
Abstract
Background
Optimal antimicrobial drug exposure in the lung is required for successful treatment outcomes for nosocomial pneumonia. Little is known about the intrapulmonary pharmacokinetics (PK) of meropenem when administered by continuous infusion (CI). The aim of this study was to evaluate the PK of two dosages of meropenem (3 g vs 6 g/day by CI) in the plasma and epithelial lining fluid (ELF) in critically ill patients with nosocomial pneumonia.
Methods
Thirty-one patients (81% male, median (IQR) age 72 (22) years) were enrolled in a prospective, randomized, clinical trial. Sixteen patients received 1 g/8 h and 15 2 g/8 h by CI (8 h infusion). Plasma and ELF meropenem concentrations were modeled using a population methodology, and Monte Carlo simulations were performed to estimate the probability of attaining (PTA) a free ELF concentration of 50% of time above MIC (50% fT>MIC), which results in logarithmic killing and the suppression of resistance in experimental models of pneumonia.
Results
The median (IQR) of meropenem AUC0–24 h in the plasma and ELF was 287.6 (190.2) and 84.1 (78.8) mg h/L in the 1 g/8 h group vs 448.1 (231.8) and 163.0 (201.8) mg h/L in the 2 g/8 h group, respectively. The penetration ratio was approximately 30% and was comparable between the dosage groups. In the Monte Carlo simulations, only the highest approved dose of meropenem of 2 g/8 h by CI allowed to achieve an optimal PTA for all isolates with a MIC < 4 mg/L.
Conclusions
An increase in the dose of meropenem administered by CI achieved a higher exposure in the plasma and ELF. The use of the highest licensed dose of 6 g/day may be necessary to achieve an optimal coverage in ELF for all susceptible isolates (MIC ≤ 2 mg/L) in patients with conserved renal function. An alternative therapy should be considered when the presence of microorganisms with a MIC greater than 2 mg/L is suspected.
Trial registration
The trial was registered in the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT-no. 2016-002796-10). Registered on 27 December 2016.
Similar content being viewed by others


Key points
This is the first article assessing the lung penetration of different doses of meropenem administered by continuous infusion in critically ill patients with nosocomial pneumonia. A dose increase leads to higher plasma and epithelial lining fluid concentrations allowing to achieve an optimal probability of target attainment for all susceptible microorganisms.
Background
The relatively high incidence, rising rates of antimicrobial resistance, and suboptimal clinical outcomes of patients with nosocomial pneumonia provide the impetus to optimize the use of existing antibiotics [1,2,3,4].
Meropenem is a carbapenem antibiotic with potent activity against many of the pathogens that cause nosocomial pneumonia [1]. It exhibits time-dependent pharmacodynamics (PD); the fraction of the dosing interval that free plasma concentrations are above the MIC (fT>MIC) is the PK/PD index that best describes its antimicrobial efficacy [2, 3]. Although the maximal bactericidal activity for meropenem has been associated with a fT>MIC of 40–50% [4], a higher pharmacodynamic target of 50–100% fT>MIC has been suggested for critically ill patients [5]. Continuous (CI) (or extended) infusion of β-lactam antibiotics increases the fT>MIC and has been linked to improved clinical outcomes in some clinical studies [6,7,8]. The emergence of resistance is another important endpoint, so regimens must be designed to provide drug exposures that minimize the development of resistance [9, 10].
Plasma drug exposures are frequently used as a proxy for effect-site concentrations. However, in some cases, they may be misleading, and measurement of antimicrobial concentrations at the site of infection might be more relevant for predicting clinical response [11]. The epithelial lining fluid (ELF) is the most clinically relevant compartment to estimate intrapulmonary drug concentrations [12, 13]. In a murine pneumonia model for meropenem, a fT>MIC of 50% in ELF has been associated with logarithmic bacterial killing and suppression of resistant subpopulation amplification [14].
In this study, we investigated the utility of CI to achieve drug exposures in the human lung which are predicted to be effective and suppress the emergence of resistance. Specifically, we sought to (1) estimate plasma and ELF concentrations of meropenem in critically ill patients with nosocomial pneumonia receiving 3–6 g/day by CI, (2) describe the population pharmacokinetics (popPK) of meropenem in this population, and (3) estimate regimens that achieve effective drug exposures in ELF against Gram-negative pathogens with MIC likely to be encountered in patients with pneumonia.
Methods
Study design, settings, and patients
This was a single-center, prospective, open-label, randomized, comparative PK clinical trial that was conducted at a tertiary surgical intensive care unit (ICU) at the Parc de Salut Mar in Barcelona, Spain, between January 2017 and February 2019. The study was approved by the local ethics committee (Comitè Etic d’Investigació Clínica del Parc de Salut Mar; approval no. 2016/7125) and the Spanish Medicines and Health Product Agency (AEMPS; registration no. 16-0774). Furthermore, the trial was registered in the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT-no. 2016-002796-10). Written informed consent was obtained from the patients or their legal representatives.
The inclusion criteria were as follows: age ≥ 18 years, diagnosis of nosocomial pneumonia and risk factors for multidrug-resistant (MDR) pathogens [Pseudomonas aeruginosa, Acinetobacter spp., and extended-spectrum β-lactamases (ESBL)-producing Gram-negative Bacilli] [15], and glomerular filtration rate (GFR) ≥ 50 mL/min/1.73 m2 (estimated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula [16]). The exclusion criteria were allergy to β-lactams, previous use of carbapenems within 15 days, GFR < 50 mL/min/1.73 m2, severely impaired liver function [cirrhosis grade C by Child-Pugh classification [17]], obesity (body mass index (BMI) > 30), pregnancy, life expectancy < 3 days, and colonization [respiratory secretions or surveillance cultures (oropharyngeal and rectal swabs)] with pathogens known to be resistant to meropenem.
The diagnosis of pneumonia was based on standard clinical and laboratory criteria and defined as a new or progressive radiological pulmonary infiltrate plus two or more of the following characteristics: temperature > 38 °C or < 35 °C, leucocyte count > 11,000 or < 4000 cells/mm3, or purulent respiratory secretions [15].
Data collection
The following data were collected at the onset of treatment: demographics, BMI, Charlson Comorbidity Index [18], Acute Physiology and Chronic Health Evaluation II (APACHE) score [19] and SOFA score, type of pneumonia [hospital-acquired (HAP) or ventilator-associated (VAP)], sepsis or septic shock [20], presence of fluid overload, and mechanical ventilation. HAP was defined as pneumonia not incubating at the time of hospital admission and occurring ≥ 48 h after admission. VAP was defined as pneumonia occurring > 48 h after endotracheal intubation [15]. Laboratory data such as renal function (GFR and serum urea at baseline and on day 3), biomarkers [C-reactive protein (CRP), pro-calcitonin (PCT)], serum albumin, and total serum proteins were collected.
Clinical cure at the end of treatment, length of ICU and hospital stay (LOS), 7- and 30-day all-cause mortality, in-hospital mortality, and microbiological data (isolated organism, MIC value, and microbiological eradication) were recorded. Clinical cure was defined as the resolution of signs and symptoms present at enrolment and the resolution or lack of progression of radiological signs of pneumonia during follow-up (7 to 10 days after treatment initiation) [21]. Microbiological eradication was defined as the eradication of the microorganisms cultured from respiratory samples at baseline and at the end of treatment [22]. Seven and 30-day all-cause mortality was considered as death from any cause during the 7 or 30 days following the end of treatment, and in-hospital mortality was defined as death occurring during the hospital stay. Antibiotic susceptibility testing of the isolated pathogens was determined using the Vitek2® automated system (Biomerieux, France) and interpreted according to EUCAST breakpoints (European Committee on Antimicrobial Susceptibility Testing) [23]. In a few cases, susceptibility was confirmed by E-test (Biomerieux, France). Adverse events potentially associated with meropenem were collected such as local (inflammation, pain, phlebitis or edema at the injection site), cutaneous (rash, pruritus), gastrointestinal (diarrhea, nausea/vomiting, constipation), neurological (headache, insomnia, agitation, delirium, confusion, dizziness, seizure, nervousness, paresthesia, hallucinations, somnolence), drug-induced liver injury (increased alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, lactate dehydrogenase, bilirubin), or Clostridioides difficile-associated diarrhea (CDAD).
Randomization
Based on previous PK-related studies [24, 25], an initial sample size of 30 patients was considered for the desired level of significance. All included patients were randomized to receive 3 g or 6 g of i.v meropenem per day. Randomization was performed by an individual not related to the study using the SISA computer program (simple interactive statistical analysis). Randomization was balanced (1:1) without blocks or stratification. All patients received meropenem (Meropenem Accordpharma®; Accord Healthcare, S.L.U. Barcelona, Spain) at an initial loading dose (LD) of 2 g (in 50 mL of 0.9% saline infused by a central line in 15 min) followed by a CI of 3 g or 6 g/day (1 g or 2 g of meropenem over 8 h every 8 h). Meropenem by CI was diluted in 100 mL of 0.9% saline solution and injected into a central venous catheter via a volumetric pump (Braum Mesulgen, Mesulgen, Germany) with an infusion dead space of < 2 mL [26]. A maximum infusion time of 8 h was chosen based on the available meropenem stability data [27] and a stability study performed in our laboratory to confirm the previous results (data not shown). All patients received empirical combination therapy with 3 MIU/8 h of nebulized colistimethate sodium (CMS) (Accord®, Accord Healthcare, Barcelona, Spain). Nebulization was performed using a vibrating-mesh nebulizer (Aeroneb Pro®, Aerogen, Galway, Ireland) as it was described in a previous study [28].
Pharmacokinetic study
Blood and ELF samples were obtained after the third or fourth day of treatment once a steady state had been achieved. Blood samples were collected pre-infusion and at 1.5, 3, 6, and 8 h after the start of meropenem infusion. ELF samples were obtained simultaneously at 6 h post-infusion by bronchoalveolar lavage (BAL) during a standardized fiberoptic bronchoscopy using a bronchoscopic BAL catheter procedure (Combicath®, Prodimed, Le Plessis Bouchard, France) by instilling three aliquots of sterile 0.9% saline (20 mL, 40 mL, and 40 mL). The time between the beginning of BAL and the total recovery of the three aliquots did not exceed 2 min for each, in order to minimize the free diffusion of urea through the alveolar epithelium, which might lead to falsely elevated concentrations of urea in the BAL fluid [29]. The liquid recovered from the first aliquot was rejected, since it is not considered representative of ELF [30]. Blood and ELF samples were centrifuged at 4 °C, and the supernatant was frozen at − 80 °C until analysis.
Bioanalytical methods
Meropenem concentrations were measured using a validated high-performance liquid chromatography (HPLC) method [31] at the Pharmacy Department of Hospital del Mar. The assay was linear from 0.5 to 80 mg/L and 0.03 to 1 mg/L in the plasma and BAL, respectively. Precision and accuracy were ≤ 15% at high, medium, and low concentrations. The limit of quantification was 0.5 and 0.03 mg/L in the plasma and BAL, respectively. Normal serum saline (0.9%) was used to prepare the standard calibrators of meropenem in BAL.
Concentrations of urea in the plasma and ELF were determined with the Urea/BUN kit (Roche® professional Diagnostics, Mannheim, Germany) being the LOQ 3 mg/dL in the plasma and 0.078 mg/dL in ELF.
Meropenem concentration in ELF (MERELF) was determined according to the following formula, using urea as an endogenous marker, to correct the meropenem concentrations in ELF following dilution from the BAL [32, 33]:
where MERBAL is the meropenem concentration measured in BAL, UreaSER is the urea concentration in the plasma, and UreaBAL is the urea concentration in BAL.
Population pharmacokinetic model
Population pharmacokinetic modeling was performed using the nonparametric adaptive grid (NPAG) approach embedded in Pmetrics (Los Angeles, CA, USA) [34, 35]. One-, two-, and three-compartment structural models were fitted to the data and evaluated. Elimination from the central compartment and intercompartmental distribution were modeled as first-order processes. Data were weighted using the inverse of the estimated assay variance, and additional process noise was modeled using gamma as a multiplicative error term.
Age, gender, actual body weight (ABW), APACHE score, serum creatinine, GFR, serum albumin, total serum proteins, serum urea, CRP, PCT, presence of septic shock, presence of fluid overload, and mechanical ventilation were evaluated as covariates using stepwise linear regression. Potential covariates were separately entered into the model and retained if their inclusion resulted in a statistically significant improvement in the log likelihood value and/or in the observed-predicted plots.
The fit of each model to the data was assessed using a linear regression of observed-predicted values both before and after the Bayesian step. The mean prediction error and the mean bias-adjusted squared prediction error were used to assess bias and imprecision, respectively. Models were compared by calculating twice the difference in the log likelihood values.
The final model was also evaluated graphically and statistically by visual predictive checks (VPCs) performed from normalized prediction distribution errors (NPDEs) [36]. One thousand datasets were simulated using the final population model parameters. For the VPCs, the 5th, 50th, and 95th percentiles of the simulated concentrations were processed using the R platform, plotted against elapsed time, and compared to observed concentrations. For a model in which random effects are well estimated, approximately 90% of the observed data are expected to be within the 5th to 95th prediction interval. NPDE results were summarized graphically by default as provided by the NPDE R package (version 1.2) using (i) a Q-Q plot (where Q is quantile) of the NPDE and (ii) a histogram of the NPDE.
Other pharmacokinetic calculations
The average AUC in the plasma and ELF for each patient was estimated using the Bayesian posterior parametric estimates from the final model using the trapezoidal rule in Pmetrics. The daily average AUC (AUC0–24) was calculated by dividing the cumulative AUC of each patient by the total time in hours and multiplying the obtained result by 24 h. The partitioning of meropenem into ELF was described using the ratio of AUC0–24 h ELF/AUC0–24 h plasma.
Monte Carlo simulations
Monte Carlo simulations (n = 1000) of plasma concentrations were employed to calculate the fT≥MIC in ELF on the third day of treatment (from 48 to 72 h post-treatment) Three different meropenem regimens (2 g of LD followed by a maintenance dose of 1 g/8 h in CI, 2 g of LD followed by a maintenance dose of 2 g/8 h in CI, and 3 g of LD followed by a maintenance dose of 3 g/8 h in CI) against a range of MIC values (0.002–16 mg/L) were examined. Human protein binding of 2% in the plasma was used to estimate free drug concentrations in the plasma [37] while measured total concentrations in ELF were regarded as equivalent to the free fraction because protein binding is expected to be negligible [38]. A probability of target attainment (PTA) ≥ 90% was considered optimal. The AUC48–72 h in the plasma and ELF with two different dosages of meropenem were also simulated.
The potential toxicity of the different regimens was estimated by calculating the probability of achieving the threshold meropenem concentrations in the plasma associated with 50% risk of developing a neurotoxicity event, which has been defined as a minimum concentration (Cmin) in the plasma ≥ 64.2 mg/L [39].
Statistical analysis
Dichotomous variables were compared using the chi-square test and Fisher’s exact test. Quantitative data were expressed as medians [interquartile range (IQR)] and compared using the Mann-Whitney U test. Correlations were analyzed using Spearman’s correlation. A P value of < 0.05 was considered statistically significant. The SPSS (SPSS, Chicago, IL, USA) version 24.0 statistical package was used throughout.
Results
Enrolment and characteristics of study patients
Thirty-one patients were enrolled: 16 in the 1 g/8 h group and 15 in the 2 g/8 h group. A subject in the 1 g/8 h group was excluded due to problems with BAL sample processing. To compensate, an additional patient was recruited and randomized (Fig. 1). The demographic and clinical characteristics of both groups are summarized in Table 1. Most patients were male (81%), with a median (IQR) age of 72 (22) years and APACHE score of 15 (8). No significant differences in any demographic or clinical variable were observed between the two groups except for a higher Charlson score in the 2 g/8 h group.

Enrolment and follow-up of the study patients
In total, 25 pathogens (20 Gram-negative, 4 Gram-positive, and 1 virus) were isolated in respiratory cultures from 17 patients (9 in the 1 g/8 h group and 16 in the 2 g/8 h group). In 14 (45%) patients, no pathogen was isolated. Five patients had a polymicrobial infection. All patients with Gram-positive and viral isolates were in the 2 g/8 h group (2 methicillin-susceptible and 2 methicillin-resistant Staphylococcus aureus and 1 influenza B virus).
More patients in the 2 g/8 h group had a positive culture in BAL fluid (10/15 (66.7%) compared to 5/16 (31.3%) in the 1 g/8 h group (P = 0.049). Five patients had positive blood cultures with Gram-negative bacteria being 3 in the 1 g/8 h group and 2 in the 2 g/8 h group with no differences between the groups (P > 0.999). Only those patients with Gram-negative isolates were included in the PK/PD analysis. The Gram-negative isolates were 3 Haemophilus influenzae, 6 Enterobacteriaceae (2 susceptible Escherichia coli and 2 ESBL-E. coli, 1 Klebsiella pneumoniae, and 1 ESBL-K. pneumoniae), and 5 P. aeruginosa (2 multi-susceptible, 1 MDR, and 2 extensively drug-resistant P. aeruginosa). Microbiological data and distributions of MICs in the two groups are shown in Table 1. All included patients initially received an empirical combination therapy of intravenous meropenem plus 3 MIU/8 h of nebulized colistimethate sodium due to the high prevalence of MDR P. aeruginosa in our unit. After having the results of the microbiological cultures, meropenem was finally used empirically in 18 patients (58%) and as targeted therapy in 13 (42%) patients. In those cases with a MDR or XDR P. aeruginosa isolation, the initial combination therapy of meropenem plus nebulized CMS was maintained and monotherapy with meropenem was used only when a more susceptible Gram-negative bacteria were isolated. When a Gram-negative bacterium resistant to meropenem was cultured (2 patients in the 2 g/8 h group and 1 patient in the 1 g/8 h group), escalation was done being ceftolozano-tazobactam plus CMS the most frequent combination.
In seven patients clinical cure was not achieved (three in the 1 g/8 h group and four in the 2 g/8 h group), but only two of them died, one in each group. In three patients, the initial treatment with meropenem was changed by ceftolozane-tazobactam with late favorable clinical response (one in the 1 g/8 h group and two in the 2 g/8 h group). In two patients, the clinical cure was achieved after a long treatment of meropenem plus nebulized CMS plus linezolid (one of each group). Finally, the other two died, one in the 1 g/8 h group due to a stroke on the eighth day of treatment with no documented clinical cure at that point and the other in the high dose group due to MRSA pneumonia.
No adverse events related to meropenem treatment were observed in any patient. The BAL procedure was well tolerated in all cases.
Pharmacokinetic data
The concentration-time profiles of meropenem in the plasma and ELF in both groups are shown in Fig. 2. Median (IQR) meropenem plasma concentrations in the 2 g/8 h group were statistically higher at all times points compared to the 1 g/8 h group. In ELF, concentrations were also higher in the 2 g/8 h group, but this difference did not reach statistical significance (6.6 (8.3) mg/L vs 3.9 (3.2) mg/L), respectively (P = 0.102). A high interindividual variability was observed in ELF exposure within both groups.

Meropenem plasma and ELF concentration-time profiles. Meropenem plasma and ELF concentration time profiles of patients receiving a loading dose of 2000 mg i.v followed by a maintenance dose of 1 g q8h and 2 g q8h i.v. Intensive sampling was performed after the second day of treatment. A statistically significant difference was observed in plasma concentrations at any time points between the two groups (meropenem plasma concentrations of 12.9 (8.4) vs 23.0 (12.8) at pre-dose; 13.4 (7.3) vs 23.9 (16.7) at 1.5 h; 13.3 (8.3) vs 21.4 (15.5) at 3 h; 11.7 (8.2) vs 22.4 (15.3) at 6 h, and 12.3 (9.1) vs 18.9 (17.4) at 8 h, in the 1 g/8 h and 2 g/8 h groups, respectively).
Population pharmacokinetic model
A total of 151 meropenem plasma concentrations and 30 ELF concentrations were included in the population analysis. A 3-compartment linear model, with zero-order input and first-order clearance from the central compartment, best described the data. Concentrations of meropenem in ELF were modeled by assuming ELF was a homogenous compartment with volume, VELF. Compartments were connected by first-order intercompartmental rate constants.
Despite different covariates, such as BMI, serum creatinine, and GFR, having a relationship with the estimated clearance, they were not included in the final model because they did not improve the goodness-of-fit. Estimates for central tendency, dispersion, and 95% credibility limits for the population PK parameters are shown in Table 2.
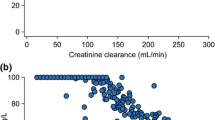
The observed-predicted values for meropenem concentrations in the plasma and ELF before and after the Bayesian step are shown in Fig. 3. After maximum a posteriori probability (MAP)-Bayesian estimation, a linear regression of the observed-vs-predicted values in the plasma had an intercept and slope of 0.0159 (CI95 − 0.685–0.717) and 1.02 (CI95 0.984–1.05), respectively, and an R2 = 0.956. The bias and imprecision were both acceptable (bias = − 0.0803 mg/L and imprecision 1.23 mg/L). For ELF, the observed-vs-predicted plot had an intercept and slope of − 0.0784 (CI95 − 0.211–0.0546) and 1.00 (CI95 0.988–1.02), respectively, and an R2 = 0.999. The bias and imprecision were both acceptable (bias = 0.114 mg/liter and imprecision 0.189 mg/L).

Population and individual predicted meropenem concentrations vs observed meropenem concentrations in the plasma and in ELF. Population (a) and individual (b) predicted meropenem concentrations vs observed concentrations of meropenem in the plasma (a and b, respectively) and in ELF (c and d, respectively). The broken line is the line of identity (observed = predicted concentrations)
Visual predictive check plots of the different doses (1 g/8 h and 2 g/8 h) in the plasma and ELF based on 1000 simulations with the final model are given in Fig. 7. The normal distribution of normalized prediction distribution errors (NPDEs) in the plasma and ELF confirmed the adequacy of the model for dosing simulations. Median (IQR) average values of AUC0–24 in the plasma and ELF estimated using the Bayesian posterior parametric estimates for each patient are shown in Table 3. Both AUC0–24, in the plasma and ELF, were statistically higher in the 2 g/8 h group than in the 1 g/8 h group. The penetration ratio was similar between the groups.
The AUC0–24 in ELF showed a moderate positive linear correlation with AUC0–24 in the plasma, age, and APACHE score (Spearman rho = 0.533, 0.575, and 0.537, respectively) (P < 0.05) and an inverse correlation with patients’ ABW, BMI, and GFR (Spearman rho = − 0.688, − 0.598, and − 0.376, P < 0.05). Figure 4 shows the comparison between the individual predicted AUC48–72 h using the Bayesian posteriors (red diamonds) and the simulated AUC48–72 h (black diamonds) in the plasma and ELF with two different dosages of meropenem.

Comparison of the individual predicted AUC48–72 h and the simulated AUC48–72 h in the plasma and ELF. Comparison of the individual predicted AUC48–72 h using the Bayesian posterior estimated concentrations (red diamonds) and the simulated AUC48–72 h (black diamonds) in the plasma and ELF with two different dosages of meropenem (2 g loading dose (LD) followed by 1 g/8 h as a continuous infusion (a) and 2 g LD followed by 2 g/8 h as a continuous infusion (b))
Probability of target attainment
The probability of target attainment (PTA) for achieving 50% fT>MIC in ELF for the three different meropenem doses on day 3 of treatment is shown in Fig. 5. With the lowest dose (2 g LD + 1 g/8 h by CI), an optimal PTA could be achieved for isolates with MICs of < 2 mg/L. With the administration of a double maintenance dose (2 g LD + 2 g/8 h), a PTA ≥ 90% in ELF could be attained for isolates with MIC up to 2 mg/L, which is the current susceptibility breakpoint [23]. A dosage increases to 3 g LD + 3 g/8 h by CI did not result in significantly greater coverage of MIC. We also estimated the dose needed for isolates with intermediate susceptibility (MIC between 2 and ≤ 8 mg/L) that was estimated to be as high as 8 g/8 h, which is four times higher than the maximum licensed meropenem dose. Figure 6 shows the simulated meropenem concentration-time profiles in ELF of each tested regimen.

Probability of target attainment (PTA) in ELF of different dosages regimens. PTA in ELF of different dosages of meropenem: 2 g loading dose (LD) followed by 1 g/8 h, 2 g LD followed by 2 g/8 h, and 3 g LD followed by 3 g/8 h; administered as a continuous infusion during the third day of treatment (from 48 to 72 h after the start of the treatment)

Comparison of the time course of meropenem concentrations simulated in ELF. Comparison of the median, percentile 5th and 95th time course of meropenem concentrations simulated in ELF during 4 days with different dosing regimens of meropenem as a continuous infusion (2 g loading dose (LD) followed by 1 g/8 h (a), 2 g LD followed by 2 g/8 h (b), and 3 g LD followed by 3 g/8 h (c))
Similar results were obtained with the administration of meropenem in an extended infusion of 4 h. The probability of target attainment (PTA) for achieving 50% fT>MIC in ELF was also assessed on day 3 of treatment for three different meropenem doses (1 g/8 h, 2 g/8 h, and 3 g/8 h) administered by extended infusion (4 h) (Fig. 7). With the lowest dose of 1 g/8 h, an optimal PTA could be achieved for isolates with MICs of < 2 mg/L, and with higher doses (2 g/8 h and 3 g/8 h), the coverage increased to a MIC up to 2 mg/L.

Visual predictive check plot for meropenem concentrations. Visual predictive check plot for meropenem concentrations at a dose of 2 g loading dose + 1 g/8 h (a) and at a dose of 2 g loading dose + 2 g/8 h (b) in the plasma and ELF (left and right, respectively). Observed concentrations (blue circles); simulated concentrations at the designated quantile given by the number on the line (lines)
From the point of view of toxicity, the probability of achieving a Cmin in the plasma of meropenem ≥ 64.2 mg/L during the first 3 days of treatment was estimated to be 0% for the two lowest doses and 1.7% for the 3 g/8 h dose. In comparison, the use of the highest dose (8 g/8 h) resulted in nearly half of the patients (49.0%) achieving this potentially toxic trough concentration.
PK/PD in ELF and clinical outcomes
Twelve patients with documented Gram-negative bacterial infections were eligible for the PK-PD sub-study.
All patients that achieved clinical cure had a fT>MIC > 50% in ELF in both groups, compared to patients who failed treatment (fT>MIC 33.3%, P = 0.045). Regarding the microbiological results, a higher proportion of patients with eradication achieved an optimal ELF target, but this difference was not statistically significant (88.9% vs 66.7%, P = 0.455). No correlation was found between the duration of mechanical ventilation (days) and meropenem ELF or plasma meropenem concentrations (data not shown).
Discussion
Meropenem is a licensed agent for the treatment of nosocomial pneumonia [15]. As for other β-lactams, the pharmacodynamics of meropenem is optimized with the use of prolonged infusions, especially CI [6,7,8]. In recent years, higher meropenem dosages are being recommended to avoid suboptimal exposure [40], but the clinical benefits are still unknown.
Several studies have identified the administration of carbapenems as an independent risk factor for the emergence of carbapenem-resistant Gram-negative bacteria in ICU patients [41, 42]. In this scenario, one proposed strategy to minimize the emergence of resistance of meropenem is the administration to achieve sufficient drug exposures to kill both susceptible and prevent the emergence of resistant subpopulations [43, 44]. Tam et al. reported that selective amplification of subpopulations of P. aeruginosa with reduced susceptibilities to meropenem was suppressed with a Cmin/MIC of ≥ 6.2 in the plasma [43]. In fact, some authors consider that no single agent can achieve an adequate exposure to provide a cell kill sufficient to allow optimal clinical outcomes and simultaneously suppress amplification of less susceptible subpopulations of organisms in infections caused by some bacteria, such as P. aeruginosa [44].
In pneumonia, a PK/PD target at the site of the infection of fT>MIC of 50% in ELF has been associated with bacterial killing and suppression of resistant subpopulation amplification in a murine model of pneumonia [14]. Unlike other PK/PD targets calculated in the plasma [45, 46], this has been more precisely defined at the site of the infection. This was the pharmacodynamic target used in this analysis and is higher than that often cited for the efficacy of the carbapenem class.
Several studies have assessed the PK/PD of meropenem in ELF [10, 47,48,49], but only two of them have focused on critically ill patients [10, 48]. Both studies assessed ELF meropenem concentration and lung penetration with the use of both intermittent and 3-h extended infusion. Although a higher penetration ratio (AUCELF/AUCplasma) of meropenem was observed with the use of an extended infusion, none of the two regimens achieved an optimal PK/PD target in ELF [48].
To our knowledge, this is the first population PK study of meropenem in critically ill patients with nosocomial pneumonia which compared two different dosages administered as a CI. Our results showed that the administration of 2 g/8 h of meropenem as a CI allowed to achieve higher meropenem concentrations in the plasma and ELF compared to the 1 g/8 h dose, although a high interindividual variability in the meropenem concentrations in ELF was observed. However, even the administration of the highest licensed dose (2 g/8 h) of meropenem by CI in patients with conserved renal function did not result in an optimal ELF target attainment for a substantial fraction of the population.
The median penetration ratio into the lungs was approximately 30% and was comparable between both groups. Similar penetration ratios (AUCELF/AUCplasma) have been reported in other contexts [10, 14, 48]. Lodise et al. simulated a penetration of 26% in patients with VAP after the administration of a single dose of 2 g meropenem given over 3 h [10], and the authors also reported a substantial variability in the lung penetration ratio (10th and 90th percentiles of 3.7% and 178.0%, respectively). In the same way, the PROMESSE study performed in 55 critically ill patients with severe pneumonia treated with 1 g/8 h reported a statistically higher AUC penetration ratio in the extended infusion group (3 h) compared to the intermittent group [mean (SD) 29 (± 3) % vs 20 (± 3) % (P = 0.047)] [48]. In our study, all ELF samples were obtained at the same time; hence, a precise estimate of the concentration-time profile of meropenem in ELF was not possible.
In our study, AUC in ELF was positively correlated with AUC in the plasma, suggesting that plasma exposures are a potential surrogate marker of lung exposures. However, the correlation was not especially strong, and plasma concentrations cannot be used to confidently predict lung concentrations. Although we acknowledge that routine measurement of drug levels in ELF is infeasible in all critically ill patients with pneumonia, direct lung measurement in those patients at risk of treatment failure (especially if pathogens with high MIC values are suspected) may be reasonable and should be considered.
In our study, a higher proportion of patients with clinical cure achieved an optimal PK/PD ratio at the infection site compared to those who failed. Although our findings suggest an association between the achievement of an optimal PK/PD of meropenem in lungs and better clinical outcomes in patients with Gram-negative respiratory infections, this was not an objective in our study. In addition, all patients received concomitant treatment with nebulized CMS, which could have influenced patients’ clinical outcomes, especially on microbiological eradication. Due to all these reasons, our results have to be confirmed in a larger prospective clinical study.
Our work is the first study assessing the achievement of a PK/PD target at the site of infection with the use of different dosages of meropenem administered by CI. Our results suggest that an optimal PTA can be achieved for MIC values < 2 mg/L with a dose of 1 g/8 h and for MIC values < 4 mg/L with a higher dose of 2 g/8 h. A lower coverage (a lower MIC dilution) was reported with the use of the same doses of meropenem administered by extended infusion (over 3 h) in the PROMESSE study. The authors used a similar target, 54% fT>MIC in ELF, a value associated with microbiological response in a clinical study of Li et al. [50], and observed that an optimal PTA could only be attained for MIC breakpoints of 0.5 mg/L and 1 mg/L with meropenem doses of 1 g/8 h and 2 g/8 h, respectively, administered by extended infusion (3 h). Similarly, Drusano et al. confirmed that even with the highest licensed meropenem dose, the 50% fT>MIC in ELF target could not be achieved even at very low MIC values (0.25 mg/L) [14]. Although the administration of meropenem by CI seems to improve drug exposure in ELF compared with extended infusion [48], it might not be sufficient to cover all intermediate Gram-negative pathogens causing nosocomial pneumonia in critically ill patients with conserved renal function. In those cases, alternative strategies may be required, especially when pathogens with high MIC values (> 2 mg/L) may be present. In fact, we estimate the meropenem dose by CI needed to achieve an optimal PTA for all considered intermediate strains (MIC between 2 and 8 mg/L) [23] that would have to be as high as 8 g/8 h, a dose that is four times higher than the highest approved meropenem dose, and that is related to a high probability of toxicity.
Our study has several limitations. Firstly, it is a relatively small single-center study. Secondly, all ELF measurements were performed at a single time point. The collection of ELF samples at different times would have allowed a more precise determination of the concentration-time profile in ELF. However, a recent study demonstrated that ELF models constructed with concentrations from sparse ELF sampling time points result in exposure estimates similar to those constructed from robustly sampled ELF profiles [51]. Thirdly, all samples were collected on the same day, so intraindividual variability during the treatment period could not be measured [52]. ELF samples were all collected in the infected lung; as distribution of inflammation is heterogeneous, the collection of samples in the clear lung could have led us to know the differences in the antibiotic diffusion. Finally, the relatively limited sample size and the use of combination therapy with nebulized CMS made difficult to correlate the PK/PD target in ELF with clinical and microbiological outcomes. Nevertheless, this study provides important and useful information about the meropenem dosages that should be used in clinical practice for treating nosocomial pneumonia caused by Gram-negative bacteria in critically ill patients, considering both the achievement of clinical cure and possibly the prevention of the emergence of resistance.
Conclusions
In conclusion, the administration of meropenem by continuous infusion improves drug exposure in the ELF, but the use of the highest licensed dose (2 g/8 h) is still needed to achieve a target attainment in ELF of > 90% for isolates with an MIC up to 2 mg/L in patients with conserved renal function. Alternative therapeutic strategies may be required for the treatment of nosocomial pneumonia caused by Gram-negative bacteria in critically ill patients when MDR strains with high MIC values are suspected.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABW:
-
Actual body weight
- APACHE:
-
Acute Physiology and Chronic Health Evaluation
- BAL:
-
Bronchoalveolar lavage
- BMI:
-
Body mass index
- CDAD:
-
Clostridioides difficile-associated diarrhea
- CI:
-
Continuous infusion
- CI95:
-
Confidence interval of 95%
- CKD-EPI:
-
Chronic Kidney Disease Epidemiology Collaboration
- C min :
-
Minimum concentration
- CMS:
-
Colistimethate sodium
- CRP:
-
C-reactive protein
- ELF:
-
Epithelial lining fluid
- ESBL:
-
Extended-spectrum beta-lactamases
- EUCAST:
-
European Committee on Antimicrobial Susceptibility Testing
- fT>MIC:
-
Percentage of time remaining concentration above MIC
- GFR:
-
Glomerular filtration rate
- HAP:
-
Hospital-acquired pneumonia
- HPLC:
-
High-performance liquid chromatography
- ICU:
-
Intensive care unit
- IQR:
-
Interquartile range
- LD:
-
Loading dose
- LOS:
-
Length of hospital stay
- MDR:
-
Multidrug-resistant
- MERBAL :
-
Meropenem concentration in BAL
- MERELF :
-
Meropenem concentration in ELF
- MIC:
-
Minimal inhibitory concentration
- PCT:
-
Pro-calcitonin
- PD:
-
Pharmacodynamic
- PK:
-
Pharmacokinetic
- popPK:
-
Population pharmacokinetic
- PTA:
-
Probability of target attainment
- SD:
-
Standard deviation
- SOFA:
-
Sequential Organ Failure Assessment
- UreaBAL :
-
Urea concentration in BAL
- UreaSER :
-
Urea concentration in the plasma
- VAP:
-
Ventilator-associated pneumonia
- XDR:
-
Extensively drug-resistant
References
Nicolau DP. Pharmacokinetic and pharmacodynamic properties of meropenem. Clin Infect Dis. 2008;47(Suppl 1):32–40.
Roberts JA, Lipman J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit Care Med. 2009;37:840–51.
Drusano GL. Antimicrobial pharmacodynamics: critical interactions of “bug and drug”. Nat Rev Microbiol. 2004;2:289–300.
Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26:1–10.
Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, Hope WW, Farkas A, Neely MN, Schentag JJ, Drusano G, Frey OR, Theuretzbacher U, Kuti JL, International Society of Anti-Infective Pharmacology and the Pharmacokinetics and Pharmacodynamics Study Group of the European Society of Clinical Microbiology and Infectious Diseases. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis. 2014;14:498–509.
Roberts JA, Abdul-Aziz MH, Davis JS, Dulhunty JM, Cotta MO, Myburgh J, Bellomo R, Lipman J. Continuous versus intermittent β-lactam infusion in severe sepsis: a meta-analysis of individual patient data from randomized trials. Am J Respir Crit Care Med. 2016;194:681–91.
Dulhunty JM, Roberts JA, Davis JS, Webb SA, Bellomo R, Gomersall C, Shirwadkar C, Eastwood GM, Myburgh J, Paterson DL, Starr T, Paul SK, Lipman J, BLING II Investigators for the ANZICS Clinical Trials Group. A multicenter randomized trial of continuous versus intermittent β-lactam infusion in severe sepsis. Am J Respir Crit Care Med. 2015;192:1298–305.
Abdul-Aziz MH, Lipman J, Akova M, Bassetti M, De Waele JJ, Dimopoulos G, Dulhunty J, Kaukonen KM, Koulenti D, Martin C, Montravers P, Rello J, Rhodes A, Starr T, Wallis SC, Roberts JA, DALI Study Group. Is prolonged infusion of piperacillin/tazobactam and meropenem in critically ill patients associated with improved pharmacokinetic/pharmacodynamic and patient outcomes? An observation from the Defining Antibiotic Levels in Intensive care unit patients (DALI) cohort. J Antimicrob Chemother. 2016;71:196–207.
Huttner A, Harbarth S, Hope WW, Lipman J, Roberts JA. Therapeutic drug monitoring of the β-lactam antibiotics: what is the evidence and which patients should we be using it for? J Antimicrob Chemother. 2015;70:3178–83.
Lodise TP, Sorgel F, Melnick D, Mason B, Kinzig M, Drusano GL. Penetration of meropenem into epithelial lining fluid of patients with ventilator-associated pneumonia. Antimicrob Agents Chemother. 2011;55:1606–10.
Ambrose PG, Bhavnani SM, Ellis-Grosse EJ, Drusano GL. Pharmacokinetic-pharmacodynamic considerations in the design of hospital-acquired or ventilator-associated bacterial pneumonia studies: look before you leap! Clin Infect Dis. 2010;51(Suppl 1):103–10.
Boselli E, Breilh D, Saux MC, Gordien JB, Allaouchiche B. Pharmacokinetics and lung concentrations of ertapenem in patients with ventilator-associated pneumonia. Intensive Care Med. 2006;32:2059–62.
Rodvold KA, George JM, Yoo L. Penetration of anti-infective agents into pulmonary epithelial lining fluid: focus on antibacterial agents. Clin Pharmacokinet. 2011;50:637–64.
Drusano GL, Lodise TP, Melnick D, Liu W, Oliver A, Mena A, VanScoy B, Louie A. Meropenem penetration into epithelial lining fluid in mice and humans and delineation of exposure targets. Antimicrob Agents Chemother. 2011;55:3406–12.
Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, Napolitano LM, O’Grady NP, Bartlett JG, Carratalà J, El Solh AA, Ewig S, Fey PD, File TM Jr, Restrepo MI, Roberts JA, Waterer GW, Cruse P, Knight SL, Brozek JL. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63:e61–111.
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–12.
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9.
Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic in longitudinal studies: development and validation. J Chronic Dis. 1987;40:373–83.
Giangiuliani G, Mancini A, Gui D. Validation of a severity of illness score (APACHE II) in a surgical intensive care unit. Intensive Care Med. 1989;15:519–22.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315:801–10.
Weiss E, Zahar JR, Alder J, Asehnoune K, Bassetti M, MJM B, Chastre J, De Waele J, Dimopoulos G, Eggimann P, Engelhardt M, Ewig S, Kollef M, Lipman J, Luna C, Martin-Loeches I, Pagani L, Palmer LB, Papazian L, Poulakou G, Prokocimer P, Rello J, Rex JH, Shorr AF, Talbot GH, Thamlikitkul V, Torres A, Wunderink RG, Timsit JF. Elaboration of consensus clinical endpoints to evaluate antimicrobial treatment efficacy in future hospital-acquired/ventilator-associated bacterial pneumonia clinical trials. Clin Infect Dis. 2019;69:1912–8.
Muller AE, Punt N, Moutona JW. Exposure to ceftobiprole is associated with microbiological eradication and clinical cure in patients with nosocomial pneumonia. Antimicrob Agents Chemother. 2014;58:2512–9.
Clinical Breakpoints-Bacteria. http://www.eucast.org/clinical_breakpoints/. Accessed 8 Jan 2019.
Roberts JA, Kirkpatrick CM, Lipman J. Monte Carlo simulations: maximizing antibiotic pharmacokinetic data to optimize clinical practice for critically ill patients. J Antimicrob Chemother. 2011;66:227–31.
Roberts JA, Kirkpatrick CM, Roberts MS, Robertson TA, Dalley AJ, Lipman J. Meropenem dosing in critically ill patients with sepsis and without renal dysfunction: intermittent bolus versus continuous administration? Monte Carlo dosing simulations and subcutaneous tissue distribution. J Antimicrob Chemother. 2009;64:142–50.
Lam WJ, Bhowmick T, Gross A, Vanschooneveld TC, Weinstein MP. Using higher doses to compensate for tubing residuals in extended-infusion piperacillin-tazobactam. Ann Pharmacother. 2013;47:886–91.
Berthoin K, Le Duff CS, Marchand-Brynaert J, Carryn S, Tulkens PM. Stability of meropenem and doripenem solutions for administration by continuous infusion. J Antimicrob Chemother. 2010;65:1073–5.
Benítez-Cano A, de Antonio-Cuscó M, Luque S, Sorlí L, Carazo J, Ramos I, Bermejo S, Campillo N, Horcajada JP, Samsó E, Grau S. Systemic pharmacokinetics and safety of high doses of nebulized colistimethate sodium in critically ill patients with hospital-acquired and ventilator-associated pneumonia. J Antimicrob Chemother. 2019;74:3268–73.
Chiu LM, Amsden GW. Intrapulmonary pharmacokinetics of antibacterial agents: implications for therapeutics. Am J Respir Med. 2002;1:201–9.
Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P, Saltini C, Selman M, Strange C, Wood B, American Thoracic Society Committee on BAL in Interstitial Lung Disease. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004–14.
Huwyler T, Lenggenhager L, Abbas M, Ing Lorenzini K, Hughes S, Huttner B, Karmime A, Uçkay I, von Dach E, Lescuyer P, Harbarth S, Huttner A. Cefepime plasma concentrations and clinical toxicity: a retrospective cohort study. Clin Microbiol Infect. 2017;23:454–9.
Dargaville PA, South M, Vervaart P, McDougall PN. Validity of markers of dilution in small volume lung lavage. Am J Resp Crit Care Med. 1999;160:778–84.
Yamazaki K, Ogura S, Ishizaka A, Oh-hara T, Nishimura M. Bronchoscopic microsampling method for measuring drug concentration in epithelial lining fluid. Am J Resp Crit Care Med. 2003;168:1304–7.
Tatarinova T, Neely M, Bartroff J, van Guilder M, Yamada W, Bayard D, Jelliffe R, Leary R, Chubatiuk A, Schumitzky A. Two general methods for population pharmacokinetic modeling: non-parametric adaptive grid and non-parametric Bayesian. J Pharmacokinet Pharmacodyn. 2013;40:189–99.
Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34:467–76.
Comets E, Brendel K, Mentré F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Prog Biomed. 2008;90:154–66.
Meropenem-Product data Sheet. https://cima.aemps.es/cima/publico/lista.html. Accessed 12 June 2019.
Kiem S, Schentag JJ. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother. 2008;52:24–36.
Imani S, Buscher H, Marriott D, Gentili S, Sandaradura I. Too much of a good thing: a retrospective study of β-lactam concentration-toxicity relationships. J Antimicrob Chemother. 2017;72:2891–7.
Roberts JA, Paul SK, Akova M, Bassetti M, De Waele JJ, Dimopoulos G, Kaukonen KM, Koulenti D, Martin C, Montravers P, Rello J, Rhodes A, Starr T, Wallis SC, Lipman J, DALI Study. DALI: defining antibiotic levels in intensive care unit patients: are current β-lactam antibiotic doses sufficient for critically ill patients? Clin Infect Dis. 2014;58:1072–83.
Armand-Lefèvre L, Angebault C, Barbier F, Hamelet E, Defrance G, Ruppé E, Bronchard R, Lepeule R, Lucet JC, El Mniai A, Wolff M, Montravers P, Plésiat P, Andremont A. Emergence of imipenem-resistant gram-negative bacilli in intestinal flora of intensive care patients. Antimicrob Agents Chemother. 2013;57:1488–95.
Mentzelopoulos SD, Pratikaki M, Platsouka E, Kraniotaki H, Zervakis D, Koutsoukou A, Nanas S, Paniara O, Roussos C, Giamarellos-Bourboulis E, Routsi C, Zakynthinos SG. Prolonged use of carbapenems and colistin predisposes to ventilator-associated pneumonia by pandrug-resistant Pseudomonas aeruginosa. Intensive Care Med. 2007;33:1524–32.
Tam VH, Schilling AN, Neshat S, Poole K, Melnick DA, Coyle EA. Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49:4920–7.
Louie A, Grasso C, Bahniuk N, Van Scoy B, Brown DL, Kulawy R, Drusano GL. The combination of meropenem and levofloxacin is synergistic with respect to both Pseudomonas aeruginosa kill rate and resistance suppression. Antimicrob Agents Chemother. 2010;54:2646–54.
Bergen PJ, Bulitta JB, Kirkpatrick CMJ, Rogers KE, McGregor MJ, Wallis SC, Paterson DL, Nation RL, Lipman J, Roberts JA, Landersdorfer CB. Substantial impact of altered pharmacokinetics in critically ill patients on the antibacterial effects of meropenem evaluated via the dynamic hollow-fiber infection model. Antimicrob Agents Chemother. 2017;61:e02642–16.
Tam VH, Chang KT, Zhou J, Ledesma KR, Phe K, Gao S, Van Bambeke F, Sánchez-Díaz AM, Zamorano L, Oliver A, Cantón R. Determining β-lactam exposure threshold to suppress resistance development in Gram-negative bacteria. J Antimicrob Chemother. 2017;72:1421–8.
Allegranzi B, Cazzadori A, Di Perri G, Bonora S, Berti M, Franchino L, Biglino A, Cipriani A, Concia E. Concentrations of single-dose meropenem (1 g iv) in bronchoalveolar lavage and epithelial lining fluid. J Antimicrob Chemother. 2000;46:319–22.
Frippiat F, Musuamba FT, Seidel L, Albert A, Denooz R, Charlier C, Van Bambeke F, Wallemacq P, Descy J, Lambermont B, Layios N, Damas P, Moutschen M. Modelled target attainment after meropenem infusion in patients with severe nosocomial pneumonia: the PROMESSE study. J Antimicrob Chemother. 2015;70:207–16.
Conte JE Jr, Golden JA, Kelley MG, Zurlinden E. Intrapulmonary pharmacokinetics and pharmacodynamics of meropenem. Int J Antimicrob Agents. 2005;26:449–56.
Li C, Du X, Kuti JL, Nicolau DP. Clinical pharmacodynamics of meropenem in patients with lower respiratory tract infections. Antimicrob Agents Chemother. 2007;51:1725–30.
Motos A, Kuti JL, Li Bassi G, Torres A, Nicolau DP. Is one sample enough? β-lactam target attainment and penetration into epithelial lining fluid based on multiple bronchoalveolar lavage sampling time points in a swine pneumonia model. Antimicrob Agents Chemother. 2019;63:e01922–18.
Zander J, Döbbeler G, Nagel D, Maier B, Scharf C, Huseyn-Zada M, Jung J, Frey L, Vogeser M, Zoller M. Piperacillin concentration in relation to therapeutic range in critically ill patients - a prospective observational study. Crit Care. 2016;20:79.
Acknowledgements
We gratefully received support from the Instituto de Salud Carlos III (ISCIII) (grant number BA18/00005), the Spanish Society of Infectious Diseases and Clinical Microbiology (SEIMC), and the Spanish Society of Hospital Pharmacy (SEFH) to do this work thanks to the research travel grant received by SL.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
AB-C made substantial contributions to the conception, study design, data collection, sample and data analysis, interpretation of the data, and drafting of the manuscript. SL made substantial contributions to the study design, sample and data analysis, interpretation of the data, and drafting of the manuscript. LS and ES made substantial contributions in revising the manuscript for intellectual content. JC and IR made substantial contributions to the conception, data collection, analysis and interpretation of the data, and drafting of the manuscript. NC and CV made substantial contributions to the study design, data collection, sample analysis, and interpretation of the data. VC and AS-F made substantial contributions to the conception, study design, data collection, sample analysis, and interpretation of the data. JPH and RA made substantial contributions in revising the manuscript for intellectual content. SB made substantial contributions to the conception, study design, data collection, and data analysis. WH and SG made substantial contributions to the concept, study designs, sample and data analysis, interpretation of the data, drafting and revising of the manuscript for intellectual content. All authors approved the final version to be published.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethics approval was obtained from the local ethics committee (Comitè Etic d’Investigació Clínica del Parc de Salut Mar; approval no. 2016/7125). Written informed consent was obtained either from the patient or their appointed legal guardian.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This manuscript will be part of a PhD thesis for the Universitat Autònoma de Barcelona.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Benítez-Cano, A., Luque, S., Sorlí, L. et al. Intrapulmonary concentrations of meropenem administered by continuous infusion in critically ill patients with nosocomial pneumonia: a randomized pharmacokinetic trial. Crit Care 24, 55 (2020). https://doi.org/10.1186/s13054-020-2763-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-020-2763-4