the placebo group, and 10% in the baricitinib group. To show a difference between the treatment groups with a 5% significance level and a 90% power, we calculated that 924 evaluable participants were needed in each group. We planned to randomise 1900 participants in total to account for expected drop-out of 2.7%.
No formal interim analysis for efficacy was planned. Safety assessment by an independent Data Monitoring Committee (DMC) was planned after prespecified number of enrolled participants. Due to external evidence provided by the RECOVERY trial [9], and updated WHO guidelines recommending baricitinib for severe/critical COVID-19 [15], the trial steering committee decided to stop enrolling immunocompetent participants from 7th March 2022, as the present trial was not expected to alter the overall accumulated evidence. This decision was supported by the chair and statistician of the DMC.
Efficacy and safety analyses were performed on the modified intent to treat analysis set, consisting of all randomised participants who received at least one dose of study drug. A sensitivity analysis was performed by censoring post-discontinuation or post-rescue observations for participants who discontinued the study intervention and/or received rescue treatment (tocilizumab or increased dose of systemic corticosteroids).
We analysed the primary endpoint with the Cochran–Mantel–Haenszel test stratified by country/group of neighbouring countries, as the sample size did not allow use of the stratification factors of randomisation (centres and disease severity). For comparison with prior baricitinib trials, a post hoc analysis of death up to day-28 was performed, using the same approach. Kaplan–Meier curves up to day 61 (60 days after inclusion) were plotted.
Additional information of analysis of secondary endpoints including safety are available in the Additional file 1: Online Appendix. The statistical analysis plan (Additional file 1: Online Appendix) was finalised and signed prior to database lock and opening of the randomisation list. All p values are 2-sided, with a significance level of 0.05. The analyses were performed using SAS version 9.4 (SAS Institute), and Stata SE version 13 (StataCorp).
Role of the funding source
The European Commission funded this research, but had no role in design, analysis, interpretation of data, or approval of the manuscript.
Results
Participant flow and recruitment
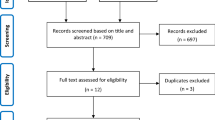
The study flowchart is shown in Fig. 1. The participants were recruited between June 3, 2021 and March 7, 2022 at 39 sites (hospital wards and intensive care units, ICUs) in Austria, Belgium, France, Germany, Ireland, Italy, Luxembourg, Norway, Portugal, and Spain (Additional file 1: Table S1). The trial was stopped before reaching the planned sample size due to evidence from the RECOVERY trial indicating survival benefit of baricitinib in the trial population [9]. In total, 299 patients were screened, 284 randomised, 9 participants did not receive any study drug or placebo, and 275 participants were included in both efficacy and safety analyses (139 in the baricitinib group and 136 in placebo).

Study Flowchart
Baseline characteristics
Median age was 60 (IQR 49–69) years, 77% were male, 72% had at least one comorbidity, 35% had received at least one dose of SARS-CoV2 vaccine, and 14% had critical disease (mechanical ventilation or ECMO) at inclusion. Systemic corticosteroids were used by 95% of participants, remdesivir by 3% and thromboprophylaxis by 90% (Table 1).
Efficacy endpoints
Efficacy results are reported in Table 2. There were 21 deaths in each group, leading to a proportion of death at day 60 of 15.1% in the baricitinib group and 15.4% in the placebo group (adjusted absolute difference and 95% confidence intervals (− 0.1% [− 8.3 to 8.0]), while at day 28 the corresponding figures were 10.1% and 13.2% respectively (− 2.9% [− 10.1 to 4.3]). Corresponding Kaplan–Meier plots are shown in Fig. 2A. Figure 2B shows Kaplan–Meier plots of sensitivity analysis when censoring post-discontinuation (6 in the baricitinib group vs 2 in placebo) and post-rescue therapy access observations (49 in the baricitinib group versus 52 in placebo), with proportion of death at day 60 being 5.8% versus 8.8% (− 3.2% [− 9.0 to 2.7]), respectively.

Kaplan–Meier plot of the probability of death within 60 days (measured at day 61 after inclusion), using the modified Intention to Treat population (mITT), consisting of all randomised participants who received at least one dose of study drug; A All observations regardless of study intervention discontinuation and/or receipt of rescue therapy; B Sensitivity analysis with participants censored at date of rescue therapy or date of discontinuation
None of the secondary efficacy endpoints (disease progression, sustained recovery, time to first hospital discharge) showed statistically significant differences between the treatment groups (Table 2). As shown in Additional file 1: Table S2, there were no significant differences for any of the PROM subscale scores (Oslo COVID-19 QLQ-PW80), and medians were below 10 for most of the domains (scores ranging from 0 to 100, 100 being the worse). Finally, none of the performed analyses comparing changes in viral loads or systemic inflammation markers were statistically significant, as shown in Additional file 1: Table S3.
Subgroup analyses
As shown in Additional file 1: Figure S1, no signal of interaction was detected for any of the pre-specified subgroup analyses, except vaccination status (not vaccinated vs at least one dose of SARS-CoV2 vaccine), with a proportion of death at day 60 of 8.0% (7/87) in the baricitinib group versus 15.9% (14/88) in the placebo group in unvaccinated participants, and 26.5% (13/49) versus 14.9% (7/47) respectively in the vaccinated participants (interaction p value = 0.0573).
Safety
The percentages of participants experiencing serious adverse events (SAEs) were similar in both groups: 46 (33.1%) participants with 148 SAEs reported in the baricitinib group, including 41 related to study drug, versus 51 (37.5%) participants with 155 SAEs in the placebo group, including 59 assessed as related to the study drug before unblinding [adjusted incidence rate ratio 0.93 (0.74 to 1.17)] (Table 3). Adverse events of special interest (AESIs) and disease-related events (DREs) were reported in both groups with similar proportions (Additional file 1: Table S4).
The results of the efficacy subgroup analysis motivated a post hoc analysis to examine the interaction between vaccination status and treatment group on safety, identifying a significant interaction for SAE occurrence: the proportion of participants that experienced SAEs was 25.3% in the baricitinib group versus 37.5% in the placebo group in unvaccinated participants and 46.9% versus 38.3% respectively in the vaccinated participants (interaction p value = 0.001). There was no significant interaction with vaccination status regarding occurrence of serious AESIs or serious DREs (Additional file 1: Table S5). The most frequent SAEs driving this difference were increased occurrence of respiratory complications and severe infections in vaccinated participants treated with baricitinib. Vaccinated participants were on average 11 years older and had more comorbidities, in particular diabetes mellitus, hypertension and chronic cardiac conditions (Additional file 1: Table S6).
Discussion
In this randomised, double-blind, placebo-controlled trial, no statistically significant difference was observed on 60-day mortality in hospitalised patients with severe/critical COVID-19 receiving SoC and either baricitinib or placebo. Of note, the trial was stopped before reaching planned sample size (n = 275 analysed versus n = 1900 planned) due to external evidence indicating survival benefit of baricitinib in the trial population. The mortality rate estimates at day 28 and day 60 are consistent with prior studies, in particular the day 28 estimate of the RECOVERY trial [5,6,7,8,9,10].
Whereas Bari-SolidAct included only patients with severe or critical COVID-19, other trials included mixed populations of mild/moderate and severe disease or only critical disease [5,6,7,8,9,10]. Background SoC, in particular remdesivir and corticosteroids have varied between trials but has not been associated with different treatment effects in subgroup analyses. In contrast to prior double-blind trials, tocilizumab was permitted as rescue therapy in accordance with updated WHO guidelines [15], and investigators were advised to stop baricitinib/placebo for patients receiving rescue therapy to avoid potential triple immunomodulation. This might have had an impact on the primary analysis, and in sensitivity analyses censoring post-rescue or post-discontinuation observations, the point estimates at day 28 and day 60 are closer to the point estimates of other trials (Fig. 2B).
Another notable difference between trial populations is the vaccination coverage. Whereas, due to timing, ACTT-2 and COV-BARRIER trials included no or very few vaccinated participants (no data reported in pubications) [5, 7, 8], the proportion of vaccinated participants was similar in Bari-SolidAct (35%) and RECOVERY (42%) [9]. In subgroup analysis, we found a signal suggesting a potential interaction between vaccination status and treatment allocation on mortality, with results indicating better survival at day 60 in unvaccinated participants treated with baricitinib while a potential opposite effect was seen in vaccinated participants. In the RECOVERY trial, no such interaction was identified [9], although populations are not directly comparable, with only severe/critical COVID-19 included in Bari-SolidAct.
In a subsequent post hoc analysis, there was a significant interaction between vaccination status and occurrence of SAEs, mainly driven by increased occurrence of respiratory complications and severe infections in vaccinated participants treated with baricitinib. In the placebo group, occurrence of SAEs was similar regardless of vaccination status. No safety signals including pulmonary embolism and other adverse events of special interest, were observed in the overall study population. While subgroup analyses must be interpreted with caution, we consider this result a potential safety signal of baricitinib in vaccinated patients. Although we lack a mechanistic explanation, vaccinated patients were on average 11 years older with more cardiometabolic comorbidities, and had lower levels of ferritin and LDH, in line with a recent observational study of vaccinated patients with breakthrough infections requiring hospitalization [16]. We hypothesise that the risk/benefit-ratio of baricitinib might be different in patients with severe/critical COVID-19 depending on SARS-CoV-2 immunisation status, and that underlying host factors such as comorbidities, older age and possibly the capacity to mount an immune response [17] could contribute to such differences.
Compared to other trials reporting 28-day mortality, participants in Bari-SolidAct were followed up to day-90 for efficacy, safety and patient reported outcomes. Pooled data from the TICO-platform reported that 20% of hospitalised patients had clinically significant post discharge-events, underscoring the need for longer follow-up [18]. Finally, Bari-SolidAct includes biobanking, however we observed no between-group differences in changes in viral load or inflammatory markers, despite the hypothesized dual action of baricitinib on viral entry and inflammatory responses [2, 19].
The main limitation of this trial is that due to its limited sample size, efficacy estimates are imprecise with wide confidence intervals. We did not achieve our target sample size because of delay in trial approval in several European countries [20], and because the trial was stopped prematurely as evidence from the RECOVERY trial indicated survival benefit of baricitinib in the trial population [9]. In addition, evolving SoC during the trial, in particular the introduction of tocilizumab, may have had an impact on the primary analysis since many participants had to discontinue intervention early due to rescue therapy. Furthermore, the subgroup analysis by vaccination status has low credibility according to the ICEMAN criteria [21]. Our study also has obvious strengths, in particular granular safety data, with routine registration of concomitant medication, safety lab and detailed follow-up of serious adverse events up to day-90.
Conclusions
In this prematurely stopped trial, we did not reach a conclusion for the primary endpoint due to lack of statistical power. In sensitivity analyses censoring observations after rescue therapy with tocilizumab or increased dose of corticosteroids, the point estimate is comparable with previous trials. We observed a significant interaction between vaccination status and treatment group on occurrence of SAEs, although this is based on a post hoc analysis and should be interpreted with caution. This potential safety signal should be explored in other trials and real-world studies, before influencing treatment decisions.
Availability of data and materials
Deidentified, individual participant data, along with a data dictionary describing variables in the dataset, will be made available to researchers whose proposed purpose of use is approved by the EU-SolidAct Trial Steering Committee. To request the dataset, please address directly to the corresponding author (marius.troseid@medisin.uio.no) or to Dominique Costagliola (dominique.costagliola@iplesp.upmc.fr) to obtain a data access form. All requests will be evaluated by the Trial Management Team and the EU-SolidAct Trial Steering Committee. For accepted requests, data will be shared after signing a data transfer agreement with the study sponsor. Data will be shared via a secure online procedure. Related documents, such as the study protocol, statistical analysis plan, and informed consent form, will be made available (with publication) on request to the corresponding author. The data will be open access for the informed consent form, protocol, and statistical analysis plan.
Abbreviations
- AE:
-
Adverse event
- AESI:
-
Adverse event of special interest
- CPAP:
-
Continuous positive airway pressure
- DRE:
-
Disease related event
- ECMO:
-
Extra corporal membrane oxygenation
- eGFR:
-
Estimated glomerular filtration rate
- EMA:
-
European Medicines Agencies
- NIV:
-
Non-invasive ventilation
- PCR:
-
Polymerase chain reaction
- PROM:
-
Patient related outcomes measures
- SAE:
-
Serious adverse event
- SARS-CoV2:
-
Severe acute respiratory coronavirus 2
- SoC:
-
Standard of care
- WHO:
-
World health organisation
References
Shi JG, Chen X, Lee F, et al. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol. 2014;54:1354–61.
Stebbing J, Phelan A, Griffin I, et al. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. 2020;20:400–2.
Kramer A, Prinz C, Fichtner F, et al. Janus kinase inhibitors for the treatment of COVID-19. Cochrane Database Syst Rev. 2022;6:Cd015209.
Selvaraj V, Finn A, Lal A, et al. Baricitinib in hospitalised patients with COVID-19: A meta-analysis of randomised controlled trials. EClinicalMedicine. 2022;49: 101489.
Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N Engl J Med. 2020.
Wolfe CR, Tomashek KM, Patterson TF, et al. Baricitinib versus dexamethasone for adults hospitalised with COVID-19 (ACTT-4): a randomised, double-blind, double placebo-controlled trial. Lancet Respir Med. 2022.
Marconi VC, Ramanan AV, de Bono S, et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med. 2021.
Ely EW, Ramanan AV, Kartman CE, et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir Med. 2022.
Baricitinib in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial and updated meta-analysis. Lancet. 2022;400:359–68.
Montejano R, de la Calle-Prieto F, Velasco M, et al. Tenofovir Disoproxil Fumarate/Emtricitabine and Baricitinib for Patients at High Risk of Severe COVID-19: The PANCOVID Randomized Clinical Trial. Clin Infect Dis. 2022.
Update to living WHO guideline on drugs for covid-19. Bmj. 2020;371:m4475.
Amdal CD, Taylor K, Kuliś D, et al. Health-related quality of life in patients with COVID-19; international development of a patient-reported outcome measure. J Patient Rep Outcomes. 2022;6:26.
Ader F, Bouscambert-Duchamp M, Hites M, et al. Remdesivir plus standard of care versus standard of care alone for the treatment of patients admitted to hospital with COVID-19 (DisCoVeRy): a phase 3, randomised, controlled, open-label trial. Lancet Infect Dis. 2022;22:209–21.
Barratt-Due A, Olsen IC, Nezvalova-Henriksen K, et al. Evaluation of the effects of remdesivir and hydroxychloroquine on viral clearance in COVID-19: a randomized trial. Ann Intern Med. 2021;174:1261–9.
Update to living WHO guideline on drugs for covid-19. Bmj. 2022;376:o80.
Lamacchia G, Mazzoni A, Spinicci M, et al. Clinical and immunological features of SARS-CoV-2 breakthrough infections in vaccinated individuals requiring hospitalization. J Clin Immunol. 2022.
Lipsitch M, Krammer F, Regev-Yochay G, et al. SARS-CoV-2 breakthrough infections in vaccinated individuals: measurement, causes and impact. Nat Rev Immunol. 2022;22:57–65.
Douin DJ, Siegel L, Grandits G, et al. Evaluating primary endpoints for COVID-19 therapeutic trials to assess recovery. Am J Respir Crit Care Med. 2022.
Richardson P, Griffin I, Tucker C, et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395:e30–1.
Diallo A, Trøseid M, Simensen VC, et al. Accelerating clinical trial implementation in the context of the COVID-19 pandemic: challenges, lessons learned and recommendations from DisCoVeRy and the EU-SolidAct EU response group. Clin Microbiol Infect. 2022;28:1–5.
Schandelmaier S, Briel M, Varadhan R, et al. Development of the Instrument to assess the Credibility of Effect Modification Analyses (ICEMAN) in randomized controlled trials and meta-analyses. CMAJ. 2020;192:E901–6.
Acknowledgements
We thank site staff for recruitment and follow-up of participants, and the patients for participating in the trial. We thank Morten Tandberg Eriksen as representative of the sponsor (OUH). We also thank Stefano Vella (chair), Stuart Pocock, Guanhua Du, Donata Medaglini, Mike Jacobs, Sylvie van der Werf, Phaik Yeong Cheah, Karen Barnes, Patrick Yeni, Tim Collier, Catia Marzolini and Dafna Yahav for supporting the trial as members of the independent DSMB. Finally, we thank all members of the EU SolidAct study group listed in the online appendix. The writing committee is listed below.
Writing Committee:
Marius Trøseid1, MD PhD, Section for Clinical Immunology and Infectious Diseases, Oslo University Hospital, Oslo, Norway, and the Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
José R. Arribas1, MD PhD, Infectious Diseases Unit, Internal Medicine Department, La Paz University Hospital, IdiPAZ, Madrid, Spain, Centro de Investigación Biomédica en Red de Enfermedades Infecciosas (CIBERINFEC) Madrid, Spain.
Lambert Assoumou, PhD, Sorbonne Université, INSERM, Institut Pierre Louis d’Épidémiologie et de Santé Publique (IPLESP), Paris, France.
Aleksander Rygh Holten, MD PhD, Department of Acute Medicine, Oslo University Hospital, Oslo, Norway, Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
Julien Poissy, MD PhD, Lille University, Lille, France/CHU Lille - Hôpital Roger Salengro, Lille, France, L’Institut national de la santé et de la recherche médicale (Inserm), Paris, France.
Vida Terzić, PhD, ANRS | Maladies Infectieuses Emergentes, F-75015 Paris, France, Institut national de la santé et de la recherche médicale, INSERM, F-75013 Paris, France.
Fulvia Mazzaferri, MD PhD, Division of Infectious Diseases, Department of Diagnostics and Public Health, University of Verona, Verona, Italy
Jesús Rodríguez Baño, MD PhD, Virgen Macarena University Hospital and Department of Medicine, Seville, Spain, University of Sevilla and Biomedicines Institute of Seville (IBiS)/CSIC, Seville, Spain, CIBERINFEC, Instituto de Salud Carlos III, Madrid, Spain.
Joe Eustace, MD PhD, University College Cork, Cork, Ireland.
Maya Hites, MD PhD, Brussels University Hospital-Erasme, Brussels, Belgium, Université Libre de Bruxelles, Brussels, Belgium.
Michael Joannidis, MD PhD, Medical University Innsbruck, Innsbruck, Austria.
José-Artur Paiva, MD PhD, Intensive Care Medicine Department, Centro Hospitalar Universitário Sao Joao, Porto, Portugal, Faculty of Medicine, University of Porto, Porto, Portugal.
Jean Reuter, MD PhD, Centre Hospitalier de Luxembourg, Service de Réanimation-Soins Intensifs, L-1210 Luxembourg, Luxembourg.
Isabel Püntmann, MD PhD, Department of Pharmacology and Toxicology, Gesundheit Nord gGmbH, Bremen, Germany.
Thale DJH Patrick-Brown, MSc, Division of Surgery, Inflammatory Diseases and Transplantation, Oslo University Hospital, Oslo, Norway and the Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
Elin Westerheim, MSc, Section for Monitoring, Clinical Trial Unit (CTU), Oslo University Hospital, Oslo, Norway.
Katerina Nezvalova-Henriksen, PhD, Department of Haematology, Oslo University Hospital and Oslo Hospital Pharmacy, Oslo, Norway.
Lydie Beniguel, PhD, Sorbonne Université, INSERM, Institut Pierre Louis d’Épidémiologie et de Santé Publique (IPLESP), Paris, France.
Tuva Børresdatter Dahl, PhD, Research Institute for Internal Medicine, Oslo University Hospital, Oslo, Norway Division of Emergencies and Critical Care, Oslo University Hospital, Oslo, Norway.
Maude Bouscambert-Duchamp, PharmD PhD, Laboratoire de Virologie, Institut des Agents Infectieux de Lyon, Centre National de Reference des Virus des Infections Respiratoires France Sud, Hospices Civils de Lyon, F-69317, Lyon, France.
Monika Halanova, MD PhD, Department of Epidemiology, Faculty of Medicine, Pavol Jozef Šafárik University in Košice, Košice, Slovakia.
Zoltán Péterfi, MD PhD, 1st Department of Internal Medicine, Division of Infectology, University of Pécs, Pécs, Hungary.
Sotirios Tsiodras, MD PhD, National and Kapodistrian University of Athens, Athens, Greece, University Hospital of Athens Attikon, Athens, Greece.
Michael Rezek, MD PhD, St. Anne University Hospital, Brno, Czech Republic
Matthias Briel, MD PhD, Swiss Clinical Trial Organisation and Department of Clinical Research, University Hospital Basel and University of Basel, Basel, Switzerland.
Serhat Ünal, MD PhD, Hacettepe University Hospital, Ankara, Turkey.
Martin Schlegel, MD PhD, Department of Anesthesiology and Intensive Care Medicine, Klinikum rechts der Isar, Technische Universität München, München, Germany.
Florence Ader, MD PhD, Hospices Civils de Lyon, Département des maladies infectieuses et tropicales, F-69004, Lyon, France, Centre International de Recherche en Infectiologie (CIRI), Inserm 1111, Université Claude Bernard Lyon 1, CNRS, UMR5308, École Normale Supérieure de Lyon, Univ Lyon, F-69007, Lyon, France.
Karine Lacombe, MD PhD, Sorbonne Université, Institut Pierre-Louis d'Épidemiologie et de Santé Publique, INSERM, F-75013, Paris, France, APHP, Hôpital Saint-Antoine, Service de Maladies Infectieuses et Tropicales, F-75012 Paris, France.
Cecilie Delphin Amdal, PhD, Research support service and Department of Oncology, Oslo University Hospital, Oslo, Norway.
Serge Rodrigues, PhD, Sorbonne Université, INSERM, Institut Pierre Louis d’Épidémiologie et de Santé Publique (IPLESP), Paris, France.
Kristian Tonby, MD PhD, Deptartment of Infectious diseases, Oslo University Hospital, Oslo, Norway Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
Alexandre Gaudet, MD PhD, Critical Care Center, Department of Intensive Care Medicine, CHU Lille, F-59000 Lille, France, Univ. Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, U1019-UMR9017-CIIL-Centre d’Infection et d’Immunité de Lille, F-59000 Lille, France.
Lars Heggelund, MD PhD, Medical Department, Drammen hospital, Vestre Viken Hospital Trust, Drammen, Norway and the Department of Clinical science, University of Bergen, Bergen, Norway.
Joy Mootien, MD PhD, Service, de réanimation médiale, GHRMSA Hopital Emile Muller, Mulhouse, France.
Asgeir Johannessen, MD PhD, Department of Infectious Diseases, Vestfold Hospital Trust, Tønsberg, Norway, Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
Jannicke Horjen Møller, MD, Intensive Care Unit, Stavanger University Hospital, Stavanger, Norway.
Beatriz Diaz Pollan, PhD, Infectious Diseases Unit, Internal Medicine Department, La Paz University Hospital, Madrid, Spain, IdiPAZ, Centro de Investigación Biomédica en Red de Enfermedades Infecciosas (CIBERINFEC), Madrid, Spain.
Anders Aune Tveita, MD PhD, Department of medicine, Vestre Viken, Bærum Hospital, Bærum, Norway.
Anders Benjamin Kildal, MD PhD, Department of Anesthesiology and Intensive Care, University hospital of North Norway, Tromsø, Norway.
Jean-Christophe Richard, MD PhD, Service de médecine intensive-réanimation, Hôpital de la Croix - Rousse - HCL Lyon, France, CREATIS INSERM U1206-CNRS UMR 5220, Lyon, France.
Olav Dalgard, MD PhD, Akershus University Hospital, Lørenskog, Norway, Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
Victoria Charlotte Simensen, MD, Norwegian Institute of Public Health, Division of Health Services, Department of Global Health, Oslo, Norway.
Aliou Baldé, PhD, Sorbonne Université, INSERM, Institut Pierre Louis d’Épidémiologie et de Santé Publique (IPLESP), Paris, France.
Lucie de Gastines, PharmD, ANRS | Maladies Infectieuses Emergentes, F-75015 Paris, France, Institut national de la santé et de la recherche médicale, INSERM, F-75013 Paris, France.
Marta del Álamo, PhD, ECRIN, Paris, France.
Burç Aydin, PhD, ECRIN, Paris, France.
Fridtjof Lund-Johansen, PhD, Department of Immunology, Oslo University Hospital, Oslo, Norway.
Mary-Anne Trabaud, PhD, Laboratoire de Virologie, Institut des Agents Infectieux de Lyon, Centre National de Reference des Virus Respiratoires France Sud, Hospices Civils de Lyon, F-69317, Lyon, France.
Alpha Diallo, MD, MPH, ANRS | Maladies Infectieuses Emergentes, F-75015 Paris, France, Institut national de la santé et de la recherche médicale, INSERM, F-75013 Paris, France.
Bente Halvorsen, PhD, Research Institute of Internal Medicine, Oslo University Hospital, Oslo, Norway and the Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
John-Arne Røttingen, MD PhD, Norwegian Institute of Public Health, Oslo, Norway.
Evelina Tacconelli2, MD PhD, Verona University Hospital, Verona, Italy, Division of Infectious Diseases, Department of Diagnostics and Public Health, University of Verona, Verona, Italy.
Yazdan Yazdanpanah2, MD PhD, Université de Paris, IAME, INSERM, F-75018 Paris, France, AP-HP, Hôpital Bichat, Service de maladies infectieuses et tropicales, F-75018 Paris, France.
Inge C. Olsen3, PhD, Department of Research Support for Clinical Trials, Oslo University Hospital, Oslo, Norway.
Dominique Costagliola3, PhD, Sorbonne Université, INSERM, Institut Pierre Louis d’Épidémiologie et de Santé Publique (IPLESP), Paris, France
Funding
EU-SolidAct is part of the European pandemic preparedness network EU RESPONSE, funded by the EU Horizon 2020 Research and Innovation programme, under grant number 101015736. EU-SolidAct has also received funding from CAPNET (France) and Klinbeforsk (Norway). The funding sources had no role in design, analysis, interpretation of data, or approval of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
The trial protocol was designed by MT, ICO, JAR, DC, JRA, AD, VT, LdG, and LA with contribution from other members of the writing committee and Eli Lilly. YY, JAR, MT, ICO, ET, JP and DC obtained funding for the study. LA, LB, AB, and SR were responsible for the data management, and AD, VT, and LdG for the pharmacovigilance. MB, MAT, FL-J, BH, and TBD were responsible for the virological and serological analysis, and biobanking. The SAP was written by LA and approved by MT, DC, JRA, and ICO. AB and LA performed the statistical analysis. This manuscript was drafted by MT, JRA, LA, ARH, JP, VT, ICO, and DC. ICO, LA, AB and DC had full access to the data. All members of the writing committee edited the manuscript for important intellectual content. All authors gave final approval of the version to be published and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The trial was conducted in accordance with ICH E6 (R2) Good Clinical Practice and the ethical principles of the Declaration of Helsinki. Informed consent by the study participant or legally authorised representative was given prior to inclusion in the study. The trial is accepted under the Clinical Trial Regulation (CTR), euclinicaltrials.eu (EU CT number 2022-500385-99-00).
Consent for publication
Not applicable.
Competing interests
MT has been member of scientific advisory board for Lilly. JRA has received advisory fees from Lilly. JP reports lecture fees from Gilead; support for attending meetings from Gilead, Eumedica, Merck Sharp & Dohme, outside the submitted work. ARH reports personal fee from Pfizer (2021) for lectures outside the submitted work. MH(it) has received funding for other trials on COVID-19 from the Federal Belgian Center for Knowledge and the joint Université Libre de Bruxelles-Fonds Erasme-COVID-19 projects (2020–21), personal fees from Gilead (2020) and Pfizer (2021) for editing and lectures outside the submitted work, and travel/congress grants from Pfizer (2020, 2021), and Gilead (2022). MJ reports consulting or speakers fees from Baxter, Gilead, CLS Behring, AM-Pharma, Novartis, Fresenius and grant support from Fresenius, Baxter, outside the submitted work. JAP reports fees for lectures and advisory boards from MSD, Pfizer, Astra-Zeneca, Jansen, Gilead, AOP Orphan Pharmaceuticals, Cepheid MB reports an unrestricted grant for Moderna (2022) outside the submitted work. MB reports an unrestricted grant for Moderna (2022) outside the submitted work. KL reports personal fees from Gilead, MSD, Janssen and ViiV Healthcare for advisory boards and lectures outside of the submitted work. JM reports personal fees from Pfizer (2017) for lectures outside the submitted work and travel fees from Pfizer (2022) and Menarini (2021). JCR reports a grant from Hamilton medical (2019–2020) outside the submitted work. FLJ reports Helse Sør-Øst grant for developing COVID-19 serology (2020–2021) and Grant from CEPI to monitor responses in patients (2021–2023). DC reports an HIV grant from Janssen (2019–2020), personal fees from Gilead (2020) and Pfizer (2022) for lectures outside the submitted work. All other authors have nothing to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Online Appendix.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Trøseid, M., Arribas, J.R., Assoumou, L. et al. Efficacy and safety of baricitinib in hospitalized adults with severe or critical COVID-19 (Bari-SolidAct): a randomised, double-blind, placebo-controlled phase 3 trial. Crit Care 27, 9 (2023). https://doi.org/10.1186/s13054-022-04205-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-022-04205-8