
Abstract
Background
Premature ovarian insufficiency refers to the loss of ovarian function before 40 years of age. The etiology is heterogeneous, and genetic factors account for 20–25% of cases. However, how to transform genetic findings to clinical molecular diagnose remains a challenge. To identify potential causative variations for POI, a next generation sequencing panel with 28 known causative genes of POI was designed, and a large cohort of 500 Chinese Han patients was screened directly. Pathogenic evaluation of the identified variants and the phenotype analysis were performed according to monogenic or oligogenic variants.
Results
A total of 14.4% (72/500) of the patients carried 61 pathogenic or likely pathogenic variants in 19 of the genes in the panel. Interestingly, 58 variants (95.1%, 58/61) were firstly identified in patients with POI. FOXL2 harbored the highest occurrence frequency (3.2%, 16/500), among whom presented with isolated ovarian insufficiency instead of blepharophimosis-ptosis-epicanthus inversus syndrome. Moreover, luciferase reporter assay confirmed variant p.R349G, which account for 2.6% of POI cases, impaired the transcriptional repressive effect of FOXL2 on CYP17A1. The novel compound heterozygous variants in NOBOX and MSH4 were confirmed by pedigree haplotype analysis, and digenic heterozygous variants in MSH4 and MSH5 were firstly identified. Furthermore, nine patients (1.8%, 9/500) with digenic or multigenic pathogenic variants presented with delayed menarche, early onset of POI and high prevalence of primary amenorrhea compared with those with monogenic variation(s).
Conclusions
The genetic architecture of POI has been enriched through the targeted gene panel in a large cohort of patients with POI. Specific variants in pleiotropic genes may result in isolated POI rather than syndromic POI, whereas oligogenic defects might have cumulative deleterious effects on the severity of POI phenotype.
Similar content being viewed by others


Background
Premature ovarian insufficiency (POI) is defined as loss of ovarian function before 40 years of age, and it is characterized by oligomenorrhea or amenorrhea, elevated follicle stimulating hormone (FSH), and decreased estrogen. Approximately 1%–5% of women under 40 years old are diagnosed with POI, and present with infertility, estrogen deficiency symptoms, and long-term complications, such as osteoporosis and cardiovascular disease [1]. The etiology of POI is heterogeneous, including genetic factors, autoimmune disorders, infections, and iatrogenic causes [2]. Genetic factors account for 20%–25% of cases [3], including chromosome abnormalities and gene mutations. However, high genetic heterogeneity exists in both isolated and syndromic patients. Currently, nearly 80 genes have been reported to be associated with POI, while only a small subset of these genes can explain more than 5% of patients [3, 4]. Due to limited gene coverage, high time consumption and expense, monogenic screening by Sanger sequencing has seldom been used in genetic studies of POI.
Recently, with the application of whole exome sequencing (WES) in POI pedigrees, vast numbers of genetic variants have been emerged in the omics era. Especially, based on the expanded genetic spectrum of POI, next-generation sequencing (NGS) of POI genes makes its genetic diagnosis possible. Fonseca et al. performed NGS covering 70 candidate genes in 12 patients with POI and found 25% of the patients carried potential mutations [5]. Bouilly et al. sequenced 19 candidate genes using NGS in 100 patients and identified a mutation frequency of 19% [6]. A higher frequency of mutation carrier (48%) has been reported in 69 POI patients through NGS of 420 candidate genes [7]. More recently, Raffaella et al. developed a 295 genes panel and screened in 64 patients, founding that the phenotypes of POI were associated with the number of variations, which supports the oligogenic nature of POI [8]. However, majority of candidate genes designed in above-mentioned panels were selected based on the phenotypes of animal models or related biological functions, lacking solid evidence in human POI. It is impractical for molecular diagnosis of POI in the absence of sufficient functional evidence. Therefore, how to improve the diagnostic efficacy of gene panel is still challenging for POI patients.
Here, we designed a targeted gene panel consisting of 28 known causative genes of human POI. A large cohort of 500 POI patients were enrolled and screened with the panel to expand the genetic spectrum and architecture of POI.
Results
Genetic analysis
In the 500 Chinese Han patients with POI, a total of 772 sequence variants were identified in the 28 genes. Among them, 226 rare variants (frequency < 0.1% in the 1000 Genomes Project and gnomAD databases) were further analyzed, including 4 nonsense, 24 splice site, 10 frameshift, and 242 missense variants. After filtering by MetaSVM, CADD, and DANN score, 22 pathogenic and 57 likely pathogenic variants were considered as potentially causative for POI (Fig. 1).

Overview of the filtering process of potential causative variants for POI. The genetic panel included 28 POI candidate genes. A total of 772 variants were identified by panel testing, of which 226 variants had rare allele frequencies (< 0.1%), including 22 pathogenic (P) variants and 57 likely pathogenic [9] variants. Among the patients carrying P or LP variants, 71 cases could be explained by monogenic variants and 9 cases had oligogenic variants located in more than one gene. Finally, 61 variants (18 pathogenic and 43 likely pathogenic) were filtered and confirmed by Sanger sequencing. MAF: minor allele frequency; P: pathogenic; LP: likely pathogenic; VUS: variant of uncertain significance; CADD: combined annotation dependent depletion; DANN: deleterious annotation of genetic variants using neural networks; SNV: single-nucleotide variant
According to classical inheritance patterns (autosomal recessive or dominant, X-linked recessive or dominant patterns), 71 patients could be explained by monogenic variants, and 8 of them carried additional P/LP variants located in one or two other genes. Intriguingly, in addition to these 71 patients, one patient carried digenic heterozygous variants in MSH4 and MSH5, which interacted to form a heterodimer during homologous recombination and were thought to be causative for POI in recessive pattern separately. Therefore, 72 patients carried pathogenic or likely pathogenic variants were screened out, and 9 of them had digenic or multigenic variants (Fig. 1). A total of 18 pathogenic and 43 likely pathogenic variants located in 19 genes (Table 1, Fig. 2), i.e. 6 meiosis genes (HFM1, SPIDR, SMC1B, MSH5, MSH4, CSB-PGBD3), 6 transcription factors (SOHLH1, POLR2C, FIGLA, NOBOX, NR5A1, FOXL2) and 7 genes involved in ligands and receptors (AMH, AMHR2, GDF9, BMP15, FSHR, BMPR2, PGRMC1). Interestingly, only three of the variants had been reported in previous studies (p.R355H in NOBOX, p.R313H in NR5A1, and p.R351G in MSH5) [10,11,12,13], whereas the other 58 variants were firstly identified in human POI (Table 1).

Prevalence of candidate gene variants identified in POI patients. The 61 pathogenic/likely pathogenic filtered variants were located in 19 genes, including 6 meiosis genes, 6 transcription factors, and 7 genes involved in steroid hormone synthesis or response pathways. Among these, the transcription factors had the highest mutation frequency (7.2%), followed by steroid hormone synthesis or receptor genes (6.2%) and meiotic genes (2.0%)
Among the 28 genes, FOXL2 harbored the highest variant occurrence frequency. Sixteen patients carried FOXL2 heterozygous variants (16/500, 3.20%), 13 of whom with variant c.1045C > G (p.R349G) (13/500, 2.60%). The frequency of p.R349G in POI patients was significantly higher than that in 1000 Genomes database (0.08%, p = 0.001) and East Asians of the ExAC database (0.24%, p < 0.001). To determine whether p.R349G influenced the transcriptional effect of FOXL2, the luciferase reporter assay was performed. The results showed that wild-type FOXL2 down-regulated the expression of CYP17A1, while the mutant FOXL2 did not present with the transcriptional repressive effect. For the transcriptional regulation of CYP19A1, wild-type FOXL2 showed similar transcriptional repression, whereas, the adverse effect of mutant FOXL2 was not significant (Fig. 3).

Variant p.R349G affected the transcriptional effect of FOXL2. The pcDNA3.1 vector, wild-type (WT) or/and mutant (MT) FOXL2 vectors and CYP17A1 (A)/CYP19A1 (B) promoter reporter were co-transfected in HEK392 cells. The results are shown as luciferase/renilla signal, and the pcDNA3.1 group was used as the control. The potential dominant-negative effect of FOXL2 p.R349G mutation was assessed by co-transfecting WT and MT vectors in 1:1 ratio. * indicated the p-value < 0.05, ** indicated the p-value < 0.01
Pedigree analysis
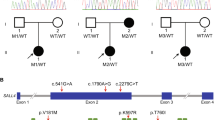
Compound heterozygous variants in NOBOX and MSH4 were identified, and haplotype analysis was further performed in the POI pedigree with two POI sisters carrying NOBOX variants and in one trio family with MSH4 variants (Fig. 4), respectively.

POI pedigrees harboring compound heterozygous variants or multigenic variants. The probands are marked with arrows, women with POI are depicted by black circles, and healthy women are depicted by white circles and healthy men by white squares. a In pedigree F254, the proband and her sister carried the compound heterozygous variation NOBOX p.L558fs and p.R355H, which was inherited from their father and mother separately. b In pedigree F191, proband POI-63 carried compound heterozygous mutation MSH4 p.M740fs and p.T792A, which was inherited from her father and mother, respectively.
In pedigree F254 (Fig. 4a), the proband POI-52 and her younger sister carried the compound heterozygous variant p.L558fs and p.R355H in NOBOX, which was inherited from their father and mother separately. The proband had menarche at 14 years of age and suffered oligomenorrhea until 25 years of age when she was diagnosed with POI. Her younger sister had secondary amenorrhea at 23 years old and was diagnosed with POI as well. A compound heterozygous variant in MSH4 (p.M740fs and p.T792A) was identified in POI-63, who had spontaneous menarche at 19 years of age and diagnosed with POI at 31 years old. The compound heterozygous variant was confirmed to be inherited paternally and maternally, respectively (Fig. 4b).
Clinical characteristics of patients carrying P or LP variants
Among the patients carrying digenic or multigenic variants, a higher percentage of primary amenorrhea (44.44% vs. 19.05%), earlier onset of POI (20.10 ± 6.81 years vs. 24.97 ± 4.67 years), and later menarche (15.82 ± 1.50 years vs. 13.95 ± 2.56 years) were observed compared with women carrying monogenic variants. However, these differences did not reach statistical significance (p > 0.05).
Discussion
In the present study, using a self-designed target panel covering 28 known POI causative genes screened in 500 Chinese Han patients, 14.40% (72/500) of patients were diagnosed with at least one pathogenic variant contributing to ovarian insufficiency. A total of 58 potential causative variants, including digenic heterozygous variants in MSH4 and MSH5, were firstly reported in POI, which not only expanded the variant spectrum of human POI, but also enriched the genetic architecture of POI pathogenesis.
Variants in pleiotropic genes result in isolated POI
Under most circumstances, the pleiotropic genes responsible for POI cause syndromic POI, which manifests with highly variable somatic abnormalities in addition to reproductive phenotypes, such as BLM for Bloom syndrome and WRN for Werner syndrome [3]. Recent genetic studies have revealed that variants in pleiotropic genes also resulted in isolated POI, such as NBN and EIF2B2, which could be explained by specific mutation sites and different types of variants [14]. In the present study, variants in pleiotropic genes, including FOXL2, NR5A1, and BMPR2, were identified in patients presenting with isolated POI, confirming that specific variants might contribute to distinct phenotypes of POI. These findings also highlighted the necessity of individualized genetic counseling and long-term healthcare follow-up in these women.
FOXL2 is preferentially expressed in the ovary, eyelids, and pituitary gland [15, 16]. Heterozygous intragenic variants of FOXL2 accounted for 71% of patients with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES), which is a dominant condition characterized by eyelid and mild craniofacial defects associated with POI (type I) or not (type II) [17]. Although more than 100 variants of FOXL2 have been found in BPES patients [17], the constitutional variants were reported in only 1.0%–2.9% of non-syndromic POI cases [18]. Through the panel test, we found that the prevalence of FOXL2 variants in isolated POI was 3.2%, which was much higher than the other genes in the panel. Intriguingly, the variant p.R349G in FOXL2 was firstly reported here and accounted for 2.6% of the cohort, which was significantly higher than the frequency in public databases. FOXL2 is involved in ovarian development by regulating the transcription of essential genes involved in steroidogenesis, including CYP17A1 and CYP19A1 [19,20,21,22]. Functional studies demonstrated that variant p.R349G impaired the transcriptional regressive effect of FOXL2 on CYP17A1, which might further influence the synthesis of estradiol and lead to folliculogenesis abnormalities [21]. Recently, somatic mutation p.C134W in FOXL2 has been found associated with GCs tumor in adult and accounted for up to 5% of ovarian malignancies [23]. Therefore, although none of the FOXL2 variation carriers in our cohort presented with eyelid malformation or ovarian tumor, the long-term follow-up is still warranted.
NR5A1 is a nuclear receptor that regulates the transcription of genes required for adrenal and reproductive development [24]. Variants in NR5A1 are associated with different reproductive phenotypes in humans, such as disorders of sex development (DSD), hypospadias, and POI. It has been reported that 0.3%–2.3% of POI patients carried NR5A1 mutations [25, 26], which is similar to the frequency in our study (1.2%). Interestingly, the variation p.R313C, locating at ligand-binding domain of NR5A1, was one of the most common variants identified in DSD patients [13]. However, the carriers of p.R313C and p.R313H in our study had normal female external genitalia, which might be explained by genetic heterogeneity during gonad differentiation.
BMPR2 is one of bone morphogenetic protein (BMP) binding soluble factors, participating in signal transduction between oocytes and GCs, which is essential for oocyte maturation [27, 28]. Most BMPR2 variants were reported previously in patients with idiopathic pulmonary arterial hypertension (IPAH), while recent NGS and functional studies have revealed that p.S987F in BMPR2 caused isolated POI by perturbing BMP15/BMPR2/SMAD signaling and GCs proliferation [29]. In this study, five variants in BMPR2 were found for the first time in seven patients with POI. It was reported that the majority of the BMPR2 variants identified in POI patients were located in cytoplasmic tail (amino acids 504–1038) of BMPR2 [30, 31]. However, three out of the five variants identified here were localized in kinase domain (amino acids 203–503). Similarly, a recent study also identified a novel heterozygous variant of POI patient in BMPR2 kinase domain (p.Val453Met) [32], suggesting that variants located in different domains of BMPR2 may have individual effects on ovarian function, highlighting the contribution of BMP signal pathway in isolated POI pathogenesis. Although no clinical features of pulmonary hypertension had been found in the time of investigation, the long-term status should be followed.
Both heterozygous and homozygous variants are pathogenic for POI
The inheritance pattern of POI includes recessive, dominant, and X-linked modes. With the accumulation of variants identified by WES and NGS, more complex inheritance patterns have been discovered. In this study, we identified a compound heterozygous variant in NOBOX, a previous dominant pathogenic gene, and digenic heterozygous variants in MSH4 and MSH5 that had never been reported in POI. Our findings provided new insights into the complexity of POI genetics.
NOBOX is an oocyte-specific transcriptional factor that plays a critical role in early folliculogenesis. Heterozygous NOBOX variants can explain up to 6.2% of POI patients via a dominant negative effect or haploinsufficiency [33]. The mutation prevalence of NOBOX in our cohort was 1.2% (6/500). Additionally, one compound heterozygous mutation p.R355H and p.L558fs was found in two POI patients in pedigree F254. The variant p.R355H was proved to disrupt the transcriptional function of NOBOX [10], while the variant p.L558fs results in a truncated NOBOX protein lacking the C-terminal 133 amino acids. However, the proband’s mother with p.R355H mutation presented with normal menstruation cycles and menopause occurred at 48 years of age. It might be explained by an incomplete penetrance effect of the causal variant. Therefore, more evidence is needed to prove the pathogenicity of heterozygous variants in NOBOX for POI.
Genes involved in meiosis are critical for early follicular development. To date, majority of mutations in meiotic genes have been found in biallelic state (homozygous or compound heterozygous), such as HFM1, BRCA2, and STAG3 [7]. MSH4 and MSH5 belong to the DNA mismatch repair gene family. The MSH4-MSH5 heterodimer plays an important role in homologous recombination repair of DNA double strand breaks, which is essential for meiosis [3]. WES in POI pedigrees has identified two homozygous variants in MSH4 and MSH5 previously [34, 35]; however, the contribution of MSH4 or MSH5 variants in the pathogenesis of sporadic POI has not been reported yet. In the present study, one homozygous variant in MSH5 and three compound heterozygous variants in MSH4 inherited in recessive pattern were identified in 5 patients, accounting for 1.0% (5/500) of patients with sporadic POI. Interestingly, patient POI-9 carried digenic heterozygous variants in MSH4 and MSH5, indicating that not only one subunit deficiency, but also dysfunctional MSH4-MSH5 interaction or cumulative haploinsufficiency of both subunits, may disrupt homologous recombination during meiosis, finally causing POI. This is the first report about digenic heterozygous variants occurred in MSH4-MSH5 heterodimer, which sheds new light on the complex genetic architecture of POI and suggests a novel mechanism of POI pathogenesis.
Digenic or multigenic variants affect the severity of POI phenotype
Previous NGS studies showed that 36%-42% of POI patients carried two or more variants in distinct genes [6, 7]. It is speculated that accumulated genetic defects or deleterious environmental exposures might aggravate the insufficient formation or accelerate the exhaustion of oocytes, resulting in diverse severity of POI phenotype. In general, menarche occurs depends on a maturing hypothalamic-pituitary-ovarian (HPO) axis. The insufficient ovarian function presents with delayed or diminished response to pituitary hormones. In this study, compared to the patients with monogenic variants, the 9 patients (1.8%) carrying digenic or multigenic variants tended to exhibit delayed age at menarche, earlier age of POI onset, and greater prevalence of primary amenorrhea. However, the above differences did not reach statistical significance, which may be due to the limitation of small sample size. Similarly, a recent study suggested that the most severe phenotypes were associated with either the major number of variations or a worse prediction in pathogenicity of variants [8]. To a certain extent, our results partially indicated that oligogenic should be considered when affected women in a family present with different phenotypes or diverse severities of POI.
One of the strengths of the present study is the largest cohort of POI patients included. Another strength is that all the candidate genes have reported evidence of confirmative pathogenicity to human POI, and that the criteria for pathogenic and likely pathogenic used to define causative variants is more strict, which is also the possible explanation for relatively lower variant frequency compared to previous studies (14.4% vs. 19% ~ 48%). There were also a few limitations. First, although the identified variants were checked in the public population data, sequencing of control women from Chinese descent is lacking. Second, the coverage of this panel did not capture all known POI-related genes. Finally, not all family members were available for co-segregation analysis or tracing variants initiation.
Conclusions
In conclusion, a self-designed targeted gene panel covering 28 causative genes of POI used in 500 patients expanded the variant spectrum and genetic architecture of POI. Specific variants in pleiotropic genes may result in isolated POI; whereas oligogenic defects could exert cumulative deleterious effect on severity of POI phenotype. A fuller understanding of POI genetics would contribute to the individualized prediction, diagnosis, and intervention for women with POI or at high risk of developing POI.
Methods
Patients
A cohort of women with POI were recruited form the Reproductive Hospital Affiliated to Shandong University from 2006. Five hundred Chinese Han patients with non-syndromic POI were selected from that cohort. All patients suffered oligomenorrhea or menopause before 40 years of age and presented with elevated FSH (> 25 IU/L) at least twice over an interval of one month. Women with chromosomal abnormalities, ovarian surgery, chemo/radiotherapy, or known autoimmune disease (such as systemic lupus erythematosus, Sjögren syndrome, rheumatoid arthritis, autoimmune thyroiditis, and so on) were excluded. The peripheral blood samples were collected at the time of enrollment. All participants signed informed consent forms. The clinical characteristics of the participants were shown in Table 2.
NGS and bioinformatics analysis
Based on the mutation frequencies identified previously [3, 18, 36], 28 causative genes with confirmative functional evidence were included in the target panel, including 11 meiosis genes (HFM1, MSH4, MSH5, SPIDR, SMC1B, SYCE1, STAG3, MCM8, MCM9, NUP107 and CSB-PGBD3), 8 ligands and receptors associated genes (AMH, AMHR2, BMP15, BMPR2, FSHR, GDF9, PGRMC1 and KHDRBS1) and 9 transcription factors preferentially expressed in the ovaries (FIGLA, FOXL2, NOBOX, NR5A1, POLR2C, SOHLH1, WT1, NANOS3, and LHX8) (Table 3). The selected genes satisfied at least one of the following two requirements: 1) pathogenic mutations of the gene has been identified in women with POI; 2) functional studies have been performed to confirm that the genes involved in ovarian function maintenance. Genomic DNA was extracted from peripheral blood, and the sequencing library was prepared using the Ion AmpliSeq Library Kit 2.0 (ThermoFisher Scientific, USA). The prepared library was sequenced on MiSeqDx (Illumina, USA) using the MiSeqDx Universal Kit V3 SBS (Illumina, USA) according to the standard protocol supplied by Illumina. Majority of the variants identified by the panel testing were single-base substitutions and micro-insertions or deletions. We referred to all nonsynonymous variants, frameshift variants, and variants affecting splicing as protein-truncating variants that might affect the function of candidate genes. Rare variants with an incidence below 0.1% in the 1000 Genomes and gnomAD databases were analyzed further.
The potential variants were classified as pathogenic (P), likely pathogenic (LP), and variants of uncertain significance (VUS) according to the guidelines proposed by the American College of Medical Genetics and Genomics [9, 37]. Pathogenic variants referred to nonsense or frameshift variants, variants located in the canonical splice site (≤ 2 intronic base pairs from the intron/exon boundary), and those previously reported to be pathogenic. Likely pathogenic variants referred to non-synonymous missense variants with a bioinformatics pathogenicity prediction of “Deleterious” by metaSVM combined with a CADD score > 3 or a DANN score > 0.95 [38, 39]. An overview of the filtering process was shown in Fig. 1.
Sanger sequencing and haplotype analysis
The filtered variants were confirmed by Sanger sequencing. The parents of POI patients carrying compound heterozygous variants or multigenic variants were also sequenced when their DNAs were available. The primers were designed by Primer Premier 5.0 (Premier Biosoft, USA). PCR products were analyzed by agarose gel electrophoresis and purified by oligogenic glycol precipitation and then sequenced on an ABI 3730XL DNA analyzer (Applied Biosystems, Forster City, CA) using the ABI-Prism Big-Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). Sequencing data were analyzed using the Sequencher 4.9 software (Gene Codes Corporation, USA).
Plasmids construction and dual-luciferase reporter assay
The human coding sequence of the FOXL2 gene was cloned into pcDNA3.1 vector with double restriction enzyme BamHI and XhoI. The mutant plasmids carrying FOXL2 p.R349G were generated by point mutation strategy using the wild-type plasmids as template. The luciferase reporter plasmids were purchased from GeneCopoeia (http://www.igenebio.com/), termed as pProDuoLuci-CYP11A1 (product ID HPRM30715), pProDuoLuci-CYP17A1 (product ID (product ID HPRM30087) and pProDuoLuci-CYP19A1 (product ID HPRM30088). All constructs were validated by Sanger sequencing.
Human embryonic kidney (HEK) 293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). The cells were plated in 24-well plates and transfected using Lipofectamine™ 3000 reagent (Invitrogen, Carlsbad, CA, USA). The total DNA concentration in each well was maintained at 500 ng, including the CYP11A1, CYP17A1 or CYP19A1 promoter double-luciferase reporter plasmids 250 ng/well, and wild-type and/or mutant FOXL2 or empty pCDNA3.1 vector 250 ng/well). After transfection for 48 h, luciferase activities were assessed via the dual-luciferase reporter assay system (Promega). Results were normalized against Renilla luciferase activity. The experiments were repeated at least three times.
Statistical analysis
SPSS v.25 software was used for the statistical analysis. Data were analyzed using one-way ANOVA tests and chi-square tests, and p-values < 0.05 were considered statistically significant. Data were shown as means ± SD.
Availability of data and materials
The variations data in the study have been submitted to ClinVar data base (https://www.ncbi.nlm.nih.gov/clinvar/) under the project accession number SCV001877109—SCV001877169. The authors affirm that all data needed to confirm the conclusions of the study are present in the current article.
References
The ESHRE Guideline Group on POI. ESHRE Guideline: management of women with premature ovarian insufficiency. Human Reprod. 2016;0:1–12. https://doi.org/10.1093/humrep/dew027.
Torrealday S, Kodaman P, Pal L. Premature ovarian insufficiency - an update on recent advances in understanding and management. F1000Res. 2017;6:2069.
Qin Y, Jiao X, Simpson JL, et al. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update. 2015;21:787–808.
França MM, Mendonca BB. Genetics of primary ovarian insufficiency in the next-generation sequencing era. J Endoc Soc. 2020;4:1–16. https://doi.org/10.1210/jendso/bvz037.
Fonseca DJ, Patiño LC, Suárez YC, et al. Next generation sequencing in women affected by nonsyndromic premature ovarian failure displays new potential causative genes and mutations. Fertil Steril. 2015;104:154-62.e2.
Bouilly J, Beau I, Barraud S, et al. Identification of multiple gene mutations accounts for a new genetic architecture of primary ovarian insufficiency. J Clin Endocrinol Metab. 2016;101:4541–50.
Patino LC, Beau I, Carlosama C, et al. New mutations in non-syndromic primary ovarian insufficiency patients identified via whole-exome sequencing. Hum Reprod. 2017;32:1512–20.
Rossetti R, Moleri S, Guizzardi F, et al. Targeted next-generation sequencing indicates a frequent oligogenic involvement in primary ovarian insufficiency onset. Front Endocrinol (Lausanne). 2021;12:664645.
Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294–300.
Qin Y, Choi Y, Zhao H, et al. NOBOX homeobox mutation causes premature ovarian failure. Am J Hum Genet. 2007;81:576–81.
Ciaccio M, Costanzo M, Guercio G, et al. Preserved fertility in a patient with a 46, XY disorder of sex development due to a new heterozygous mutation in the NR5A1/SF-1 gene: evidence of 46, XY and 46, XX gonadal dysgenesis phenotype variability in multiple members of an affected kindred. Horm Res Paediatr. 2012;78:119–26.
Wu X, Xu Y, Feng K, et al. MutS homologue hMSH5: recombinational DSB repair and non-synonymous polymorphic variants. PLoS ONE. 2013;8:e73284.
Song Y, Fan L, Gong C. Phenotype and molecular characterizations of 30 children from China with NR5A1 mutations. Front Pharmacol. 2018;9:1224.
Tucker EJ, Grover SR, Robevska G, et al. Identification of variants in pleiotropic genes causing “isolated” premature ovarian insufficiency: implications for medical practice. Eur J Hum Genet. 2018;26:1319–28.
Laven JSE, Visser JA, Uitterlinden AG, et al. Menopause: genome stability as new paradigm. Maturitas. 2016;92:15–23.
Shi J, Zhang B, Choi JY, et al. Age at menarche and age at natural menopause in East Asian women: a genome-wide association study. Age (Dordr). 2016;38:513–23.
Verdin H, De Baere E. FOXL2 impairment in human disease. Horm Res Paediatr. 2012;77:2–11.
Jiao X, Ke H, Qin Y, et al. Molecular genetics of premature ovarian insufficiency. Trends Endocrinol Metab. 2018;29:795–807.
Yang X, He W, Gong F, Li W, Li X, Zhong C, et al. Novel FOXL2 mutations cause blepharophimosis-ptosis-epicanthus inversus syndrome with premature ovarian insufficiency. Mol Genet Genom Med. 2018;6:261–7.
Kuo FT, Bentsi-Barnes IK, Barlow GM, Pisarska MD. Mutant Forkhead L2 (FOXL2) proteins associated with Premature Ovarian Failure (POF) dimerize with wild-type FOXL2, leading to altered regulation of genes associated with granulosa cell differentiation. Endocrinology. 2011;152:3917–29.
Taniguchi F, Couse JF, Rodriguez KF, Emmen JMA, Poirier D, et al. Estrogen receptor-α mediates an intraovarian negative feedback loop on thecal cell steroidogenesis via modulation of CYP17A1 (cytochrome P450, steroid 17α-hydroxylase/17,20 lyase) expression. FASEB J. 2007;21(2):586–95.
Elzaiat M, Todeschini AL, Caburet S, Veitia RA. The genetic make-up of ovarian development and function: the focus on the transcription factor FOXL2. Clin Genet. 2017;91:173–82.
Jamieson S, Butzow R, Andersson N, et al. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod Pathol. 2010;23:1477–85.
Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–90.
Sun Q-Y, Jiao X, Qin Y, et al. Novel NR5A1 missense mutation in premature ovarian failure: detection in Han Chinese indicates causation in different ethnic groups. PLoS ONE. 2013;8:e74759.
Jaillard S, Sreenivasan R, Beaumont M, et al. Analysis of NR5A1 in 142 patients with premature ovarian insufficiency, diminished ovarian reserve, or unexplained infertility. Maturitas. 2020;131:78–86.
Otsuka F. Multifunctional bone morphogenetic protein system in endocrinology. Acta Med Okayama. 2013;67:75.
Shimasaki S, Moore RK, Otsuka F, Erickson GF. The bone morphogenetic protein system in mammalian reproduction. Endocr Rev. 2004;25:72–101.
Patino LC, Silgado D, Laissue P. A potential functional association between mutant BMPR2 and primary ovarian insufficiency. Syst Biol Reprod Med. 2017;63:145–9.
Girerd B, Coulet F, Jais X, et al. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest. 2015;147:1385–94.
Heddar A, Ogur C, Da Costa S, et al. Genetic landscape of a large cohort of primary ovarian insufficiency: new genes and pathways and implications for personalized medicine. EBioMedicine. 2022;84:104246.
Franca MM, Funari MFA, Lerario AM, et al. Screening of targeted panel genes in Brazilian patients with primary ovarian insufficiency. PLoS ONE. 2020;15:e0240795.
Bouilly J, Bachelot A, Broutin I, et al. Novel NOBOX loss-of-function mutations account for 6.2% of cases in a large primary ovarian insufficiency cohort. Hum Mutat. 2011;32:1108–13.
Carlosama C, Elzaiat M, Patiño LC, Mateus HE, Veitia RA, Laissue P. A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Hum Mol Genet. 2017;16:3161–6.
Guo T, Zhao S, Zhao S, et al. Mutations in MSH5 in primary ovarian insufficiency. Hum Mol Genet. 2017;26:1452–7.
De Vos M, Devroey P, Fauser BC. Primary ovarian insufficiency. Lancet. 2010;376:911–21.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Kim J, Field A, Schultz KAP, et al. The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer. 2017;141:2030–6.
Acknowledgements
The authors thank all of the participants involved in this study. T.G. designed the genetic panel; W.L. performed the NGS data analysis and candidate variants classification with the help of H.K.; Y.Q. and T.G. collected participants’ samples and clinical data; T.G. and W.L wrote the manuscript; and Y.Q. supervised the article. All authors contributed to this work, discussed the results, and critically reviewed and revised the manuscript.
Funding
This work was supported by the National Key Research & Developmental Program of China [2022YFC2703800, 2022YFC2703000]; the National Natural Science Foundation of China [82125014, 32070847]; Basic Science Center Program of NFSC [31988101]; Program for Excellent Young Scholars of Shandong Province [ZR2022YQ69]; the Natural Science Foundation of Shandong Province [ZR2022QH313], Taishan Scholars Program for Young Experts of Shandong Province [tsqn202211371], Jinan Shizhong District Key Industry Leading Talents Project, Scientific Research Foundation of Shandong Provincial Hospital [2021FY035].
Author information
Authors and Affiliations
Contributions
T.G. designed the genetic panel; W.L. performed the NGS data analysis and candidate variants classification with the help of H.K.; Y.Q. and T.G. collected participants’ samples and clinical data; T.G. and W.L wrote the manuscript; and Y.Q. supervised the article. All authors contributed to this work, discussed the results, and critically reviewed and revised the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institutional Review Board of Reproductive Medicine of Shandong University, and written informed consents were obtained from all participants prior to blood sample collection and molecular studies.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, W., Ke, H., Tang, S. et al. Next-generation sequencing of 500 POI patients identified novel responsible monogenic and oligogenic variants. J Ovarian Res 16, 39 (2023). https://doi.org/10.1186/s13048-023-01104-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-023-01104-6