
Abstract
This article is one of ten reviews selected from the Annual Update in Intensive Care and Emergency Medicine 2015 and co-published as a series in Critical Care. Other articles in the series can be found online at http://ccforum.com/series/annualupdate2015. Further information about the Annual Update in Intensive Care and Emergency Medicine is available from http://www.springer.com/series/8901.
Similar content being viewed by others


Introduction
Renal disease is a global phenomenon with the incidence of both acute and chronic renal insufficiency continuing to rise [1,2]. Acute kidney injury (AKI) is a known independent predictor of hospital mortality despite its multifactorial nature. After an episode of AKI, there are four potential outcomes [3]:
-
I.
full recovery of renal function to baseline;
-
II.
incomplete recovery of renal function resulting in chronic kidney disease (CKD);
-
III.
exacerbation of pre-existing CKD accelerating progression towards end-stage renal failure (ESRF);
-
IV.
non-recovery of function leading to ESRF.
It was previously assumed that those who recovered kidney function after an episode of AKI were faced with a relatively benign course with favorable outcomes. However, there is now increasing concern that this is not neccesarily the case and these individuals may be at risk of poor long-term outcomes through the development of CKD (including ESRF), further episodes of AKI and an increased risk of premature death. In the following review, we will describe the main pathogenetic links between AKI and CKD and introduce some potential key players.
Long-term outcomes after acute kidney injury
The observation that AKI and CKD may be intimately linked has been the subject of several recent studies [4-8]. However, as is often the case, demonstration of a clear association does not necessarily confer causation. Indeed, epidemiological studies often struggle to identify accurate pre-morbid and post-AKI renal function in order to precisely interpret long-term data. For example, in retrospective studies follow‐up data may be missing or may have been captured at times of intercurrent illness, hence blunt endpoints, such as dialysis dependence or mortality, are used. In addition, serum creatinine and the derived estimated glomerular filtration rate (eGFR) are the only markers of renal function used in routine clinical practice. Their limitations are well known, and they may not accurately reflect renal function. Critical illness in particular, may be associated with significant decreases in serum creatinine through many potential mechanisms and these changes may persist through to hospital discharge hence confounding assessment of renal function [9]. Moreover, elevated serum creatinine levels at hospital discharge may represent pre-existing CKD rather than non-recovery, depending on the completeness of data availability.
Early studies suggesting a link between AKI and CKD were hindered by sample size as well as selection of population groups but recent studies are based on larger cohorts with longer follow-up data. For example, Lo et al. retrospectively analyzed more than 500,000 patients with a baseline pre-admission eGFR > 45 ml/min2 who survived a stay in hospital [7]: 343 patients with dialysis-dependent AKI survived their ICU stay and were still dialysis-free at 30 days. Comparison between this cohort and patients without dialysis-require AKI demonstrated an increased risk of CKD stage 4 or 5 of 1.7/100 person-years in the non-AKI group and 47.9/100 person-years in the AKI group (adjusted hazard ratio [HR] 28.1; 95% confidence interval [CI] 21.1–37.6). Of note, 41 patients developed long-term dialysis dependency and all stemmed from the AKI group. Similarily, Wald et al. compared 3,769 adults who received renal support after an episode of AKI to 13,598 matched controls who did not require acute renal replacement therapy (RRT) [10]. After a median follow-up of 3 years, the incidence of chronic dialysis in the AKI cohort was 2.63/100 person-years compared to 0.91/100 person-years among control participants (adjusted HR, 3.23; 95% CI, 2.70–3.86).
Interrogation of large databases continues to support the hypothesis that an AKI event heralds an increased risk of CKD. Using the Medicare database in the US, Ishani et al. identified patients ≥ 67 years old over a 2-year period [11]. More than 200,000 patients who survived to hospital discharge were included with patients categorized as having AKI alone, CKD alone, AKI on background of CKD, or neither. The development of ESRF at 2 years was identified by cross-reference with the US Renal Data System. Predictably, when compared to patients with neither CKD nor AKI, the highest risk of ESRF was for those with acute-on-chronic kidney disease (adjusted HR 41.19; 95% CI 34.58–49.08). Interestingly, patients with AKI alone had a significantly higher risk of developing ESRF than patients with CKD alone (adjusted HR 13.00; 95% CI 10.57–15.99 versus adjusted HR 8.43; 95% CI 7.39–9.61). However, this study is limited in that it relied on administrative diagnostic coding, which may not have been sufficiently sensitive. For example, the absence of a coded diagnosis for CKD does not reliably indicate normal baseline function.
Existing evidence suggests that the relationship between AKI and risk of CKD depends on the presence and also the severity of AKI. Chawla et al. analyzed the data of 5,351 patients in a Veterans Affairs cohort with normal baseline function admitted with AKI [12]. They developed a number of models to predict the likelihood of developing CKD stage 4 or worse following hospital discharge and showed by multivariate analysis that severity of AKI, whether by RIFLE (Risk – Injury – Failure – Loss – End stage) classification or mean serum creatinine, was a strong predictor of CKD stage 4. Advanced age, low serum albumin and the presence of diabetes were also predictive.
In a meta-analysis of 13 retrospective studies including those cited above, the pooled incidences of CKD and ESRF post-AKI were 25.8/100 person-years and 8.6/100 person-years, respectively [13]. Compared to patients without AKI the adjusted HRs were 8.8 for developing CKD (95% CI 3.1–25.5), 3.1 for ESRF (95% CI 1.9–5.0) and 2.0 for mortality (95% CI 1.3–3.1). Furthermore, ‘recovery’ of AKI as defined by a recorded eGFR within 90 days post-hospitalization that was at least 90% of the baseline eGFR was still associated with the development of CKD [8]. Cohort patients met strict criteria, including a baseline eGFR > 60 ml/min, no history of renal disease (including proteinuria) and an increase of at least 50% in serum creatinine during their index admission. In this single center study, 1,610 patients were matched with 3,652 controls. The risk of de novo CKD was nearly doubled (adjusted HR 1.9; 95% CI 1.75–2.09).
To assess these important observations in more detail, there are several ongoing prospective studies focusing on the link between AKI and CKD. The Assessment, Serial Evaluation and Subsequent Sequelae of Acute Kidney Injury (ASSESS-AKI) study is a North American multicenter project including adult and pediatric cohorts [14]. Detailed annual reviews will be conducted for up to 4 years with blood and urinary biomarkers. Similarly, the At Risk in Derby (ARID) study is a UK, single center, case–control study aiming to recruit 1,084 hospitalized patients, again with blood and urine samples collected at designated time points [ISRCTN25405995]. The results of these studies are awaited with interest.
Potential mechanisms underlying the progression of AKI to CKD
In AKI, several processes are initiated in both injured and regenerating tissues, including premature cell-cycle arrest, secretion of bioactive molecules, recruitment of infiltrating inflammatory and stem cells, and activation of myofibroblasts and fibrocytes [4]. Some of these pathways are directly linked to processes that are believed to cause progression of CKD.
Common risk factors/pre-existing comorbidities
There is intuitively an overlap between risk factors for AKI and progressive CKD. In many patients, the factors that predispose to AKI continue to exist after the episode of AKI has finished. Important risk factors for progressive CKD leading to ESRF include pre-existing CKD and proteinuria. Both signify significant structural and functional changes within glomeruli, tubulo-interstitial compartments and the renal vasculature, which may leave the kidney particularly vulnerable to further injury in the presence of nephrotoxins or intercurrent illness. Importantly, in CKD, the increase in serum creatinine for a given fall in GFR is greater than in patients with normal baseline renal function due to the non-linear relationship between serum creatinine and GFR. As a consequence, the diagnosis of AKI is more likely to be made using conventional consensus criteria.
The importance of proteinuria is apparent in the results described in a prospective cohort of 11,200 participants in the Atherosclerosis Risk in Communities (ARIC) study. The association between baseline urine albumin-to-creatinine ratio and eGFR with hospitalizations or death with AKI was examined [15]. Using a urine albumin-to-creatinine ratio < 10 mg/g as a reference, the relative hazards of AKI after an average of 8-years follow-up, adjusted for age, sex, race, cardiovascular risk factors, and categories of eGFR were 1.9 (95% CI 1.4–2.6), 2.2 (95% CI 1.6–3.0), and 4.8 (95% CI 3.2–7.2) for urine albumin-to-creatinine ratio groups of 11–29 mg/g, 30–299 mg/g, and ≥ 300 mg/g, respectively. There was a similar correlation in risk of AKI with decreasing eGFR groups. The impact of pre-existing CKD and proteinuria was the focus of a Canadian study that retrospectively analyzed outcomes of 920,985 patients who had had their eGFR and urine dipstick recorded between 2002 and 2007 [16]. The authors not only demonstrated that the risk of AKI rose cumulatively with worsening CKD and increased proteinuria but that this risk continued post-AKI with an increased chance of reaching the combined endpoint of ESRF or doubling of the serum creatinine. Harel et al. followed survivors of dialysis-dependent AKI who had recovered renal function [17]. They showed that pre-existing CKD (HR 3.86; 95% CI 2.99–4.98), hypertension (HR 1.82; 95% CI 1.28–2.58) and a higher Charlson comorbidity index score (HR 1.10; 95% CI 1.05–1.15/per unit) were significantly associated with risk of progression to ESRF.
What is clear, is that there is homogeneity among many of the risk factors for both AKI and CKD. For example, the baseline characteristics of patients who develop AKI are often significantly different to those who do not. Hsu et al. compared 1,746 dialysis-requiring AKI patients with 600,820 controls and found that the traditional risk factors for CKD progression (pre-existing CKD, proteinuria, hypertension and diabetes) were all found to be independently associated with risk of severe AKI [18]. Bucaloiu et al. reported that patients with AKI had a significant preponderance of other ‘traditional renal risk factors’, such as a history of hypertension, coronary artery disease, vascular disease, chronic heart failure, dyslipidemia, chronic lung or liver disease, cancer and hypoalbuminemia [8]. These conditions per se, as well as their potential treatments, have the potential to contribute to a decline in kidney function together with, as well as independently of, AKI.
Glomerular hyperfiltration
In many models of acute renal disease, a loss of nephron mass and resultant hyperfiltration in the remaining glomeruli have been described. Similar to the sequelae following subtotal nephrectomy, it has been postulated that this results in hypertrophy of the residual glomeruli through increased work [4,6]. As a result, tubular workload and O2 consumption increase because of the increased flow. This can lead to hypoxic signaling and stimulation of tubulo-interstitial fibrosis, the latter of which is a significant component in the development of CKD [4,6].
Mitochondrial dysregulation
Recent findings have revealed striking morphological changes within mitochondria during cell injury. In health, mitochondria constantly undergo fission and fusion [19]. During cell injury, the dynamics are shifted to fission, i.e., the production of short mitochondrial rods or spheres. This type of mitochondrial fragmentation is associated with damage in the outer and inner membranes of the organelles, membrane leakage, decreased function and consequent cell death. Emerging evidence has suggested a pathogenic role of mitochondrial fragmentation in AKI [19,20]. This may be related to an increase in non-compartmentalized reactive oxygen species (ROS) formation coupled with a loss of competent antioxidant systems. The blockade of mitochondrial fragmentation has a renoprotective effect in both ischemic and cisplatin-induced AKI [20].
While cell death is the predominant effect of mitochondrial dysregulation, mitochondrial fragmentation may have a less dramatic chronic impact under certain circumstances. For example, Funk and Schnellmann demonstrated a persistent disruption of mitochondrial homeostasis after AKI, which in turn may result in suboptimal cellular respiration, reduction in cellular adenosine triphosphate (ATP) and consequent tissue dysfunction, all contributing to the development of chronic damage [21]. It may well be that targeting mitochondrial dynamics for the therapy of AKI and prevention of CKD has a potential role but more preclinical studies are necessary to test this hypothesis.
Endothelial injury and reduced capillary density
Several different animal models have demonstrated diminished vascular density after an episode of AKI, especially in foci of tubulo-interstitial fibrosis [4,22-24]. Such vascular rarefaction leads to the activation of hypoxia-inducible pathways and promotion of pro-inflammatory and pro-fibrotic processes [6]. In a vicious circle, capillary rarefaction, hypoxic signaling and tissue hypoxia may mutually reinforce each other leading to further damage and fibrosis.
Tubulo-interstitial inflammation/fibrosis
Tubulo-interstitial fibrosis is a predominant feature of CKD following AKI. Tubular hypertrophy and reduced capillary density play an important role in the pathogenesis. In addition, inflammation has been shown to be a key process in both ischemic and septic AKI, characterized by interstitial neutrophil infiltration during the acute phase and monocytic-lymphocytic infiltration in later stages [4,6]. Monocyte infiltration potentiates injury as well as promoting fibroblast proliferation and consequent fibrosis [6]. Such pro-fibrotic processes are initiated and maintained by ongoing production and secretion of a variety of peptides, including cytokines and growth factors. Although they are necessary for repair and tubule regeneration, these bioactive molecules also have a stimulating effect on perivascular fibroblasts and initiate fibrosis [6].
These cellular and paracrine processes combined with changes in tissue architecture lead to altered anatomical relationships between important structures further promoting fibrosis.
Potential key regulators
Transforming growth factor β
Transforming growth factor-β (TGF-β) is a key profibrotic cytokine that exerts a broad range of actions in the kidney in both health and disease [25]. AKI is a pro-inflammatory condition involving a complex interaction of cytokines, various renal cell types and infiltrating leukocytes [26,27]. TGF-β is upregulated in AKI and has a direct, detrimental effect via initiation of renal tubular apoptosis and extracellular matrix deposition [28,29]. Up-regulation of TGF-β continues into the recovery phase. Animal research using a bilateral ischemia/reperfusion model demonstrated recovery of renal function and normal histology at 4 and 8 weeks post-injury but clear evidence of tubulo-interstitial fibrosis and high levels of TGF-β expression at 40 weeks [30]. Urinary TGF-β levels reflect renal production and are elevated in a wide range of renal disease. Although TGF-β may have a role in AKI, its role in predicting the risk of CKD post-AKI has yet to be defined [31].
Endothelin-1
The kidney is both an important target as well as a source of the potent vasoconstrictor and mitogen, endothelin-1 (ET-1), which is mainly produced by endothelial cells. ET receptors are widely distributed within the human kidney and are present as two sub-types [32]. ET A receptors are localized to vascular smooth muscle notably in the glomeruli, vasa recta and arcuate arteries, and ET B receptors are predominantly localized in the medulla. In AKI, circulating and tissue ET-1 levels rise and ET receptor gene expression increases resulting in both endothelial dysfunction and enhanced vasoconstriction in different vascular beds. Studies which included ET-1 gene deletion, or blockade of the ET receptor, mitigated the initiation phase of ischemic, endotoxemic, or rhabdomyolysis-induced AKI [33-35].
However, data are conflicting. At least five studies have shown that ET-1 receptor blockade either conferred no functional protection, or worsened post-ischemic AKI [36-40]. In a more recent ischemia-reperfusion model in mice undergoing unilateral ischemia without contralateral nephrectomy, an increase in intrarenal ET-1 production was observed, along with increased expression of the ET A receptor and evidence of ET-1 gene activation alongside progressive histological changes and a 40% loss of renal mass [41]. Treatment with atrasentan, an ET A receptor antagonist ameliorated microvascular injury and abrogated the loss in renal mass.
The mechanisms underlying the effects of ET-1 and ET receptor blockers remain unclear. ET-1 is known to alter intrarenal vascular tone but may also change systemic hemodynamics and affect oxidative stress and inflammatory processes [32,42,43]. Future research may determine the role of ET A and B receptor blockers, either alone or in combination.
Galectin-3
Galectin-3 is a β-galactoside–binding lectin that has emerged as a key regulator of inflammation and fibrosis. It is highly evolutionarily conserved and plays an important role in several diverse biological processes and disease states [44]. Galectin-3 is strongly linked to the development of organ fibrosis in multiple sites [45-49]. The common pathways involve macrophage activation, TGF-β upregulation, fibroblast proliferation and collagen deposition. Galectin-3 knockout mice are resistant to the development of fibrosis, including that in the kidney [45,47,50-52].
A retrospective analysis of 2,450 patients who participated in the Framingham Offspring study demonstrated that elevated levels of plasma galectin-3 were associated with increased risks of rapid GFR decline and of incident CKD in the community [53].
There has been intense interest in the setting of chronic heart failure in which galectin-3 has been shown to have an emerging role in the prediction, diagnosis and prognosis of this condition, presumably due to its pathogenic role in cardiac fibrosis [54-62]. Heart failure studies also demonstrated that galectin-3 levels were inversely correlated to GFR [57,63-65].
The effects of galectin-3 in AKI are far from clear. One group studied two models of AKI in the rat (ischemic and nephrotoxic) and found that galectin-3 was intensely upregulated and prevented chronic tubular injury by limiting apoptosis, enhancing matrix remodeling and attenuating fibrosis [66]. However, another group using an ischemia-reperfusion model in wild-type versus knockout mice demonstrated that in early AKI the knockout mice seemed protected, with lower levels of interleukin-6, fewer ROS, less macrophage infiltration and lower peak concentrations of urea [67]. Using modified citrus pectin to reduce galectin-3 expression in mice, the severity of AKI observed was reduced following nephrotoxic insult [68].
These observations make galectin-3 an attractive candidate molecule to explain the demonstrable link between AKI and CKD. It is upregulated in AKI and serum levels appear to rise with renal impairment. Furthermore, it has pro-fibrotic actions up stream to TGF-β. More research in this area is awaited.
Endothelial hypoxia-inducible transcription factor (HIF)
Chronic renal hypoxia may also play a role in progressive renal disease, in part due to vasoconstriction and reduced capillary density. During periods of renal hypoxia, the kidneys initiate adaptive processes to facilitate endurance and maintain renal oxygenation in order to preserve tubular integrity. Hypoxia also affects the expression of potentially protective genes, which participate in tissue oxygenation, cell metabolism and survival [69]. Proximal tubular cells are highly sensitive to hypoxia because they are principally dependent on oxidative catabolism [69]. In contrast, distal tubular cells are able to use glycolysis and endure severe hypoxic challenges better, provided that transport diminishes [69,70].
Hypoxia-inducible factors (HIFs) are key regulators of gene expression in response to declining PO2 [71]. Upon hypoxia, HIF dimers translocate into the nucleus where they activate various genes involved in the relevant adaptive responses. HIF-mediated genes act to ameliorate hypoxia, counteract oxidative stress and improve cell survival. Although HIF protects the kidney against AKI and more than 100 HIF target genes have already been identified, intrinsic HIF activation is submaximal in AKI [72]. There is also some evidence that excessive activation of HIF may be deleterious and induce interstitial fibrosis and cyst formation, suggesting a complex interaction between AKI and CKD via hypoxia and HIF activation [72,73].
Conclusion
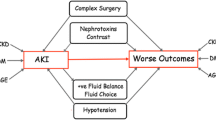
There is a strong intimacy between AKI and CKD. By mutually reinforcing the severity of the other, complex processes lead to the acceleration of disease progression (Figure 1). Much of the burden of poor outcomes is related to co-morbid disease, which in itself needs correct management. Other important pathogenic mechanisms that pave the road from AKI to CKD include glomerular hyperfiltration and hypertrophy, mitochondrial dysregulation, cellular infiltration and paracrine actions of bioactive molecules, reduced capillary density and promotion of tubulo-interstitial fibrosis. Interestingly, these processes are independent of the original insult or cause of AKI. Endothelin-1, TGF-β, serum galectin-3 and HIF appear to play important roles in these pathways and may be promising target molecules for future intervention studies.

Pathophysiological processes involved in the acute kidney injury (AKI)-chronic kidney disease (CKD) pathway. HIF: hypoxia-inducible factor; TGF: transforming growth factor.
The hope is that future prospective studies will provide further information on the specific risks of CKD after AKI, identify markers of poor outcomes and inform potential preventative strategies. The optimal follow-up and management of patients surviving an episode of AKI have no evidence base to-date. However, measuring a true post-recovery serum creatinine, quantifying degree of proteinuria and identifying any factors that pose a risk of recurrent AKI or progression of CKD seem prudent.
Currently, management is limited to optimization of co-morbid conditions (e.g., diabetes, heart failure, hypertension, fluid balance) and avoidance of nephrotoxic insults. Where impaired eGFR or proteinuria is present, referral to a nephrologist may be appropriate.
Abbreviations
- AKI:
-
Acute kidney injury
- CKD:
-
Chronic kidney disease
- eGFR:
-
estimated glomerular filtration rate
- ESRF:
-
End-stage renal failure
- ET-1:
-
Endothelin-1
- HIF:
-
Hypoxia-inducible factor
- RIFLE:
-
Risk – Injury – Failure – Loss – End stage
- ROS:
-
Reactive oxygen species
- TGF-β:
-
Transforming growth factor-β
References
Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol. 2013;24:37–42.
Hsu CY, Vittinghoff E, Lin F, Shlipak MG. The incidence of end-stage renal disease is increasing faster than the prevalence of chronic renal insufficiency. Ann Intern Med. 2004;141:95–101.
Okusa MD, Chertow GM, Portilla D. The nexus of acute kidney injury, chronic kidney disease, and World Kidney Day 2009. Clin J Am Soc Nephrol. 2009;4:520–2.
Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371:58–66.
Leung KC, Tonelli M, James MT. Chronic kidney disease following acute kidney injury-risk and outcomes. Nat Rev Nephrol. 2012;9:77–85.
Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298:F1078–94.
Lo LJ, Go AS, Chertow GM, et al. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009;76:893–9.
Bucaloiu ID, Kirchner HL, Norfolk ER, Hartle JE, Perkins RM. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012;81:477–85.
Prowle JR, Kolic I, Purdell-Lewis J, Taylor R, Pearse RM, Kirwan DJ. Serum creatinine changes associated with critical illness and detection of persistent renal dysfunction after AKI. Clin J Am Soc Nephrol. 2014;9:1015–23.
Wald R, Quinn RR, Luo J, et al. University of Toronto Acute Kidney Injury Research Group: Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA. 2009;302:1179–85.
Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223–8.
Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011;79:1361–9.
Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81:442–8.
Go AS, Parikh CR, Ikizler TA. The assessment, serial evaluation, and subsequent sequelae of acute kidney injury (ASSESS-AKI) study: design and methods. BMC Nephrol. 2010;11:22.
Grams ME, Astor BC, Bash LD, Matsushita K, Wang Y, Coresh J. Albuminuria and estimated glomerular filtration rate independently associate with acute kidney injury. J Am Soc Nephrol. 2012;21:1757–64.
James MT, Hemmelgarn BR, Wiebe N, et al. Alberta Kidney Disease Network: Glomerular filtration rate, proteinuria, and the incidence and consequences of acute kidney injury: a cohort study. Lancet. 2010;376:2096–103.
Harel Z, Bell CM, Dixon SN, et al. Predictors of progression to chronic dialysis in survivors of severe acute kidney injury: a competing risk study. BMC Nephrol. 2014;15:114.
Hsu C, Ordonez J, Chertow G, Fan D, McCulloch C, Go A. The risk of acute renal failure in patients with chronic kidney disease. Kidney Int. 2008;74:101–7.
Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83:568–81.
Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–85.
Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol. 2012;302:F853–64.
Horbelt M, Lee SY, Mang HE. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293:F688–95.
Basile DP, Friedrich JL, Spahic J. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300:F721–33.
Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–301.
Loeffler I, Wolf G. Transforming growth factor-beta and the progression of renal disease. Nephrol Dial Transplant. 2014;29 Suppl 1:i37–45.
Kinsey GR, Okusa MD. Role of leukocytes in the pathogenesis of acute kidney injury. Crit Care. 2012;16:214.
Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109:e102–7.
Gentle ME, Shi S, Daehn I. Epithelial cell TGFbeta signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol. 2013;24:787–99.
Gewin L, Vadivelu S, Neelisetty S. Deleting the TGF-beta receptor attenuates acute proximal tubule injury. J Am Soc Nephrol. 2012;23:2001–11.
Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281:F887–99.
Tsakas S, Goumenos DS. Accurate measurement and clinical significance of urinary transforming growth factor-beta1. Am J Nephrol. 2006;26:186–93.
Dhaun N, Webb DJ. The road from AKI to CKD: the role of endothelin. Kidney Int. 2013;84:637–8.
Mino N, Kobayashi M, Nakajima A, et al. Protective effect of a selective endothelin receptor antagonist, BQ-123, in ischemic acute renal failure in rats. Eur J Pharmacol. 1992;221:77–83.
Lopez-Farre A, Gomez-Garre D, Bernabeu F, Lopez-Novoa JM. A role for endothelin in the maintenance of post-ischaemic renal failure in the rat. J Physiol. 1991;444:513–22.
Clozel M, Ramuz H, Clozel JP, et al. Pharmacology of tezosentan, new endothelin receptor antagonist designed for parenteral use. J Pharmol Exp Ther. 1999;290:840–6.
Abu-Saleh N, Ovcharenko E, Awad H, et al. Involvement of the endothelin and nitric oxide systems in the pathogenesis of renal ischemic damage in an experimental diabetic model. Life Sci. 2012;91:669–75.
Forbes JM, Hewitson TD, Becker GJ, Jones CL. Simultaneous blockade of endothelin A and B receptors in ischemic acute renal failure is detrimental to long-term kidney function. Kidney Int. 2001;59:1333–41.
Forbes JM, Leaker B, Hewitson TD, Becker GJ, Jones CL. Macrophage and myofibroblast involvement in ischemic acute renal failure is attenuated by endothelin receptor antagonists. Kidney Int. 1999;55:198–208.
Fenhammar J, Andersson A, Forestier J, et al. Endothelian receptor A antagonism attenuates renal medullary blood flow impairment in endotoxemic pigs. PLoS One. 2011;6:e21534.
Gellai M, Jugus M, Fletcher T, DeWolf R, Nambi P. Reversal of postischemic acute renal failure with a selective endothelin A receptor antagonist in the rat. J Clin Invest. 1994;93:900–6.
Zager RA, Johnson ACM, Andress D, Becker K. Progressive endothelin-1 gene activation initiates chronic/end-stage renal disease following experimental ischemic/reperfusion injury. Kidney Int. 2013;84:703–12.
Arfian N, Emoto N, Vignon-Zellweger N, Nakayama K, Yagi K, Hirata K. ET-1 deletion from endothelial cells protects the kidney during the extension phase of ischemia/reperfusion injury. Biochem Biophys Res Commun. 2012;425:443–9.
Gulmen S, Kiris I, Narin C, et al. Tezosentan reduces the renal injury induced by abdominal aortic ischemia-reperfusion in rats. J Surg Res. 2009;157:e7–e13.
Cummings RD, Liu F-T, et al. Galectins. In: Varki A, Cummings RD, Esko JD, editors. Essentials of Glycobiology. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 2009. p. 475–88.
Calvier L, Miana M, Reboul P, et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol. 2013;33:67–75.
Lalancette-Hebert M, Swarup V, Beaulieu JM, et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–95.
Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. 2006;103:5060–5.
Nishi Y, Sano H, Kawashima T, et al. Role of galectin-3 in human pulmonary fibrosis. Allergol Int. 2007;56:57–65.
Lippert E, Falk W, Bataille F, et al. Soluble galectin-3 is a strong, colonic epithelial-cell-derived, lamina propria fibroblast-stimulating factor. Gut. 2007;56:43–51.
Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol. 2008;172:288–98.
Mackinnon AC, Gibbons MA, Farnworth SL, et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012;185:537–46.
Sasaki S, Bao Q, Hughes RC. Galectin-3 modulates rat mesangial cell proliferation and matrix synthesis during experimental glomerulonephritis induced by anti-Thy1.1 antibodies. J Pathol. 1999;187:481–9.
O’Seaghdha CM, Hwang SJ, Ho JE, Vasan RS, Levy D, Fox CS. Elevated galectin-3 precedes the development of CKD. J Am Soc Nephrol. 2013;24:1470–7.
Chen K, Jiang RJ, Wang CQ, et al. Predictive value of plasma galectin-3 in patients with chronic heart failure. Eur Rev Med Pharmacol Sci. 2013;17:1005–11.
Fermann GJ, Lindsell CJ, Storrow AB, et al. Galectin 3 complements BNP in risk stratification in acute heart failure. Biomarkers. 2012;17:706–13.
Gopal DM, Kommineni M, Ayalon N, et al. Relationship of plasma galectin-3 to renal function in patients with heart failure: effects of clinical status, pathophysiology of heart failure, and presence or absence of heart failure. J Am Heart Assoc. 2012;1:e000760.
Ho JE, Liu C, Lyass A, et al. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol. 2012;60:1249–56.
Lok DJ, Klip IT, Lok SI, et al. Incremental prognostic power of novel biomarkers (growth-differentiation factor-15, high-sensitivity C-reactive protein, galectin-3, and high-sensitivity troponin-T) in patients with advanced chronic heart failure. Am J Cardiol. 2013;112:831–7.
Tsai TH, Sung PH, Chang LT, et al. Value and level of galectin-3 in acute myocardial infarction patients undergoing primary percutaneous coronary intervention. J Atheroscler Thromb. 2012;19:1073–82.
van der Velde AR, Gullestad L, Ueland T, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH. Circ Heart Fail. 2013;6:219–26.
Weir RA, Petrie CJ, Murphy CA, et al. Galectin-3 and cardiac function in survivors of acute myocardial infarction. Circ Heart Fail. 2013;6:492–8.
de Boer RA, Yu L, van Veldhuisen DJ. Galectin-3 in cardiac remodeling and heart failure. Curr Heart Fail Rep. 2010;7:1–8.
Tang WH, Shrestha K, Shao Z, et al. Usefulness of plasma galectin-3 levels in systolic heart failure to predict renal insufficiency and survival. Am J Cardiol. 2011;108:385–90.
Lok DJ, Van Der Meer P, de la Porte PW, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study. Clin Res Cardiol. 2010;99:323–8.
Shah RV, Chen-Tournoux AA, Picard MH, Kimmenade RR, Januzzi JL. Galectin-3, cardiac structure and function, and long-term mortality in patients with acutely decompensated heart failure. Eur J Heart Fail. 2010;12:311–9.
Nishiyama J, Kobayashi S, Ishida A, et al. Up-regulation of galectin-3 in acute renal failure of the rat. Am J Pathol. 2000;157:815–23.
Fernandes Bertocchi AP, Campanhole G, Wang PH, et al. A Role for galectin-3 in renal tissue damage triggered by ischemia and reperfusion injury. Transpl Int. 2008;21:999–1007.
Kolatsi-Joannou M, Price KL, Winyard PJ, Long DA. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS One. 2011;6:e18683.
Heyman SN, Evans RG, Rosen S, Rosenberger C. Cellular adaptive changes in AKI: mitigating renal hypoxic injury. Nephrol Dial Transplant. 2012;27:1721–8.
Epstein FH. Oxygen and renal metabolism. Kidney Int. 1997;51:381–5.
Gunaratnam L, Bonventre JV. HIF in kidney disease and development. J Am Soc Nephrol. 2009;20:1877–87.
Nangaku M, Rosenberger C, Heyman SN, Eckardt KU. Regulation of hypoxia-inducible factor in kidney disease. Clin Exp Pharmacol Physiol. 2013;40:148–57.
Schietke RE, Hackenbeck T, Tran M, et al. Renal tubular HIF-2α expression requires VHL inactivation and causes fibrosis and cysts. PLoS One. 2012;7:e31034.
Declarations
Publication of this article was funded by departmental funds.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Varrier, M., Forni, L.G. & Ostermann, M. Long-term sequelae from acute kidney injury: potential mechanisms for the observed poor renal outcomes. Crit Care 19, 102 (2015). https://doi.org/10.1186/s13054-015-0805-0
Published:
DOI: https://doi.org/10.1186/s13054-015-0805-0