
Abstract
Background
Ovarian cancer (OCA), the fifth leading deaths cancer to women, is famous for its low survival rate in epithelial ovarian cancer cases, which is very complicated and hard to be diagnosed from asymptomatic nature in the early stage. Thus, it is urgent to develop an effective genetic prognostic strategy.
Methods
Current study using the Database for Annotation, Visualization and Integrated Discovery tool for the generation and analysis of quantitative gene expression profiles; all the annotated gene and biochemical pathway membership realized according to shared categorical data from Pathway and Kyoto Encyclopedia of Genes and Genomes; correlation networks based on current gene screening actualize by Weighted correlation network analysis to identify therapeutic targets gene and candidate bio-markers.
Results
3095 differentially expressed genes were collected from genome expression profiles of OCA patients (n = 53, 35 advanced, 8 early and 10 normal). By pathway enrichment, most genes showed contribution to cell cycle and chromosome maintenance.1073 differentially expression genes involved in the 4 dominant network modules are further generated for prognostic pattern establish, we divided a dataset with random OCA cases (n = 80) into 3 groups efficiently (p = 0.0323, 95 % CIs in Kaplan-Meier). Finally, 6 prognosis related genes were selected out by COX regression analysis, TFCP2L1 related to cancer-stem cell, probably contributes to chemotherapy efficiency.
Conclusions
Our study presents an integrated original model of the differentially expression genes related to ovarian cancer progressing, providing the identification of genes relevant for its pathological physiology which can potentially be new clinical markers.
Similar content being viewed by others

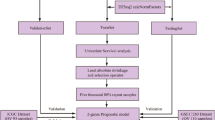

Background
As the fifth leading cause of cancer related to deaths, ovarian cancer just have only 30–40 % with a five year survival rate in women [1]. In American, there were 219,800 new cases and around 142,700 women succumb to this fatal disease in 2014 [2], and the lifetime risk of epithelial ovarian cancer is one of 72 women [3], what is much more worse in developing countries [4, 5]. Ovarian tumors can be classified into epithelial (60 %), germ cell (30 %) and sex-cord stromal tumors (8 %) [6], among which the vast majority of malignant ovarian cancers (80 %-85 %) [7] began in the ovarian epithelium. Unfortunately, ovarian cancer is highly asymptomatic at early stages, the epithelial ovarian cancer (EOC) is hard to be diagnosed due to the multitude of clinical and histopathological aspects [8], lack of precursor lesions [9] and their evolution [10], thus most patients with EOC are diagnosed at advanced stage and have a poor prognosis. It is reported that only 30 % in stage I or II could be cured by surgery with five-year survival rate of 90 %, in contrast, EOC in stages III or IV could spread throughout widely with 5-year survival of less than 30 % [11, 12]. Besides, malignant ovarian germ cell tumor is hard to be distinguished by medical image and always happen to young women below the age of 20.
Recently, more and more reports declared that ovarian cancer has home history, about 10 % of EOC cases are related to inherited genes like BCRA1and BCRA2 [13, 14], and ovarian germ cell tumors can be cancerous or non-cancerous tumors depend on genome difference [15–17]. Actually, cancer is a disease single of genomes or networks of molecular interaction and control, advanced ovarian cancer with a high relapse rate related to the acquirement of chemo resistance, due to it’s ability to converting the tumor cells back into a stem cell-like state. Luckily, several existing drugs [18, 19, 14] can attack the pathway and reverse the cellular transformation, thus ‘re-sensitizing’ the tumor to treatment. For these reasons, it is urgently to develop effective strategies to stratify early and advance stage patients.
Correlation networks are increasingly being used in bioinformatics applications like generating modules (clusters) of highly correlated genes, summarizing such modules using an intra-modular hub gene or the eigengene, and analysis of modules’ networks or calculating module membership measures, which can be used to identify candidate biomarkers or therapeutic targets. Currently, we use weighted correlation network analysis (WGCNA) to correlate networks facilitate network based standardized and screened gene, aim at establish an a feasible genetic method to prognostic of outcome of individual’s ovarian carcinoma, especially the bottleneck problem of epithelial ovarian cancer and malignant ovarian germ cell cancer, therefore, making an advantage to choose the most suitable chemotherapy for a certain patient.
Materials
As the paper did not involve any human or animal study, there was no need for any ethical approval.
Literature selecting and building
For analysis of differential genome-wide expression between patients in different cancer stages, we selected GSE12470 dataset [20], including gene array data from 35 advanced ovarian cancer patients, 8 early ovarian cancer patients and 10 non-cancer persons.
For prognostic analysis on different types of ovarian cancer, GSE14764 data set [21] was selected, which includes genome-wide expression data from 80 ovarian cancer patients. In addition, GSE63885 dataset [22] and GSE49997 dataset [23] were tested to verify the established prognostic analysis model, these two data sets are consisted of genome-wide expression data from 101 candidates with differential ovarian cancer and mRNA expression data from 204 candidates suffered from ovarian cancer respectively.
Database search
Gene Expression Omnibus [24] functional genomics repository was searched for the relationships between the probe in the platforms used in the selected datasets and corresponding genes. One probe set (contain several probes, N ≥1) matching one target gene, therefore average value [25] of different corresponding probe IDs is represent one gene expression level. Skew distribution of gene expression was transformed to skew normal distribution by log2 transformed and final probe set level data was generated through Robust Multichip Analysis [26] (a model-based algorithms) with default parameters [27].
Screening of differentially expressed genes
After expression data for post-processing of standardization, we directly employed a more mature significance analysis of microarrays (SAM) algorithm [28]. Differentially expressed genes were screened by using t-test and analysis of variance, if N is large number of our genes, it will generate a lot of false positives, then use controlling the FDR (false discovery rate) values corrected for multiple testing in the false-positive rate. Calculate the relative difference statistic d:
d, statistic measures the relative differences in gene expression are corrected d statistics. X1 ‘represents the average expression level of a state under genes, X2’ represents the average expression levels of gene, s represents the variance of the gene.
Construction of co-expression network and module-mining
Construction of co-expression network mining based on the differentially expression value, weighted correlation network analysis (WGCNA) [29] was used for finding modules of correlated differential genes, summarizing such modules, relating modules to one another, and weighting module’s membership and contributed genes. All the genes used in WGCNA methods had been screened as previously described.
Screening of differentially expressed module
Specific gene regulatory network module were screened in two conditions, and then determine the gene for each module in the two states within the overall expression differences, using a global analysis of variance method [30], Global-Ancova method based on correlation analysis of variance test set and a set of functional gene phenotype, P value tested with less than 0.05 network modules selected as differentially expressed module. The method may be R language Global-Ancova package implementation.
Enrichment analysis of gene function
For a group identified gene sets, we used DAVID [31] tool - a software is based on the hyper geometric enrichment test methods of distribution test, to achieve function and the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/) [32] pathway enrichment analysis.
Survival analysis
By statistical analysis, we able to achieve some network modules consisted of differential selected genes with some chemotherapy related regulation factors. In view of these differential genes, we classified dataset GSE14764, GSE63885 and GSE49997 into subgroups, all the candidates are treated by chemotherapy. Prognosis analyses were conducted by SURVICAL package in R environment, and Kaplan-Meier estimates of overall survival (OS) respective 95 % confidence intervals (CIs) were provided for each cluster. In addition, for each dataset, Cox regression modeling [33] was used to control and assess for statistically significant prognostic factors, included adjustments for age, histology, and stage. Then the Pairwise comparisons between clusters were carried out in Cox model, based on calculated p-values, genes with p < 0.05 are considered to be relevant to the clinical characteristics and prognosis of ovarian cancer.
Results
Overall gene-expression profiling standardization
As previously described, we generated original genome expression profiling and mRNA expression data got from each data set (GSE12470, GSE14764, GSE49997 and GSE63885), after using GEO database matching the probe ID in the platform to Gene Symbols, corresponding genes and gene’s IDs were collected from these data sets respectively. A quantitative genome expression distributions map are showed in type of box-plots (see Fig. 1), values from each dataset were linearized when provided as logarithms, raw files were converted into pre-processed data by RMA with default parameters [27].

Box-plots of the distribution of gene expression values for analysis of ovarian cancer gene expression profiling (with a p-value of 0.05 and FC of 2.0). The abscissa represents each candidate IDs, while the vertical axis marks the data of genome expression of related patients, all the datein genome expression profiling are the mean value of many experiment locations. a GSE12470 dataset, 13356 genes in 53 samples (10 normal, 35 advanced, 8 early), (b) GSE14764dataset, 80 samples in different types of ovarian cancer and 13046 genes, C) GSE49997 dataset, 204 samples in various epithelial ovarian cancer and 16150 genes, D) GSE63885 dataset, 101 samples in different ovarian cancer and mRNA and 20693 genes. a, b, c are consisted of gene-expression data, while (d) reveals mRNA expression levels. Apparently, most genes’ expression values are approximately in each sample
Ovarian cancer’s genetic screening and pathways analysis
According to the procedure adopted by Dai et al. [26], pre-processed data of 53 samples (Fig. 1a) were analyzed by SAM in R environment, samples including data from patients in various stage and non-cancer individuals. Lists of 3095 differentially expressed genes are collected (Accompanying Table 1), showing (i.e., fold change (FC) equals 2.0) were generated at SAM p-value thresholds of 5 %.
To identify the biological processes associated with these 3095 differential expressed genes, we explore the DAVID; http://david.abcc.ncifcrf.gov/). Compared with online human genome database, the top 10 enriched clusters with the 511 genes mainly distributed at cell cycle including mitosis, deposition of nucleosomes at the centromere, Chromosome Maintenance including Chromosome, telomere maintenance and nucleosome assembly, Regulation of RNA transcription level including RNA polymerase I (Table 1, Accompanying Table 2).
Based on these 511 genes related to top 10 pathways, overall 80 candidates were completely clustered by principal component analysis (PCA), which indicates a high-performance of differences genetic screening (Fig. 2).

Clustering map base on 511 screened differential genes. Red spot indicate healthy individual, spot in blue indicate patient suffering from ovarian cancer, spots in different colors are effectively separated from each other
Differences genetic screening and pathways analysis on ovarian cancer in different stages
By using WGCNA software in R language, gene co-expression networks (Accompanying Table 3) are established from 3095 differential expression genes (Accompanying Table 1). Each gene was weighted and ranked by calculating the network edges, top 10 are showed in Table 2, Gene RACGAP1 [34], RAD51AP1 [35], RAE1 [36], NEK2 [37] had been reported as ovarian cancer related genes, while the others are newly defined related gene. In addition, these 3095 genes were divided into 17 modules (Table 3) by the block-wise, Modules function of WGCNA package. After further screening on Global-Ancova package using R language and comparing original gene-expression data set GSE12470, 4 network modules of differentially expressed cancer genes were identified (Table 4, details showed in Accompanying Table 4) as the representative module to apply function analyses because most of genes in the network are expressed in the candidate who suffered from cancer.
GO and KEGG analysis on these 4 modules (Table 4) shows blue modules is mainly take part in female metabolism regulation and controlling: Androgen and estrogen metabolism and Steroid hormone biosynthesis which straightly related to ovarian functions, Aminoacyl-tRNA biosynthesis which play a key role in protein synthesis [38] and has been suggested to be associated with the progression of various ovarian cancers [39, 40], most interested is porphyrin and chlorophyll metabolism pathways also be involved into ovarian cancer progression, porphyrin was reported as treatment elements for ovarian cancer [41], while chlorophyll as important grapevine iron nutrition for blood [42, 43] which most females are short for it [44], besides, some reporter illustrated cancer resistance protein can against the porphyrin and chlorophyll metabolism [45], thus, blue module may potentially denotes the progress of ovarian cancer and support to our subsequence prognosis analysis. Besides, gene UMPS ranked second was involved in pathway of aminoacyl-tRNA biosynthesis, further suggests that UMPS could be related to a certain ovarian cancer. And gene IARS belongs to drug metabolism pathway in blue module ranked eighth in Table 2, suggesting that this gene maybe important for applicability of drug treatment in specific case.
Greenyellow module is mainly related to PPAR signaling pathway, which is involved in ovarian follicle development [46] and ovarian cancers progress [47]. Grey module is mainly devoted to melanogenesis. Presently, no representation shows melanogenesis is related to cancer progression, but melanogenesis is regarded as a potential instruction for understanding of complex diseases [48]. In currently study, we select the modules to evaluate ovarian cancer in different stages and various types, thus, this module probably take an important part in subsequence prognosis analysis for patients in various conditions, beside, the other functions of this modules also help to analysis cancer proceeding like amino acids metabolism and energy homeostasis. Tan module is mainly devoted to carbohydrates and sucrose metabolism, and this is a risk factor for many cancer [49] and female ovarian health [50], also very important to diagnosis of advanced ovarian cancer patients [51, 52].
All these supported researches and relevance data illustrated that we had generated network modules from differential expression genes of various ovarian cancers successfully, and these networks are competent for predict ovarian cancer’ subgroups, also potentially indicate the proceeding of ovarian cancer in different patients.
Prognostic analysis of subgroups of ovarian cancers
1073 differential expression genes involved in the 4 dominant network modules were generated from GSE12470 expression dataset as previous described. By using SUVIVLE package in R basing on these differential genes, GSE14764 dataset composed by various ovarian cancer patients’ gene expression profiles (n = 80) were classified into 3 subgroups (Fig. 3a). Pair wise comparisons between clusters based on p-values were carried out by Kaplan-Meier estimates of OS respective 95 % confidence intervals (CIs). Kaplan-Meier estimates of Fig. 3a has been showed in Fig. 3b with P = 0.0323.

Cluster analysis: Heat map profiles of ovarian cancer patients with 1073 extracted differential genes from GSE12470 data set (n = 53). a Heat map profiles of extracted differentiated genes and various ovarian cancer patients from GSE14764 dataset (genome expression, n = 80), the Kaplan-Meier curves are with respect to (b) overall survival (OS) rite at non-significant P = 0.0323, (c) Heat map profiles of extracted differentiated genes and various ovarian cancer patients from GSE49997 dataset (mRNA expression, n = 204), corresponding Kaplan-Meier curves (d) with a non-significant P = 1.02e - 05, (e) Heat map profiles of extracted differentiated genes and various subtypes of epithelial ovarian cancer patients from GSE63885 dataset (genome expression, n = 101), the Kaplan-Meier curves are with respect to (f) overall survival (OS) rite at non-significant P =0.0781,A) is for prognosis trials, (c, e) are used to verify the availability of selected modules and extracted differential expression genes. All estimates of OS respective 95 % confidence intervals (Cis)
In order to verify the availability of the prognostic functions of these 4 modules, we useGSE49997 and GSE63885 datasets to repeat the same experiment. GSE49997 [23] is composed by mRNA expression data from epithelial ovarian cancer patients (n = 204) while GSE63885 datasets are consisted by genome expression data from various ovarian cancers. According to the original articles, candidates in GSE49997 [23] dataset are classified into the clinic-pathologic parameters of the histological serous and non-serous tumor subtypes, each subtypes can be divided into 2 subclasses derived from International Federation of Gynecology and Obstetrics stage-directed supervised classification approach (IFGO). One group’s (subclass2) conditions deteriorated extremely from a certain time point and appear much lower livability in both serous and non-serous histological subtypes than another (subclass1)’s, as revealed by univariate analysis (hazard ratios [HR] of 3.17 and 17.11, respectively; P 0.001) and in models corrected for relevant clinic pathologic parameters (HR 2.87 and 12.42, respectively; P 0.023). Similarly, candidates in GSE63885 [22] datasets adapt the same classification approach(IFGO), and they discovered that histological type could be a confusing factor and gene expression exploration of ovarian carcinomas should be performed on histologically homogeneous groups to direct the prognostic analysis on chemotherapy. In their experiment, clinical endpoints like overall survival, disease-free survival, tumor response to chemotherapy are not confirmed by validation either on the same group or on the independent group of patients, just CLASP1 gene with BRCA1 mutation status related to one ovarian cancer subclass which tend to deteriorate easily.
Comparatively, heat map profiles in current researches (Fig. 3c and Fig. 3e showed) showed the samples from GSE49997 and GSE63885 dataset had been efficiently divided into 2 groups base on the same differential expressed genes and 4 network modules used in Fig. 3a, which are identical with the original dataset information. In Kaplan-Meier estimates of OS respective 95 % confidence intervals (CIs) were provided for these two heat maps with p equals to 1.02e-05 and p equals to 0.0781 respectively. According to these two verification models and similarities in classification to original data sources we described above, the selected 1073 different genes in 4 majority network modules is competent to classify ovarian cancer into subtypes that are prognostic of different chemotherapy outcome, especially for epithelial ovarian cancer and ovarian germ cell cancer (especially for stage 4 and stage 5), which are notorious for diagnosis and distinction at the early stage with analogous morphological characteristics. In addition, the modules we established may prefer much more accuracy and practicability, as GSE63885 [22] datasets with less stringent criteria for gene selection (FDR <10%and uncorrected p-value <0.001).
For further extraction and prognosis of genes directly related to ovarian cancer survival, we used univariate COX regression method to calculate the correlation between genes and survival prognosis within the module, GSB14764 dataset genes associated with prognosis in a total of 35 genes; GSE49997 dataset and prognosis related genes, a total of 47 genes (Additional file 1: Table S5); GSE63885 dataset and prognosis with a total area of Venn diagram with 57 genes (Additional file 1: Table S6). View these three ovarian cancer prognostic gene intersection situations, find the intersection between any two relatively small (Fig. 4), the intersection of the six genes LRRC8D, TTC304, TFCP2L1, LIBRINEPOR, PAR52. Outstandingly, dysregulation of this EPOR may affect the growth of certain tumors [53, 54].

Three data sets COX univariate regression analysis were screened for ovarian cancer prognostic gene Venn diagram
Discussion
As previously described, ovarian cancer like epithelial ovarian cancer and ovarian germ cell cancer has difference subclasses, but it is hard to distinguish the malignant from carcinoid tumors due to the multitude of clinical and history pathological aspects [8], lack of precursor lesions [9] and their evolution [10], which cause the bad one with a low survival rate and complicated due to frequent development of resistance to standard therapies and asymptomatic nature of the early stage. Thus, recently, more and more researches are focus on genome level analysis aim at recognize collaborative gene and relatively network modules, which will bring out some newly efficiently diagnoses, and help to the cancer prevent and treatment to individuals base on targeted chemotherapy.
Current established genetic ovarian carcinomas prognostic pattern contains 1073 difference expression genes involved in the 4 dominant network modules successfully divided a dataset with random OCA cases (n = 80) into 3 groups (p = 0.0323, 95 % CIs in Kaplan-Meier). Two other previously reported datasets verified this classification is available and can be used in both genome (n = 204, p =1.02e-05, 95 % CIs in Kaplan-Meier) and mRNA (n = 101, p =0.0781, 95 % CIs in Kaplan-Meier) profiles, also demonstrated that this pattern can be used to distinguish epithelial ovarian cancer and ovarian germ cell cancer subclasses that trend todevelopmalignantly.6 prognosis related genes were selected by COX regression analysis (LRRC8D, TTC30A, TFCP2L1, LMBR1, EPOR and PARS2), these difference genes regulate modules through the whole work, rather than a few genes play a prognostic classification, which can make the outcome much more convincing. Beyond them, EPOR is famous for its affection to tumor growth [53, 54], support the function to divide the malignant epithelial ovarian cancer or ovarian germ cell cancer from carcinoid tumors;TTC30A and LRRC8D are rarely reported before, but recent statistics shows that these two gene related to immune system, and may have regulation ability to host protein [55–57], these can be considered in chemotherapy methods choosing. In addition, corresponding to earlier pathway analysis (Aminoacyl-tRNA biosynthesis in blue module, Table 4), PARS2 encodes a putative member of the class II family of aminoacyl-tRNA synthetases, further suggested a highly correlated gene networks in currently generated modules. What is importantly is that TFCP2L1 probably contribute to the differentiation of cancer stem cells, as embryonic stem cell self renewal pathways converge on the transcription factor Tfcp2l1 [58], and this never been reported before.
The present study describes a validation analysis of a previously defined gene signature to establish its relevance as a clinically useful prognostic factor. While the accuracy of prognostic outcome restricted by two elements, the routine use of recently published new prognostic factors in clinical practice has had limited success, and the updated gene databases.
References
Chaudhury S, Maheshwari A, Ray P. Ovarian Cancer: An ever challenging malady. Biomed Res J. 2014;34.
Tewari K, Monk B. Introduction to ovarian cancer. The 21st Century Handbook of Clinical Ovarian Cancer. Berlin: Springer; 2015. 1–10.
Eisenhauer E, Salani R, Copeland L. Epithelial ovarian cancer. Clinical Gynecologic Oncology. 8th ed. Philadelphia: Elsevier Saunders; 2012. p. 285–328.
Huang Z, Gao Y, Wen W, Li H, Zheng W, Shu XO, et al. Contraceptive methods and ovarian cancer risk among Chinese women: A report from the Shanghai Women’s Health Study. Int J Cancer. 2015;137:607–14.
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29.
Kurman RJ, Cancer IAfRo, World Health Organization. WHO classification of tumours of female reproductive organs. International Agency for Research on Cancer. 2014.
Devouassoux-Shisheboran M, Genestie C. Pathobiology of ovarian carcinomas. Chin J Cancer. 2015;34:50–5.
Mostofi FK, Sesterhenn IA. Histological typing of testis tumours. Hong Kong: Springer Science & Business Media; 2012
Tolcher MC, Swisher EM, Medeiros F, Lima JF, Hilderbrand JL, Donovan JL, et al. Characterization of Precursor Lesions in the Endometrium and Fallopian Tube Epithelium of Early-Stage Uterine Serous Carcinoma. Int J Gynecol Pathol. 2015;34:57–64.
Fotopoulou C, Zang R, Gultekin M, Cibula D, Ayhan A, Liu D, et al. Value of tertiary cytoreductive surgery in epithelial ovarian cancer: an international multicenter evaluation. Ann Surg Oncol. 2013;20:1348–54.
Gadducci A, Cosio S, Zola P, Sostegni B, Fuso L, Sartori E. Prognostic factors and clinical outcome of patients with recurrent early-stage epithelial ovarian cancer: an Italian multicenter retrospective study. Int J Gynecol Cancer. 2013;23:461–8.
Cliby WA, Powell MA, Al-Hammadi N, Chen L, Philip Miller J, Roland PY, et al. Ovarian cancer in the United States: Contemporary patterns of care associated with improved survival. Gynecol Oncol. 2015;136:11–7.
Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012;307:382–9.
Funes JM, Henderson S, Kaufman R, Flanagan JM, Robson M, Pedley B, et al. Oncogenic transformation of mesenchymal stem cells decreases Nrf2 expression favoring in vivo tumor growth and poorer survival. Mol Cancer. 2014;13:20.
van der Zwan YG, Biermann K, Wolffenbuttel KP, Cools M, Looijenga LH. Gonadal Maldevelopment as Risk Factor for Germ Cell Cancer: Towards a Clinical Decision Model. Eur Urol. 2014;67:692–701.
Lin KY, Bryant S, Miller DS, Kehoe SM, Richardson DL, Lea JS. Malignant ovarian germ cell tumor—Role of surgical staging and gonadal dysgenesis. Gynecol Oncol. 2014;134:84–9.
Hennes E, Zahn S, Lopes L, Schönberger S, Leuschner I, Göbel U, et al. Molecular genetic analysis of bilateral ovarian germ cell tumors. Klin Padiatr. 2012;224:359–65.
Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim Biophys Acta-Mol Basis Dis. 2013;1832:606–17.
Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse ML, et al. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013;32:4825–35.
Yoshihara K, Tajima A, Komata D, Yamamoto T, Kodama S, Fujiwara H, et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009;100:1421–8. doi:10.1111/j.1349-7006.2009.01204.x.
Denkert C, Budczies J, Darb‐Esfahani S, Györffy B, Sehouli J, Könsgen D, et al. A prognostic gene expression index in ovarian cancer—validation across different independent data sets. J Pathol. 2009;218:273–80.
Lisowska KM, Olbryt M, Dudaladava V, Pamuła-Piłat J, Kujawa K, Grzybowska E, et al. Gene expression analysis in ovarian cancer - faults and hints from DNA microarray study. Front Oncol. 2014;4:6. doi:10.3389/fonc.2014.00006.
Pils D, Hager G, Tong D, Aust S, Heinze G, Kohl M, et al. Validating the impact of a molecular subtype in ovarian cancer on outcomes: a study of the OVCAD Consortium. Cancer Sci. 2012;103:1334–41. doi:10.1111/j.1349-7006.2012.02306.x.
Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, et al. NCBI GEO: mining millions of expression profiles--database and tools. Nucleic Acids Res. 2005;33:D562–566. doi:10.1093/nar/gki022.
Ma H, Schadt EE, Kaplan LM, Zhao H. COSINE: COndition-SpecIfic sub-NEtwork identification using a global optimization method. Bioinformatics. 2011;27:1290–8. doi:10.1093/bioinformatics/btr136.
Carter SL, Eklund AC, Mecham BH, Kohane IS, Szallasi Z. Redefinition of Affymetrix probe sets by sequence overlap with cDNA microarray probes reduces cross-platform inconsistencies in cancer-associated gene expression measurements. BMC Bioinformatics. 2005;6:107.
Ferrari F, Bortoluzzi S, Coppe A, Sirota A, Safran M, Shmoish M, et al. Novel definition files for human GeneChips based on GeneAnnot. BMC Bioinformatics. 2007;8:446. doi:10.1186/1471-2105-8-446.
Larsson O, Wahlestedt C, Timmons JA. Considerations when using the significance analysis of microarrays (SAM) algorithm. BMC Bioinformatics. 2005;6:129. doi:10.1186/1471-2105-6-129.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi:10.2202/1544-6115.1128.
Mansmann U, Meister R. Testing differential gene expression in functional groups. Goeman’s global test versus an ANCOVA approach. Methods Inf Med. 2005;44:449–53. doi:10.1267/METH05030449.
Dennis Jr G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3.
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205.
Chan AT, Leung S, Ngan RK, Teo PM, Lau WH, Kwan WH, et al. Overall survival after concurrent cisplatin–radiotherapy compared with radiotherapy alone in locoregionally advanced nasopharyngeal carcinoma. J Natl Cancer Inst. 2005;97:536–9.
Lu KH, Patterson AP, Wang L, Marquez RT, Atkinson EN, Baggerly KA, et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004;10:3291–300. doi:10.1158/1078-0432.CCR-03-0409.
Miles GD, Seiler M, Rodriguez L, Rajagopal G, Bhanot G. Identifying microRNA/mRNA dysregulations in ovarian cancer. BMC Res Notes. 2012;5:164. doi:10.1186/1756-0500-5-164.
Barber A, Zhang T, DeMars LR, Conejo-Garcia J, Roby KF, Sentman CL. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67:5003–8. doi:10.1158/0008-5472.CAN-06-4047.
Liu X, Gao Y, Lu Y, Zhang J, Li L, Yin F. Upregulation of NEK2 is associated with drug resistance in ovarian cancer. Oncol Rep. 2014;31:745–54. doi:10.3892/or.2013.2910.
Mirande M. Aminoacyl-tRNA synthetase family from prokaryotes and eukaryotes: structural domains and their implications. Prog Nucleic Acid Res Mol Biol. 1991;40:95–142.
Kim S, You S, Hwang D. Aminoacyl-tRNA synthetases and tumorigenesis: more than housekeeping. Nat Rev Cancer. 2011;11:708–18.
Choi JW, Lee J-W, Kim JK, Jeon HK, Choi JJ, Kim DG, et al. Splicing variant of AIMP2 as an effective target against chemoresistant ovarian cancer. J Mol Cell Biol. 2012;4:164–73.
Luo T, Wilson BC, Lu Q-B. Evaluation of one-and two-photon activated photodynamic therapy with pyropheophorbide-a methyl ester in human cervical, lung and ovarian cancer cells. J Photochem Photobiol B Biol. 2014;132:102–10.
Tessarin P, Ingrosso E, Rombolà A, Boliani A, Covarrubias J, Yunta F, editors. Improvement of grapevine iron nutrition by a bovine blood-derived compound. VII International Symposium on Mineral Nutrition of Fruit Crops 984. 2012.
Qu J, Ma L, Zhang J, Jockusch S, Washington I. Dietary chlorophyll metabolites catalyze the photoreduction of plasma ubiquinone. Photochem Photobiol. 2013;89:310–3.
Hefferon KL. Nutritionally enhanced food crops; progress and perspectives. Int J Mol Sci. 2015;16:3895–914.
Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm. 2012;9:3236–45.
Dupont J, Reverchon M, Cloix L, Froment P, Ramé C. Involvement of adipokines, AMPK, PI3K and the PPAR signaling pathways in ovarian follicle development and cancer. Int J Dev Biol. 2012;56:959–67.
Van Langendonckt A, Romeu L, Ambroise J, Amorim C, Bearzatto B, Gala JL, et al. Gene expression in human ovarian tissue after xenografting. Mol Hum Reprod. 2014;20:514–25.
Milenković T, Memišević V, Ganesan AK, Pržulj N. Systems-level cancer gene identification from protein interaction network topology applied to melanogenesis-related functional genomics data. J R Soc Interface. 2010;7:423–37.
Drake I, Sonestedt E, Gullberg B, Ahlgren G, Bjartell A, Wallström P, et al. Dietary intakes of carbohydrates in relation to prostate cancer risk: a prospective study in the Malmö Diet and Cancer cohort. The American journal of clinical nutrition. 2012;96(6):1409–18.
Chaffin CL, Latham KE, Mtango NR, Midic U, VandeVoort CA. Dietary sugar in healthy female primates perturbs oocyte maturation and In vitro preimplantation embryo development. Endocrinology. 2014;155:2688–95.
Shim S-H, Lee SJ, Kim S-O, Kim SN, Kim DY, Lee JJ, et al. Nomogram for predicting incomplete cytoreduction in advanced ovarian cancer patients. Gynecol Oncol. 2015;136:30–6.
Okamoto T, Mandai M, Matsumura N, Yamaguchi K, Kondoh H, Amano Y, et al. Hepatocyte nuclear factor‐1β (HNF‐1β) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol Carcinog. 2015;54:35–49.
Paragh G, Kumar SM, Rakosy Z, Choi SC, Xu X, Acs G. RNA interference-mediated inhibition of erythropoietin receptor expression suppresses tumor growth and invasiveness in A2780 human ovarian carcinoma cells. Am J Pathol. 2009;174:1504–14. doi:10.2353/ajpath.2009.080592.
Poniewierska-Baran A, Schneider G, Ratajczak J, Kucia M, Ratajczak MZ. Novel evidence that neuroblastoma and rhabdomyosarcoma, Two types of small round blue cell tumors, frequently infiltrate bone marrow and express functional erythropoietin receptor (EpoR)—therapeutic implications. Blood. 2014;124:4019–9.
Kubota K, Kim JY, Sawada A, Tokimasa S, Fujisaki H, Matsuda-Hashii Y, et al. LRRC8 involved in B cell development belongs to a novel family of leucine-rich repeat proteins. FEBS Lett. 2004;564:147–52.
Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–6.
Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, et al. CBFβ stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell. 2013;49:632–44.
Ye S, Li P, Tong C, Ying QL. Embryonic stem cell self‐renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. 2013;32:2548–60.
Acknowledgements
This study was supported by Shanghai Science Foundation of Health and Family-planning Bureau (grant number: 201440362) and “1255” Science Development Project of Changhai Hospital (grant number: CH125540900, CH125510105)
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors’ contributions
SYC and MJX carried out all the operations and designed and applied the technique. They had also critically reviewed the final draft of the manuscript. TY and LL collected the data and wrote the manuscript. YC and JCW conceived of the study and participated in its design and coordination and found and organized the literature. All authors read and approved the final manuscript.
Authors’ information
Sheng-Yun Cai, Tian Yang and Yu Chen co-first authors.
Sheng-Yun Cai, Tian Yang and Yu Chen contributed equally to this work.
Additional file
Additional file 1:
Tables S5 and S6.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cai, SY., Yang, T., Chen, Y. et al. Gene expression profiling of ovarian carcinomas and prognostic analysis of outcome. J Ovarian Res 8, 50 (2015). https://doi.org/10.1186/s13048-015-0176-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-015-0176-9