
Abstract
Background
Cytochrome P450 oxidoreductase deficiency (PORD) is a rare disease exhibiting a variety of clinical manifestations. It can be difficult to differentiate with other diseases such as 21-hydroxylase deficiency (21-OHD), polycystic ovary syndrome (PCOS) and Antley–Bixler syndrome (ABS). Nearly 100 cases of PORD have been reported worldwide. However, the genetic characters and clinical management are still unclear, especially in China.
Case presentation
In this study, we report a 27-year-old female Chinese patient who first presented with amenorrhea and recurrence of large ovary cyst. She was misdiagnosed with PCOS and non-classical 21-OHD due to ovary cysts and elevated 17-hydroxy-progesterone. The patient’s complaining of a mild difficulty of bending the metacarpophalangeal joints reminded us to consider PORD, which usually presents with skeletal deformities and sexual dysfunction. The diagnosis of PORD was confirmed by genetic analyses, which showed the patient harboring a homozygous missense mutation in the POR gene (R457H) and her parents carrying the heterozygous mutation. The patient was treated with low-dose corticosteroids and estrogen/progesterone sequential therapy, and her ovarian cyst gradually reduced with regular menstruation in the follow-up. Moreover, the clinical and genetic characteristics of 104 previously reported PORD cases were also summarized and analyzed.
Conclusions
PORD is a very rare disease which can be easily misdiagnosed in mild cases. Clinicians should keep in mind of this disease in patients with sexual dysfunction, especially combined with special skeletal deformities. Our data could provide a consciously understanding of this disease for clinic practicers. Low-dose corticosteroids combined with estrogen/progesterone sequential therapy will be effective in PORD patients with recurrence of large ovary cyst. The fact that the reported PORD patients in China carrying an identical variant R457H in POR gene also give us a viewpoint that R457H mutation in POR gene maybe important in causing PORD in Chinese as same as in Japanese.
Similar content being viewed by others


Background
Cytochrome P450 oxidoreductase (POR) is encoded by the POR gene. POR transfers electrons from NADPH to cytochrome P450 enzymes for drug and toxin metabolism as well as steroid hormone synthesis (Fig. 1) [1]. Since P450 enzymes are involved in the catalysis of many substrates, POR deficiency (PORD) (OMIM: 613571 and OMIM: 201750) can lead to disorders of steroid hormone, drug and toxin metabolism, to various extents. PORD resulting from mutations in the POR gene has been reported by different teams since 2004 [2–6]. Currently, nearly 100 cases have been reported worldwide (Table 1). However, the treatment method of this rare disease is not clear due to the limited number of patients.

POR involvement in various biological pathways. POR donates electrons to all P450 enzymes, and at the same time, provides electrons for some proteins and small molecules, which involved in numerous biological activities such as biosynthesis, cholesterol metabolism, sex hormone metabolism and the metabolism of drugs and toxins
PORD can cause reduced activities of multiple enzymes participating in the synthesis of both adrenal and gonadal steroid hormones, which further leads to the dysfunction of the sex hormones and a decrease in glucocorticoids. Therefore, the disease is a type of special congenital adrenal hyperplasia (CAH). PORD is very rare and exhibits a variety of clinical manifestations, such as oligomenorrhea and ovarian cysts which resemble polycystic ovary syndrome (PCOS), elevated 17-hydroxy-progesterone (17OHP) which resembles 21-hydroxylase deficiency (21-OHD), and skeletal deformities which resemble Antley–Bixler syndrome (ABS). Therefore, in some cases, it can be difficult to differentiate PORD from PCOS, 21-OHD and ABS. However, PORD usually doesn’t exhibit hyperandrogenism which differs from PCOS and 21-OHD. In this study, we report a female adult Chinese PORD case who was misdiagnosed many times, and analyze the clinical presentation and genetic character of the patient and her parents.
Case presentation
Patient characteristics
A 27-year-old female (46,XX) patient with normal cognition visited the Endocrinology Clinic of 1st affiliated hospital of China Medical University (Shenyang) for amenorrhea with an unknown etiology. The patient had labial fusion when she was born and underwent plastic surgery afterward. During puberty, the patient underwent another surgery for an ovarian cyst. The patient was treated in another hospital as PCOS for several times resulting with mild remission of amenorrhea and recurrence of ovarian cysts. The patient was suspected of having 21-OHD due to increased 17OHP levels and came to our hospital for a genetic test for CYP21A2 mutations. The patient did not have fatigue, loss of appetite or other symptoms and was the only child in the family. Her parents appeared to be normal. She was born by vaginal delivery, and the mother had no complaints of abnormal manifestations during the perinatal period or history of reproductive system diseases. Her parents are nonconsanguineous. Physical examination of the patient revealed the following characteristics: a height of 165 cm and weight of 55 kg; no hyperandrogenism symptoms like hirsutism or acne; and no purple stripes. The patient complained of difficulty of bending the metacarpophalangeal joints from childhood (Fig. 2a-c). There were no obvious abnormalities in the genitals.

The appearance of the hands of the patient, hand DR and adrenal CT scans. a-c shows patient’s difficulty of bending the metacarpophalangeal joints, however her hand DR (d) suggested there were no bone abnormalities. The adrenal CT scan (e) showed there were no obvious hyperplasia changes
Clinical examination and testing
The ultrasound showed the presence of a 1.2 × 1.4 cm fluid area in the left ovary, and there was a 9.5 × 6.3 × 4.3 cm cyst in the right ovary behind the uterus. Digital radiography (DR) suggested there were no bone abnormalities (Fig. 2d). The adrenal CT scan showed there were no obvious hyperplasia changes (Fig. 2e). The laboratory test results of the patient and her parents are shown in Table 1. The levels of progesterone and 17-hydroxy progesterone of the patient were significantly increased. Androgen levels were not increased, and the basal level of cortisol in the morning was within the lower limit of the normal level. Cortisol was stimulated by the ACTH stimulation test. There were no abnormalities in the other measurements or in those of the parents.
Gene testing
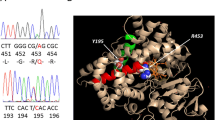
CYP21A2 (NM_000500) was tested first due to the suspicion of 21-OHD, but no mutations were found in the patient. Then the patient’s complaint about a mild difficulty of bending the metacarpophalangeal joints reminded us to consider PORD, which usually presents with skeletal deformities and sexual dysfunction, so POR (NM_000941) gene was tested secondly. Exon 11 of PORharbored a homozygous mutation (c.1370G > A) which leads to a conversion of arginine at amino acid position 457 to histidine (R457H). The patient’s parents were both heterozygous carriers of this variant (Fig. 3). Since the disease-causing homozygous mutations in POR gene were found, no further genetic analysis was performed.

POR gene testing results of the patient and her parents. Exon 11 of POR harbored a homozygous mutation (c.1370G > A) which leads to a conversion of arginine at amino acid position 457 to histidine (R457H). The patient’s parents were both heterozygous carriers of this variant
Treatment and follow up
Our patient once underwent surgery to remove a large ovarian cyst; however, since the diagnosis was unclear and hormone replacement therapy was not given, the ovarian cyst soon recurred. The patient was given low-dose corticosteroids twice a day (hydrocortisone 5 mg in the morning and 2.5 mg in the afternoon) and estrogen/progesterone sequential therapy in our hospital, and her ovarian cysts gradually decreased in size (the biggest ovarian cyst shrank to 3.5 × 3.3 × 2.3 cm) with regular menstruation in the following visits. The serum levels of ACTH, LH, 17OHP and P were declined accordingly (Table 2). No adverse effect of corticosteroids was found in the follow-up.
Discussion
PORD is a rare variant of CAH disease. As early as 1985, a CAH case which had characteristics of both 21-OHD and 17α-hydroxylase deficiency (17-OHD) was reported; however, no mutations were found in the patient in CYP21A2 or CYP17A1, two genes known to underlie these diseases [7]. In 2004, Fluck et al. reported that this new CAH disease was caused by a mutation in the POR gene, and they also showed that the mutation of POR could lead to a decrease in the activity of P450 enzymes including CYP21A2, CYP17A1 and CYP19A1 [6]. POR donates electrons to all P450 enzymes, and at the same time, provides electrons for some proteins and small molecules (Fig. 1). POR plays an important role in biological activities such as biosynthesis, cholesterol metabolism, sex hormone metabolism and the metabolism of drugs and toxins. In addition, the loss of POR enzyme activity varies with POR mutation type; thereby, PORD has a variety of clinical manifestations from menstrual disorders to severe hermaphroditism and skeletal malformations, and even fetal death. At present, around 100 PORD cases have been reported worldwide (Table 1). The proportion of female and male cases is roughly equal (54:46). Most of the reported patients are of Japanese or European descent (38:46). The majority are newborn or fetal cases (76%), and adults account for 24% reported cases. The primary clinical manifestations include abnormal hormone levels at birth or developmental delay after puberty (89.2%), hermaphroditism (75%), maternal virilization (40.8%), ABS-like skeletal abnormalities (82.7%), underlying adrenal insufficiency (74.6%) and ovary cysts in females (46.7%). Clinicians should always keep in mind of this disease in patients with sexual dysfunction, especially combined with special skeletal deformities.
Most of PORD patients presented with ambiguous genitalia (75%). The pathogenic mechanisms of this manifestation are associated with impaired syntheses of testosterone, dihydrotestosterone and conversions of the androgens to estrogens. These syntheses and conversions depend on CYP17A1, CYP19A1, CYP21A2, which require POR [8, 9]. In addition, maternal virilization (40.8%) is described in mothers during pregnancies of PORD fetus, and it is also due to placental aromatase (CYP19A1) deficiency [10].
Similar to 21-OHD, despite that the basal levels of ACTH and cortisol are generally normal in PORD patients, the ACTH stimulation test showed that most of the patients (74.6%) have insufficient adrenocortical reserve. Activities of different POR mutants for target enzymes would have a certain role in variability of adrenocortical insufficiency [11–13]. Patients with insufficient adrenocortical reserve may need long-term glucocorticoid replacement therapy [14], especially during disease, inflammation, stress or surgery. Mineralocorticoid dysfunction is not apparent in PORD patients. However, PORD patients who are homozygous for an A287P mutation have been found to have increased blood pressure (caused by an increase in deoxycorticosterone), which is similar to that seen in 17-OHD [15]. Thus, regular blood pressure monitoring is required for PORD patients.
Ovarian cysts were found in this patient and in other adult female patients (46.7%). If patients have a history of amenorrhea or anovulation, the disease is easily misdiagnosed as PCOS at primary medical units. The decrease in estrogen causes hypergonadotropic hypogonadism, which stimulates the growth of the ovaries. On the other hand, mutations in POR lead to the reduced activity of CYP51A1, thus reducing the synthesis of meiosis-activating sterol and further inducing a dysfunction in meiosis and the maturation of the oocytes. Under the above-mentioned “double hit”, compared with ovarian cysts associated with other hormone synthesis disorders, those in PORD are more difficult to control. Surgical treatment combined with corticosteroid and sex hormone replacement therapy is often required to prevent recurrence [14]. Our patient responded well to this therapy during follow-up.
Another important clinical feature of PORD is that there are different degrees of Antley-Bixler syndrome-like skeletal deformities (82.7%). Skeletal deformities include midface hypoplasia, craniosynostosis, foot and hand deformity, bony union of the large joints and femur bending. Some researchers have attempted to quantify these skeletal deformities and assess the severity according to quantitative indices. They found that compound heterozygous and homozygous mutations in the POR gene are associated with more serious skeletal deformities. This may also result from the effect of various mutations on the activity of related enzymes [16]. Our patient harbored a homozygous mutation but only had mild metacarpophalangeal joint contracture, which is not a typical skeletal deformity, showing the heterogeneity of this symptom in PORD. The pathogenic mechanisms of PORD-associated skeletal deformities may be associated with lanosterol 14α demethylase (CYP51A1) which involved in cholesterol biosynthesis [17] and retinoic acid which metabolized by microsomal CYP26 proteins [18].
P450 liver enzymes and heme oxygenase involving in drug metabolism need POR to provide electrons, therefore, the mutation of POR gene or associated SNPs may affect drug metabolism in human body [19, 20] and affect the progression of some diseases such as malaria and sepsis [21]. For example, the R457H mutation in this patient can inactivate the P450 enzymes including CYP1A2, CYP2C19, CYP2D6 and CYP3A4 which are important for drug metabolism [12, 22, 23]. Thus, there may be possible risks if the PORD patients are administered medications metabolized by these enzymes.
It has been reported that there are nearly 200 POR mutations and single nucleotide polymorphisms (SNPs) [24], including more than 60 missense mutations (http://www.cypalleles.ki.se/por.htm). According to the cases reported in the literature, the most common mutation type in the Japanese population is R457H (51/78, 65.4%), and it is A287P (39/92, 42.4%) in Europeans [6, 11, 13, 16, 25–33], which shows significant ethnic differences. R457H was once thought to be a founder mutation in Japan [34], but this mutation has also been reported in Europeans and other ethnicity [35]. There are few PORD case reports in China. By searching the literature published in Chinese, we found that only two PORD cases have been reported, and both cases were compound heterozygous carriers including R457H. The patient we report here is the third Chinese PORD patient to be described, and she is homozygous for R457H, with parents who are heterozygous, nonconsanguineous carriers. These genetic characters suggest R457H maybe common in the Chinese PORD patients, which are similar with Japanese.
Conclusions
In conclusion, PORD is a very rare disease which can be easily misdiagnosed in mild cases. Clinicians should keep in mind of this disease in patients with sexual dysfunction, especially combined with special skeletal deformities. Our data could provide a consciously understanding of this disease for clinic practicers. Low-dose corticosteroids combined with estrogen/progesterone sequential therapy will be effective in PORD patients with recurrence of large ovary cyst. The fact that the reported PORD patients in China carrying an identical variant R457H in POR gene also give us a viewpoint that R457H mutation in POR gene maybe also important in causing PORD in Chinese, which is similar with Japanese.
Abbreviations
- 17-OHD:
-
17α hydroxylase deficiency
- 17OHP:
-
17-hydroxy-progesterone
- 21-OHD:
-
21-hydroxylase deficiency
- ABS:
-
Antley–Bixler syndrome
- CAH:
-
Congenital adrenal hyperplasia
- CT:
-
Computer tomography
- PCOS:
-
Polycystic ovary syndrome
- POR:
-
Cytochrome P450 oxidoreductase
- PORD:
-
Cytochrome P450 oxidoreductase deficiency
References
Fluck CE, Nicolo C, Pandey AV. Clinical, structural and functional implications of mutations and polymorphisms in human NADPH P450 oxidoreductase. Fundam Clin Pharmacol. 2007;21:399–410.
Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A. Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am J Med Genet A. 2004;128A:333–9.
Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EM, et al. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet. 2004;363:2128–35.
Pandey AV, Fluck CE, Huang N, Tajima T, Fujieda K, Miller WL. P450 oxidoreductase deficiency: a new disorder of steroidogenesis affecting all microsomal P450 enzymes. Endocr Res. 2004;30:881–8.
Miller WL, Huang N, Fluck CE, Pandey AV. P450 oxidoreductase deficiency. Lancet. 2004;364:1663.
Fluck CE, Tajima T, Pandey AV, Arlt W, Okuhara K, Verge CF, Jabs EW, Mendonca BB, Fujieda K, Miller WL. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet. 2004;36:228–30.
Peterson RE, Imperato-McGinley J, Gautier T, Shackleton C. Male pseudohermaphroditism due to multiple defects in steroid-biosynthetic microsomal mixed-function oxidases. A new variant of congenital adrenal hyperplasia. N Engl J Med. 1985;313:1182–91.
Fukami M, Homma K, Hasegawa T, Ogata T. Backdoor pathway for dihydrotestosterone biosynthesis: implications for normal and abnormal human sex development. Dev Dyn. 2013;242:320–9.
Fluck CE, Pandey AV. Clinical and biochemical consequences of p450 oxidoreductase deficiency. Endocr Dev. 2011;20:63–79.
Shackleton C, Marcos J, Arlt W, Hauffa BP. Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): a disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype. Am J Med Genet A. 2004;129a:105–12.
Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, Jabs EW, Van Vliet G, Sack J, Fluck CE, Miller WL. Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005;76:729–49.
Agrawal V, Huang N, Miller WL. Pharmacogenetics of P450 oxidoreductase: effect of sequence variants on activities of CYP1A2 and CYP2C19. Pharmacogenet Genomics. 2008;18:569–76.
Fukami M, Nishimura G, Homma K, Nagai T, Hanaki K, Uematsu A, Ishii T, Numakura C, Sawada H, Nakacho M, et al. Cytochrome P450 oxidoreductase deficiency: identification and characterization of biallelic mutations and genotype-phenotype correlations in 35 Japanese patients. J Clin Endocrinol Metab. 2009;94:1723–31.
Idkowiak J, O’Riordan S, Reisch N, Malunowicz EM, Collins F, Kerstens MN, Kohler B, Graul-Neumann LM, Szarras-Czapnik M, Dattani M, et al. Pubertal presentation in seven patients with congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2011;96:E453–62.
Tomalik-Scharte D, Maiter D, Kirchheiner J, Ivison HE, Fuhr U, Arlt W. Impaired hepatic drug and steroid metabolism in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. Eur J Endocrinol. 2010;163:919–24.
Krone N, Reisch N, Idkowiak J, Dhir V, Ivison HE, Hughes BA, Rose IT, O’Neil DM, Vijzelaar R, Smith MJ, et al. Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2012;97:E257–67.
Debeljak N, Fink M, Rozman D. Many facets of mammalian lanosterol 14alpha-demethylase from the evolutionarily conserved cytochrome P450 family CYP51. Arch Biochem Biophys. 2003;409:159–71.
Laue K, Pogoda HM, Daniel PB, van Haeringen A, Alanay Y, von Ameln S, Rachwalski M, Morgan T, Gray MJ, Breuning MH, et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011;89:595–606.
Subramanian M, Agrawal V, Sandee D, Tam HK, Miller WL, Tracy TS. Effect of P450 oxidoreductase variants on the metabolism of model substrates mediated by CYP2C9.1, CYP2C9.2, and CYP2C9.3. Pharmacogenet Genomics. 2012;22:590–7.
Pandey AV, Fluck CE. NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol Ther. 2013;138:229–54.
Pandey AV, Fluck CE, Mullis PE. Altered heme catabolism by heme oxygenase-1 caused by mutations in human NADPH cytochrome P450 reductase. Biochem Biophys Res Commun. 2010;400:374–8.
Fluck CE, Mullis PE, Pandey AV. Reduction in hepatic drug metabolizing CYP3A4 activities caused by P450 oxidoreductase mutations identified in patients with disordered steroid metabolism. Biochem Biophys Res Commun. 2010;401:149–53.
Sandee D, Morrissey K, Agrawal V, Tam HK, Kramer MA, Tracy TS, Giacomini KM, Miller WL. Effects of genetic variants of human P450 oxidoreductase on catalysis by CYP2D6 in vitro. Pharmacogenet Genomics. 2010;20:677–86.
Huang N, Agrawal V, Giacomini KM, Miller WL. Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc Natl Acad Sci U S A. 2008;105:1733–8.
Parween S, Roucher-Boulez F, Fluck CE, Lienhardt-Roussie A, Mallet D, Morel Y, Pandey AV. P450 oxidoreductase deficiency: loss of activity caused by protein instability from a novel L374H mutation. J Clin Endocrinol Metab. 2016;101:4789–98.
Bonamichi BD, Santiago SL, Bertola DR, Kim CA, Alonso N, Mendonca BB, Bachega TA, Gomes LG. Long-term follow-up of a female with congenital adrenal hyperplasia due to P450-oxidoreductase deficiency. Arch Endocrinol Metab. 2016;60:500–4.
Tzetis M, Konstantinidou A, Sofocleous C, Kosma K, Mitrakos A, Tzannatos C, Kitsiou-Tzeli S. Compound heterozygosity of a paternal submicroscopic deletion and a maternal missense mutation in POR gene: Antley-bixler syndrome phenotype in three sibling fetuses. Birth Defects Res A Clin Mol Teratol. 2016;106:536–41.
Koika V, Armeni AK, Georgopoulos NA. Delayed diagnosis of disorder of sex development (DSD) due to P450 oxidoreductase (POR) deficiency. Hormones (Athens). 2016;15:277–82.
Fluck CE, Mallet D, Hofer G, Samara-Boustani D, Leger J, Polak M, Morel Y, Pandey AV. Deletion of P399_E401 in NADPH cytochrome P450 oxidoreductase results in partial mixed oxidase deficiency. Biochem Biophys Res Commun. 2011;412:572–7.
Sahakitrungruang T, Huang N, Tee MK, Agrawal V, Russell WE, Crock P, Murphy N, Migeon CJ, Miller WL. Clinical, genetic, and enzymatic characterization of P450 oxidoreductase deficiency in four patients. J Clin Endocrinol Metab. 2009;94:4992–5000.
Ko JM, Cheon CK, Kim GH, Yoo HW. A case of Antley-Bixler syndrome caused by compound heterozygous mutations of the cytochrome P450 oxidoreductase gene. Eur J Pediatr. 2009;168:877–80.
Hershkovitz E, Parvari R, Wudy SA, Hartmann MF, Gomes LG, Loewental N, Miller WL. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20-lyase deficiency. J Clin Endocrinol Metab. 2008;93:3584–8.
Scott RR, Gomes LG, Huang N, Van Vliet G, Miller WL. Apparent manifesting heterozygosity in P450 oxidoreductase deficiency and its effect on coexisting 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:2318–22.
Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, Okuyama T, Nakai H, Soneda S, Tachibana K, et al. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab. 2005;90:414–26.
Adachi M, Asakura Y, Matsuo M, Yamamoto T, Hanaki K, Arlt W. POR R457H is a global founder mutation causing Antley-Bixler syndrome with autosomal recessive trait. Am J Med Genet A. 2006;140:633–5.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets during and/or analyzed during the current study available from the corresponding author on reasonable request.
Authors’ contributions
YB, JHL and XLW carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. JHL participated in the sequence alignment. XLW participated in the design of the study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
The patient and her family provided written informed consent for publication of their data.
Ethics approval and consent to participate
The Hospital Ethics Committee of the First Hospital of China Medical University approved the study.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bai, Y., Li, J. & Wang, X. Cytochrome P450 oxidoreductase deficiency caused by R457H mutation in POR gene in Chinese: case report and literature review. J Ovarian Res 10, 16 (2017). https://doi.org/10.1186/s13048-017-0312-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-017-0312-9