
Abstract
Ovarian cancer is the most lethal of all gynecologic malignancies because women commonly present with advanced stage disease and develop chemotherapy refractory tumors. While cytoreductive surgery followed by platinum based chemotherapy are initially effective, ovarian tumors have a high propensity to recur highlighting the distinct need for novel therapeutics to improve outcomes for affected women. The Notch signaling pathway plays an established role in embryologic development and deregulation of this signaling cascade has been linked to many cancers. Recent genomic profiling of serous ovarian carcinoma revealed that Notch pathway alterations are among the most prevalent detected genomic changes. A growing body of scientific literature has confirmed heightened Notch signaling activity in ovarian carcinoma, and has utilized in vitro and in vivo models to suggest that targeting this pathway with gamma secretase inhibitors (GSIs) leads to anti-tumor effects. While it is currently unknown if Notch pathway inhibition can offer clinical benefit to women with ovarian cancer, several GSIs are currently in phase I and II trials across many disease sites including ovary. This review will provide background on Notch pathway function and will focus on the pre-clinical literature that links altered Notch signaling to ovarian cancer progression.
Similar content being viewed by others


Background
Ovarian cancer represents the most lethal gynecologic malignancy in the United States. In 2014 alone, approximately 22,000 women are estimated to be diagnosed with ovarian cancer and more than 14,000 deaths attributed to the disease are projected to occur [1]. This high mortality is explained in part by the advanced disease stage at the time of diagnosis with approximately 75% of the patients presenting with stage III - IV disease [2]. Therapeutic strategies include cytoreductive surgery followed by six cycles of platinum and taxane based chemotherapy [3],[4]. Despite the fact that the majority of patients achieve complete clinical remission following this treatment regimen, the prognosis of ovarian cancer remains unfavorable with a 5-year survival rate of approximately 50% [4]. This poor outcome is mainly attributed to the development of recurrent disease that is often resistant to chemotherapy [5],[6]. Treatment options for recurrent ovarian cancer are currently limited and not curative, warranting the development of novel therapeutic strategies.
Epithelial ovarian malignancies comprise heterogeneous tumors, both on a cellular and a molecular level. A recently developed dualistic model divides the different ovarian cancer subtypes into type I and type II carcinomas [7]. Type I tumors comprise low-grade serous, low-grade endometrioid, clear cell and mucinous ovarian cancers. They are characterized by indolent, genetically stable tumors that arise from a precursor lesion, often present at an early stage and are relatively resistant to cytotoxic therapy. In contrast, the more prevalent type II tumors are highly aggressive, present at an advanced stage, typically harbor TP53 mutations that lead to genetic instability, and are initially more sensitive to chemotherapeutic agents. While high-grade serous carcinomas account for the vast majority of type II ovarian cancers, other subtypes include high-grade endometrioid ovarian carcinoma and carcinosarcomas [8],[9]. In recent years, multiple genetic and epigenetic abnormalities as well as changes in molecular pathways have been identified that are often characteristic for specific histologic subtypes [10],[11]. Therapeutic targeting of the molecular aberrations and cellular signaling pathways involved in tumor progression may provide novel treatment options for women with recurrent ovarian cancer. This review will focus on the role of the Notch signaling cascade in high-grade serous ovarian cancer and the potential therapeutic effectiveness of Notch pathway inhibition in this disease.
The Notch signaling pathway
Functions of Notch signaling
The evolutionary conserved Notch pathway was first discovered in Drosophila a century ago, when flies with a mutation in the Notch gene were found to have wing deformities [12]. The functional significance of the Notch signaling cascade has been well established in neural development [13],[14] and has since been established in multiple cellular processes, during embryonic development and in self-renewing adult tissues [15],[16]. The Notch pathway functions through cell-to-cell contact and is involved in the regulation of proliferation, differentiation and apoptosis, depending on the cellular context [17],[18]. In adult tissues, Notch signaling acts to control tissue homeostasis and stem cell maintenance.
Notch receptors and ligands
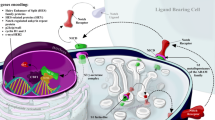
Thus far, four Notch receptors (Notch1-4) and five ligands have been identified in mammals. Three ligands belong to the Delta-like family (Dll1, 3 and 4) and two ligands (Jagged1 (Jag1) and Jagged2 (Jag2)) are Serrate-like [19]-[23]. Notch receptors as well as their ligands are single-pass transmembrane proteins with extracellular domains that consist of multiple epidermal growth factor (EGF)-like repeats [24],[25]. The receptors are synthesized as inactive precursors in the endoplasmic reticulum that are proteolytically cleaved by furin-like convertases in the trans-Golgi compartment [26]. This first cleavage, termed S1, results in an extracellular N-terminal fragment and a transmembrane C-terminal fragment that also includes the Notch intracellular domain (NICD). Finally, non-covalent binding between the two fragments forms the mature Notch heterodimeric receptor [27] (Figure 1). During the process of Notch receptor synthesis, the extracellular fragment is glycosylated by Fringe glycosyltransferases, which modifies the binding affinity between the receptor and its ligands [28],[29].

The Notch signaling cascade is activated by cell-cell interaction.
Signaling cascade
As shown in Figure 1, Notch signaling is activated by a receptor-ligand binding between two neighboring cells, leading to a conformational change of the Notch receptor and exposure of a cleavage site (S2) in its extracellular domain [30],[31]. S2 cleavage by A Disintegrin And Metalloprotease (ADAM)10 or 17 produces an intermediate transmembrane fragment termed NEXT (Notch extracellular truncation) which is accessible to gamma-secretase for S3 cleavage [32]. The gamma-secretase complex consists of four subunits: the catalytic subunit presenilin, nicastrin, APH-1 and PEN-2 [33]. S3 cleavage by gamma-secretase leads to release of the NICD, which translocates to the nucleus and binds to the DNA bound CBF-1/Su(H)/Lag-1 protein complex (CSL, also known as RBP-jκ) that constitutively represses transcription in the absence of NICD [34],[35]. The NICD displaces a co-repressor complex from CSL and recruits co-activators such as Mastermind-like 1 (MAML1), allowing the transcription of Notch target genes [34],[36].
Notch target genes
The most well-known Notch target genes are transcription factors of the Hairy/Enhancer of Split (Hes) and Hes-related (Hey) families [37]. Hes and Hey members are helix-loop-helix proteins, forming homo-or heterodimers that regulate transcription of genes involved in cell fate determination [37]-[39]. Other Notch pathway targets include cell cycle regulators cyclin D1 and p21, NF-κB family members, c-Myc and Deltex [40]-[43].
Crosstalk with other signaling pathways
The Notch pathway is part of a complex network of developmental signaling pathways that also includes the Hedgehog, Wnt, receptor tyrosine kinases (RTK), transforming growth factor-β (TGF-β) and Janus kinase/signal transducers and activators of transcription (Jak/STAT) pathways [44],[45]. The Notch pathway has also been implicated as interacting with the EGFR/HER2 receptor tyrosine kinase family, as well as phosphatidylinositol 3-kinase/AKT/mTOR signaling cascade, two central growth pathways in the physiologic and neoplastic setting [46]. Although integration of Notch signaling with signals from other pathways has been a focus of investigation, the exact mechanisms of this crosstalk remain largely unknown. The interaction of Notch signaling with other pathways highly depends on the cellular context, and differs from one cellular environment to another. For example, the Notch and Wnt pathways were found to cooperate in maintaining undifferentiated hematopoietic stem cells [47], whereas an antagonistic interaction between Wnt and Notch has been reported in Drosophila development [48]. Similarly, opposed functions in distinct cellular contexts have been reported for the Notch and Ras pathways. Notch signaling was shown to complement the Ras pathway during Drosophila eye development [49], while inhibition of the Ras pathway by Notch signaling has been demonstrated in C. elegans hermaphrodite vulval development [50],[51]. Activation of Sonic Hedgehog (Shh) led to upregulation of Notch signaling and determination of arterial cell fate in zebrafish [52], and induction of Shh has been observed in murine somatic and human embryonic stem cells following Notch receptor activation [53]. The phosphatidylinositol 3-kinase (PI3K)-AKT and Notch signaling cascades were found to interact agonistically during murine hematopoietic stem cell differentiation and in a variety of human cell types including T-cells and neurons [54],[55]. Negative crosstalk between the EGFR and Notch pathways has been described in Drosophila wing development [56],[57], while Notch signaling was shown to activate Her2 (ERBB2) in human embryonic kidney cells [58].
Notch signaling in cancer
Oncogenic Notch signaling was first described in T-cell acute lymphoblastic leukemia (T-ALL), in which a chromosomal translocation event generates a constitutively active variant of Notch1 [59]. Later studies have shown that Notch1 activating mutations occur in the majority of T-ALLs [60]. Deregulation of Notch receptors, ligands or targets has since been described in a variety of solid and hematological tumors including breast [61],[62] pancreatic [63], brain [64], lung [65], ovarian [11], head and neck [66] and colorectal [67] cancer, as well as leukemia [60],[68], lymphomas [69],[70] and multiple myeloma [71]. While these and other studies demonstrate Notch pathway involvement in tumor initiation and progression, tumor suppressive roles of Notch signaling have also been reported [72],[73]. Most notably, Notch functions as a tumor suppressor in the skin, and loss-of-function mutations in Notch receptors have been identified in cutaneous squamous cell carcinoma [74],[75]. Although little is known about the mechanisms behind these contradictory actions of the Notch pathway in cancer, it is generally assumed that the various outcomes of Notch signaling depend on interactions with the microenvironment and crosstalk with other signaling pathways [72].
Notch signaling in serous ovarian cancer
In ovarian cancer, a role for Notch signaling was first discovered in two studies aimed at identifying potential diagnostic markers of epithelial ovarian cancer. Gene expression of human ovarian cancer samples or cell lines was evaluated and compared to normal ovarian surface epithelium samples or cell lines, respectively [76],[77]. One study reported upregulation of the Notch3 gene in all analyzed ovarian cancers [76], while the second study reported increased Jag2 gene expression in ovarian cancer cell lines compared with benign controls [77]. Subsequently, Park et al. used a single nucleotide polymorphism (SNP) array to analyze DNA copy number alterations in high-grade serous ovarian carcinomas and identified an amplicon corresponding to the Notch3 locus in 20% of cases. Notch3 gene amplification correlated with Notch3 protein overexpression, as determined by fluorescent in situ hybridization and immunohistochemistry (IHC) [78]. These results were confirmed by others, who demonstrated amplification of the Notch3 gene using a SNP array in 21% of analyzed ovarian cancers [79]. Moreover, recent large-scale genomic and epigenomic analyses of high-grade serous ovarian carcinomas by The Cancer Genome Atlas (TCGA) Network revealed altered Notch signaling in 22% of cases with alterations in Notch3 occurring in 50% of those cases [11]. In a study of Notch ligands, Jag1 was shown to be the most highly expressed ligand in both ovarian cancer cells and surrounding peritoneal mesothelial cells with interaction of Jag1 and Notch3 resulting in activation of the signaling cascade and promotion of cell proliferation and adhesion [80]. In addition, Pbx1 and DLGAP5 have been identified as direct target genes of Notch3 in ovarian cancer, and knockdown of either target with shRNAs led to reduced cell proliferation and, in the case of Pbx1 knockdown, impaired tumor formation [81],[82]. More recently, the TCGA data generated from 488 ovarian cancer patients were used to analyze epigenetic changes of genes of the Notch superfamily. Inverse correlations were found between DNA methylation and expression of the Notch pathway target genes CCND1 and PPARG and the Notch-interacting gene RUNX1. Additionally, an inverse correlation in expression was established between CCND1, PPARG and RUNX1 and specific miRNAs that regulate each of those genes. Subsequent survival analyses revealed significantly poorer overall survival rates of patients with high CCND1, PPARG and RUNX1 gene expression and low methylation or low levels of the relevant miRNAs compared with patients with low gene expression and high methylation or high miRNA levels [83]. Furthermore, several groups have linked Notch3 expression to the clinical prognosis of ovarian cancer. Using quantitative reverse transcription polymerase chain reaction (RT-PCR) and IHC, Jung et al. observed elevated mRNA levels of Notch3, Jag1 and Jag2 as well as higher Notch3 and Jag2 protein expression in serous ovarian cancer samples as compared to benign controls. High Notch3 mRNA expression correlated significantly with worse overall survival and clinical chemoresistance, and Notch3 protein overexpression was significantly associated with the prognostic parameters advanced stage disease, lymph node metastases and distant metastases [84]. Moreover, elevated nuclear Notch3 immunostaining has been found in recurrent serous ovarian carcinoma specimens as compared to primary ovarian cancer samples from the same patients. A significant association between high Notch3 mRNA or nuclear Notch3 protein levels and worse overall and progression-free survival rates was also described [85],[86]. Ectopic expression of Notch3 following transduction with a Notch3 intracellular domain (NICD3) in ovarian surface epithelium and low-grade serous ovarian cancer cell lines with low endogenous Notch3 expression led to increased resistance to carboplatin in vitro. Inversely, shRNA-mediated knockdown of Notch3 in OVCAR3 cells resulted in higher sensitivity to carboplatin, compared with OVCAR3 cells transduced with a non-specific control shRNA [85].
The role of Notch1 in ovarian cancer was first studied by Hopfer et al., [87] who evaluated mRNA expression of Notch pathway members in ovarian adenocarcinomas, borderline tumors and adenomas and demonstrated more frequent expression of Jag2 and DLL1 in adenocarcinomas as compared to adenomas. Although quantitative RT-PCR and Western blot analyses revealed similar Notch1 expression levels in ovarian adenocarcinomas and adenomas, stable transfection of A2780 ovarian cancer cells with the intracellular domain of Notch1 (NICD1) increased cell proliferation and enhanced colony-formation capacity, suggesting a role for Notch1 signaling in ovarian tumor growth [87]. Analyses of the NICD1 protein by other investigators revealed high NICD1 levels in OVCAR3, SKOV3 and CaOV3 cell lines. Additionally, NICD1 was expressed in 76% of primary human serous ovarian cancer samples, as assessed by Western blotting. Subsequent siRNA downregulation of NICD1 in all three ovarian cancer cell lines resulted in inhibition of cell proliferation [88]. Results obtained from PCR, immunoblotting or functional studies of Notch1 have been easily interpreted. In contrast, IHC analyses of Notch1 expression in ovarian cancer have produced variable results. Wang and colleagues used IHC to study Notch1 levels in ovarian cancer specimens of various histological grades as well as patient-matched contralateral benign ovarian samples and normal ovarian tissues. Notch1 immunostaining was observed in 95% of analyzed serous ovarian cancer specimens versus 8% and 6% of matched benign controls and normal ovarian samples, respectively. Notch1 immunostaining was predominantly found in the cell membrane and cytoplasm. Positivity scores correlated with histological grade and clinical disease stage, and the IHC findings were confirmed by Western blotting and quantitative RT-PCR [89]. An IHC protocol was recently developed to detect the gamma-secretase cleaved NICD1. In contrast to the findings of the full-length Notch1 IHC studies, NICD1 was not expressed in any of the 147 analyzed ovarian cancer specimens although a subset of samples from other cancer types showed nuclear NICD1 immunostaining with this method [90]. In another study, IHC analysis of 10 serous ovarian tumors by others detected expression of Notch1, Jag1 and Dll1 in both cytoplasm and nucleus, and the observed Notch1 protein levels correlated significantly with metastasis in this small cohort [91].
Notch signaling in serous ovarian cancer stem cells
A few investigations have shown elevated Notch levels in recurrent or chemoresistant ovarian cancer. In ovarian cancer and many other malignancies, the development of recurrent disease has been attributed, at least in part, to a tumorigenic and chemotherapy resistant sub-population of cancer cells called tumor-initiating or cancer stem cells (CSCs) [92]-[94]. Consistent with this hypothesis, increased expression of CSC markers and enrichment of the side population (cells showing increased efflux of Hoechst dye as identified by flow cytometry, reviewed in Foster et al., [95]) have been observed in ovarian cancer samples following platinum based chemotherapy, compared with chemotherapy naive cells [96],[97]. Similarly, increased resistance to cisplatin and paclitaxel has been demonstrated in sphere forming primary serous ovarian cancer cells, cultured under stem cell-selective conditions, compared with the same cells cultured under differentiating conditions. These spheroid ovarian CSCs also showed elevated levels of the CSC marker proteins CD44 and CD117 as well as an increase in mRNA levels of Notch1 and other stem cell genes, compared with differentiated cells and parental bulk tumor cells [98]. Retrovirus-mediated overexpression of the Notch3 intracellular domain (NICD3) in ovarian surface epithelium and low-grade serous ovarian cancer cell lines with low endogenous Notch3 levels led to upregulation of the stem cell associated genes Nanog and Oct4, further suggesting a role of the Notch pathway in CSC function [85]. Furthermore, gene microarray analyses of serous ovarian cancer side population cells from ascites samples showed upregulation of three genes involved in Notch signaling, compared with main population cells. These genes, ADAM19, FPTG and ST3GAL6, were also found to be overexpressed in recurrent ovarian cancer specimens, compared with matched primary samples [99]. Supporting these findings, Steg and colleagues have reported increased transcript levels of stem cell pathway genes, including the Notch pathway member presenilin 2 (PSEN2), in recurrent ovarian cancer samples compared with matched primary tumors [96]. Collectively, these studies provide evidence suggesting that Notch signaling is enhanced in ovarian CSCs, as compared to tumor bulk cells.
Notch signaling and epithelial-to-mesenchymal transition in serous ovarian cancer
In addition to its role in CSCs, the Notch pathway has been implicated in epithelial-to-mesenchymal transition (EMT) and may thereby promote tumor invasiveness and metastasis [100]. The process of EMT has been associated with chemoresistance and stem cell-like characteristics in several cancers [101], including ovarian cancer [102]-[104]. A recent study has demonstrated that overexpression of NICD3 in OVCA429 serous ovarian cancer cells induces EMT, as confirmed by a fibroblast-like cell morphology as well as upregulation of the mesenchymal markers Slug, Snail and smooth muscle α-actin and down regulation of the epithelial marker E-cadherin. The OVCA429/NICD3 cells showed increased resistance to carboplatin-induced apoptosis compared with OVCA429 control cells [105]. Although further investigation of the role of the Notch pathway in ovarian EMT is needed, these findings suggest that activation of Notch can induce EMT in serous ovarian carcinomas.
Notch signaling and angiogenesis in serous ovarian cancer
While Notch signaling has been linked extensively to tumor angiogenesis [106]-[108] in many malignancies, few studies have described a similar role of the pathway in ovarian tumor angiogenesis [109],[110]. Lu and colleagues investigated gene expression differences between endothelial cells from high-grade serous ovarian carcinomas and endothelial cells from benign ovaries using gene microarrays. Overexpression of 23 genes was found in ovarian tumor endothelial cells as compared to benign ovarian endothelial cells. Among these upregulated genes was Jagged1, and silencing of this gene with a siRNA decreased tube formation and migration of endothelial cells [109]. Other investigators observed Dll4 overexpression in tumor and endothelium in 72% of ovarian cancer samples analyzed by IHC, and increased Dll4 levels were associated with worse overall survival when compared to samples with low Dll4 expression. When DLL4 was silenced in vivo using nanoparticle delivery of a DLL4 specific siRNA to mice harboring A2780 or SKOV3ip1 cell line derived xenografts, the targeting of both tumor cells and tumor-associated mouse endothelial cells inhibited tumor growth and deregulated angiogenesis. Moreover, tumor growth was further inhibited when the vascular endothelial growth factor inhibitor bevacizumab was added to this treatment regimen [110] suggesting synergy between anti-VEGF treatment and Dll4 targeting. In summary, the described studies provide evidence to suggest that the Notch pathway is not only involved in epithelial ovarian tumor growth, but also plays a role in endothelial cell function and angiogenesis of serous ovarian tumors.
Therapeutic targeting of the Notch pathway in serous ovarian cancer
The growing body of evidence regarding the role of Notch signaling in cancer has led to the development of different Notch pathway inhibitors, a number of which are in current clinical trials (see Table 1). Several steps in the pathway can be targeted, and established classes of inhibitors include monoclonal antibodies against Notch ligands or receptors, receptor decoys, gamma-secretase inhibitors (GSIs), peptides that block the nuclear transcriptional complex, and natural compounds [111],[112]. GSIs are the most widely studied Notch pathway targeting agents. A variety of GSIs with distinct chemical structures but similar biological activity have been developed and each [113] inhibits signaling through all four Notch paralogs by preventing formation of the active NICD. Pre-clinical studies in a variety of cancers have shown inhibition of tumor growth or cell proliferation by GSIs [114]-[118]. In early phase clinical trials of GSIs, promising anti-tumor effects in several solid malignancies have been observed [119]-[122] despite dose-limiting toxicities evidenced mainly by gastrointestinal events. Over 40 clinical trials investigating the efficacy of GSI therapy in solid or hematologic cancers are ongoing or have recently been completed (see Table 1 for current list of ongoing trials). A subset of these trials use GSI in combination with standard chemotherapy or other targeting agents (([112]) https://clinicaltrials.gov/).
Preclinical studies of Notch inhibition with GSI in serous ovarian cancer: in vitro models
Several groups have studied the in vitro effectiveness of GSIs in serous ovarian cancer. A reduction of cell proliferation and induction of apoptosis have been reported in OVCAR3 and A2780 ovarian cancer cell lines following administration of the compound GSI-1, compared with DMSO controls [78]. The same GSI was used by others who showed decreased cell proliferation post-treatment in A2780 and cisplatin resistant KFr13 serous ovarian cancer cell lines which both express high levels of NICD3 [86]. Moreover, it was shown that treatment of A2780 cells with the GSI DAPT decreased cell proliferation in a dose- and time-dependent manner, inhibited colony formation and induced cell cycle arrest and apoptosis while reducing Notch1 and Hes1 mRNA and protein levels [123]. In contrast with these findings, a recent study showed unchanged proliferation rates of OVCAR3, SKOV3 and several other ovarian cancer cell lines following treatment with the GSIs Compound E, DAPT or dibenzazepine (DBZ) [94].
Preclinical studies of Notch inhibition with GSI in serous ovarian cancer: in vivo models
Limited studies have investigated Notch inhibition in vivo for the treatment of ovarian cancer. Using in vitro and in vivo ovarian cancer models, McAuliffe and colleagues have investigated both the anti-tumor efficacy of GSI-1 as a single agent and in combination with cisplatin, and the effects of these treatments on ovarian CSC sub-populations. These investigators observed a reduction in cell viability following GSI treatment and a synergistic response to combined GSI and cisplatin therapy in established cell lines and cultured cells derived from ascites samples of platinum resistant and platinum sensitive patients. Sensitivity to GSI was found to correlate with Notch3 protein levels, as assessed by Western blotting. siRNA-mediated knockdown of Notch3 increased sensitivity to cisplatin in PA-1 and OVCAR3 cell lines and had no effect in SKOV3 cells which do not express detectable levels of Notch3. The described synergy of GSI and cisplatin was confirmed in vivo, using xenografts derived from these established cell lines. In addition, in vitro treatment with GSI or GSI + cisplatin decreased the side population and in vivo GSI therapy reduced both the side population and mRNA levels of the CSC markers CD44 and ALDH1, suggesting GSI is effective at eliminating CSCs [124]. The in vivo effect of single agent GSI or GSI in combination with standard chemotherapy in a patient derived xenograft (PDX) model was explored by our group using cohorts of mice harboring PDX tumors derived from either clinically platinum sensitive or clinically platinum resistant primary ovarian serous tumors. Three of four platinum sensitive tumors and one of three platinum resistant tumors showed a decrease in tumor growth following single agent GSI treatment. Moreover, combination treatment with GSI and paclitaxel led to a markedly greater reduction in tumor growth compared with paclitaxel alone and GSI alone in all platinum resistant tumors. The combination of GSI with paclitaxel and carboplatin was not more effective than paclitaxel/carboplatin alone in platinum sensitive tumors [125]. These findings highlight a potential role for Notch pathway inhibition in addition to cytotoxic therapy in the recurrent, platinum resistant setting in serous ovarian cancer.
Clinical studies of Notch inhibition with GSI in serous ovarian cancer
To date, early clinical trials have provided little data regarding the efficacy of GSIs in ovarian cancer patients. A recent phase I clinical trial using the GSI RO4929097 in a range of advanced solid tumors reported prolonged stable disease in three of nine ovarian cancer patients [122]. In other phase I studies, no clinical benefit of RO4929097 in combination with gemcitabine was found in two of two ovarian cancer patients included in the trial [119], and the GSI MK-0752 was clinically ineffective as single agent therapy in three of three ovarian tumors [120]. Tables 1 and 2 highlight ongoing trials of various GSIs. While the only phase II trial exclusively in ovarian carcinoma assesses RO4929097 as a single agent, many other disease sites are testing GSI in combination with other biologics or cytotoxic chemotherapies. In the reported trial literature, the most clinical benefit has been observed in those trials that combine GSI with other agents suggesting a supporting therapeutic role for GSI. The fact that monotherapy has resulted in lower published clinical benefit rates stems in part from phase I trial designs that are powered to detect toxicity, not efficacy. However, an additional contributing factor is that durable tumor control from Notch inhibition requires careful selection for tumors innately dependent on Notch signaling for proliferation.
To date, no trials testing Notch pathway inhibition in patients pre-selected based on alterations in the Notch pathway have been reported. Significant pre-clinical data, however, seem to support such stratification as a promising approach. Future clinical studies will need to employ clinical as well as scientific endpoints to understand better the molecular characteristics of tumors that respond to therapy. Without a clear biomarker, specific biologic therapies such as GSI, are unlikely to become a mainstay of therapy. In addition to biomarker discovery, more trials investigating combination of GSI with conventional cytotoxics, such as taxanes needs to be conducted. Pre-clinical rationale exists for this combination approach as tumor cell resistance to cytotoxics and other biologic therapies, such as taxanes and anti-HER2 therapies, has been linked to heightened Notch signaling [131],[132]. Since Notch may be an important escape pathway that reconstitutes the oncogenic potential of a tumor cell, Notch inhibition may be a key adjunct to conventional therapies known to be effective [133].
Other Notch inhibitors in serous ovarian cancer
In addition to targeting of gamma-secretase activity, alternative methods of Notch pathway inhibition have been studied in ovarian cancer. Inhibition of Jag1 using siRNA constructs in the IGROV-AF1 cell line and the taxane resistant SKOV3Trip2 cell lines resulted in a reduction of cell viability as well as sensitization to docetaxel in SKOV3Trip2 cells. The same cell lines were used to generate intraperitoneal tumors in mice, and the effects of targeting Jag1 in stromal and malignant cells were evaluated by treatment of mice with either mouse specific Jag1 siRNA, human specific Jag1 siRNA or both siRNA constructs. Either siRNA alone reduced tumor growth, and the combination of mouse and human Jag1 siRNAs showed a synergistic effect in tumors derived from both cell lines. Combination treatment of SKOV3Trip2-derived tumors with human and mouse specific Jag1 siRNAs and docetaxel led to the highest decrease in tumor weight, compared with the siRNAs alone or docetaxel alone. Microvessel densities were reduced after treatment with anti-murine Jag1 siRNA, suggesting this therapy induced anti-angiogenic effects [134]. Finally, the natural compounds xanthohumol and withaferin A inhibited cell growth and induced apoptosis and cell cycle arrest in ovarian cancer cell lines through downregulation of Notch1 (withaferin A and xanthohumol) and Notch3 (withaferin A) [135],[136].
Conclusions
Considerable evidence supports an important oncogenic role of Notch signaling in high-grade serous ovarian cancer. Perturbation in normal regulation of Notch1 and Notch3 as well as Notch ligands, target genes and other members of the Notch pathway has been described. The many biological and clinical aspects of ovarian tumorigenesis in which aberrant Notch signaling appears to be involved include tumor initiation and progression, metastasis, resistance to chemotherapy, CSC activity, angiogenesis and EMT. Despite its functional complexity and crosstalk with other signaling cascades, the Notch pathway represents an attractive therapeutic target in ovarian cancer. Pre-clinical analysis of Notch inhibition suggests Notch targeting agents such as GSIs hold promise as potential treatment strategies for ovarian cancer patients, most notably in the setting of recurrence and chemoresistance. In addition, combination regimens with conventional cytotoxic therapy as well as other targeted therapies warrant further investigation.
Authors’ contribution
JG and BR conceived the idea for the review, WG, RF, RV and BR participated in the review of the literature, and drafting of the manuscript. All authors read and approved the final manuscript.
References
Siegel R, Ma J, Zou Z, Jemal A: Cancer statistics, 2014. CA Cancer J Clin 2014, 64: 9–29.
Hennessy BT, Coleman RL, Markman M: Ovarian cancer. Lancet 2009, 374: 1371–1382.
Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R: Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2003, 21: 3194–3200.
Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA: Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 2006, 354: 34–43.
Cooke SL, Brenton JD: Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol 2011, 12: 1169–1174.
Pinato DJ, Graham J, Gabra H, Sharma R: Evolving concepts in the management of drug resistant ovarian cancer: dose dense chemotherapy and the reversal of clinical platinum resistance. Cancer Treat Rev 2013, 39: 153–160.
Kurman RJ, Shih Ie M: Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer–shifting the paradigm. Hum Pathol 2011, 42: 918–931.
Lim D, Oliva E: Precursors and pathogenesis of ovarian carcinoma. Pathology 2013, 45: 229–242.
Kurman RJ: Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol 2013,24(Suppl 10):x16–21.
Banerjee S, Kaye SB: New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res 2013, 19: 961–968.
Research CGA: Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474: 609–615.
Mohr OL: Character Changes Caused by Mutation of an Entire Region of a Chromosome in Drosophila. Genetics 1919, 4: 275–282.
Giniger E: Notch signaling and neural connectivity. Curr Opin Genet Dev 2012, 22: 339–346.
Artavanis-Tsakonas S, Delidakis C, Fehon RG: The Notch locus and the cell biology of neuroblast segregation. Annu Rev Cell Biol 1991, 7: 427–452.
Koch U, Lehal R, Radtke F: Stem cells living with a Notch. Development 2013, 140: 689–704.
Liu J, Sato C, Cerletti M, Wagers A: Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol 2010, 92: 367–409.
Bray SJ: Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 2006, 7: 678–689.
Perdigoto CN, Bardin AJ: Sending the right signal: Notch and stem cells. Biochim Biophys Acta 1830, 2013: 2307–2322.
Lindsell CE, Shawber CJ, Boulter J, Weinmaster G: Jagged: a mammalian ligand that activates Notch1. Cell 1995, 80: 909–917.
Shawber C, Boulter J, Lindsell CE, Weinmaster G: Jagged2: a serrate-like gene expressed during rat embryogenesis. Dev Biol 1996, 180: 370–376.
Dunwoodie SL, Henrique D, Harrison SM, Beddington RS: Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development 1997, 124: 3065–3076.
Ikeuchi T, Sisodia SS: The Notch ligands, Delta1 and Jagged2, are substrates for presenilin-dependent "gamma-secretase" cleavage. J Biol Chem 2003, 278: 7751–7754.
D'Souza B, Miyamoto A, Weinmaster G: The many facets of Notch ligands. Oncogene 2008, 27: 5148–5167.
Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S: Nucleotide sequence from the neurogenic locus Notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 1985, 43: 567–581.
Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S: Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell 1991, 67: 687–699.
Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A: The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A 1998, 95: 8108–8112.
Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S: Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 1997, 90: 281–291.
Haines N, Irvine KD: Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol Molecular cell biology 2003, 4: 786–797.
Stanley P, Okajima T: Roles of glycosylation in Notch signaling. Curr Top Dev Biol 2010, 92: 131–164.
Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R: A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell 2000, 5: 197–206.
Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A: A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell 2000, 5: 207–216.
Saxena MT, Schroeter EH, Mumm JS, Kopan R: Murine Notch homologs (N1–4) undergo presenilin-dependent proteolysis. J Biol Chem 2001, 276: 40268–40273.
Tolia A, De Strooper B: Structure and function of gamma-secretase. Semin Cell Dev Biol 2009, 20: 211–218.
Kovall RA: More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene 2008, 27: 5099–5109.
Kopan R, Ilagan MX: The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 2009, 137: 216–233.
Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD: MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for Notch receptors. Nat Genet 2000, 26: 484–489.
Iso T, Kedes L, Hamamori Y: HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 2003, 194: 237–255.
Stifani S, Blaumueller CM, Redhead NJ, Hill RE, Artavanis-Tsakonas S: Human homologs of a Drosophila Enhancer of split gene product define a novel family of nuclear proteins. Nat Genet 1992, 2: 119–127.
Maier MM, Gessler M: Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun 2000, 275: 652–660.
Ronchini C, Capobianco AJ: Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol 2001, 21: 5925–5934.
Oswald F, Liptay S, Adler G, Schmid RM: NF-kappaB2 is a putative target gene of activated Notch1 via RBP-Jkappa. Mol Cell Biol 1998, 18: 2077–2088.
Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, Li Y, Wolfe MS, Shachaf C, Felsher D, Blacklow SC, Pear WS, Aster JC: c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev 2006, 20: 2096–2109.
Choi JW, Pampeno C, Vukmanovic S, Meruelo D: Characterization of the transcriptional expression of Notch1 signaling pathway members, Deltex and HES-1, in developing mouse thymocytes. Dev Comp Immunol 2002, 26: 575–588.
Hurlbut GD, Kankel MW, Lake RJ, Artavanis-Tsakonas S: Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol 2007, 19: 166–175.
Brechbiel J, Miller-Moslin K, Adjei AA: Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev 2014, 40: 750–759.
Yamaguchi H, Chang SS, Hsu JL, Hung MC: Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene 2014, 33: 1073–1081.
Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T: Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol 2005, 6: 314–322.
Axelrod JD, Matsuno K, Artavanis-Tsakonas S, Perrimon N: Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science 1996, 271: 1826–1832.
Cooper MT, Bray SJ: R7 photoreceptor specification requires Notch activity. Curr Biol: CB 2000, 10: 1507–1510.
Berset T, Hoier EF, Battu G, Canevascini S, Hajnal A: Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science 2001, 291: 1055–1058.
Sundaram MV: The love-hate relationship between Ras and Notch. Genes Dev 2005, 19: 1825–1839.
Lawson ND, Vogel AM, Weinstein BM: Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 2002, 3: 127–136.
Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD: Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442: 823–826.
McKenzie G, Ward G, Stallwood Y, Briend E, Papadia S, Lennard A, Turner M, Champion B, Hardingham GE: Cellular Notch responsiveness is defined by phosphoinositide 3-kinase-dependent signals. BMC Cell Biol 2006, 7: 10.
Cornejo MG, Mabialah V, Sykes SM, Khandan T, Lo Celso C, Lopez CK, Rivera-Munoz P, Rameau P, Tothova Z, Aster JC, DePinho RA, Scadden DT, Gilliland DG, Mercher T: Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood 2011, 118: 1264–1273.
de Celis JF, Bray S, Garcia-Bellido A: Notch signalling regulates veinlet expression and establishes boundaries between veins and interveins in the Drosophila wing. Development 1997, 124: 1919–1928.
Culi J, Martin-Blanco E, Modolell J: The EGF receptor and N signalling pathways act antagonistically in Drosophila mesothorax bristle patterning. Development 2001, 128: 299–308.
Chen Y, Fischer WH, Gill GN: Regulation of the ERBB-2 promoter by RBPJkappa and NOTCH. J Biol Chem 1997, 272: 14110–14114.
Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J: TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66: 649–661.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC: Activating mutations of Notch1 in human T cell acute lymphoblastic leukemia. Science 2004, 306: 269–271.
Stylianou S, Clarke RB, Brennan K: Aberrant activation of notch signaling in human breast cancer. Cancer Res 2006, 66: 1517–1525.
Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, Kim R, Tang J, Montie JE, Chinnaiyan AM, Rubin MA, Aster JC: JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res 2004, 64: 6854–6857.
Mullendore ME, Koorstra JB, Li YM, Offerhaus GJ, Fan X, Henderson CM, Matsui W, Eberhart CG, Maitra A, Feldmann G: Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin Cancer Res 2009, 15: 2291–2301.
Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, Sundaresan T, Pastorino S, Park JK, Mikolaenko I, Maric D, Eberhart CG, Fine HA: Expression of Notch1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res 2005, 65: 2353–2363.
Westhoff B, Colaluca IN, D'Ario G, Donzelli M, Tosoni D, Volorio S, Pelosi G, Spaggiari L, Mazzarol G, Viale G, Pece S, Di Fiore PP: Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A 2009, 106: 22293–22298.
Sun W, Gaykalova DA, Ochs MF, Mambo E, Arnaoutakis D, Liu Y, Loyo M, Agrawal N, Howard J, Li R, Ahn S, Fertig E, Sidransky D, Houghton J, Buddavarapu K, Sanford T, Choudhary A, Darden W, Adai A, Latham G, Bishop J, Sharma R, Westra WH, Hennessey P, Chung CH, Califano JA: Activation of the NOTCH pathway in head and neck cancer. Cancer Res 2014, 74: 1091–1104.
Fre S, Pallavi SK, Huyghe M, Lae M, Janssen KP, Robine S, Artavanis-Tsakonas S, Louvard D: Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci U S A 2009, 106: 6309–6314.
Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, Grunn A, Fangazio M, Capello D, Monti S, Cresta S, Gargiulo E, Forconi F, Guarini A, Arcaini L, Paulli M, Laurenti L, Larocca LM, Marasca R, Gattei V, Oscier D, Bertoni F, Mullighan CG, Foa R, Pasqualucci L, Rabadan R, Dalla-Favera R, Gaidano G: Analysis of the chronic lymphocytic leukemia coding genome: role of Notch1 mutational activation. J Exp Med 2011, 208: 1389–1401.
Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H, Dorken B: Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood 2002, 99: 3398–3403.
Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, Gunawardana J, Jenkins C, Cochrane C, Ben-Neriah S, Tan K, Morin RD, Opat S, Sehn LH, Connors JM, Marra MA, Weng AP, Steidl C, Gascoyne RD: Whole transcriptome sequencing reveals recurrent Notch1 mutations in mantle cell lymphoma. Blood 2012, 119: 1963–1971.
Jundt F, Probsting KS, Anagnostopoulos I, Muehlinghaus G, Chatterjee M, Mathas S, Bargou RC, Manz R, Stein H, Dorken B: Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 2004, 103: 3511–3515.
Ranganathan P, Weaver KL, Capobianco AJ: Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 2011, 11: 338–351.
South AP, Cho RJ, Aster JC: The double-edged sword of Notch signaling in cancer. Semin Cell Dev Biol 2012, 23: 458–464.
Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F: Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 2003, 33: 416–421.
Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, Pennypacker S, Zou Y, Sharma M, North JP, Vemula SS, Mauro TM, Neuhaus IM, Leboit PE, Hur JS, Park K, Huh N, Kwok PY, Arron ST, Massion PP, Bale AE, Haussler D, Cleaver JE, Gray JW, Spellman PT, South AP, et al.: Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A 2011, 108: 17761–17766.
Lu KH, Patterson AP, Wang L, Marquez RT, Atkinson EN, Baggerly KA, Ramoth LR, Rosen DG, Liu J, Hellstrom I, Smith D, Hartmann L, Fishman D, Berchuck A, Schmandt R, Whitaker R, Gershenson DM, Mills GB, Bast RC Jr: Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res 2004, 10: 3291–3300.
Euer NI, Kaul S, Deissler H, Mobus VJ, Zeillinger R, Weidle UH: Identification of L1CAM, Jagged2 and Neuromedin U as ovarian cancer-associated antigens. Oncol Rep 2005, 13: 375–387.
Park JT, Li M, Nakayama K, Mao TL, Davidson B, Zhang Z, Kurman RJ, Eberhart CG, Shih Ie M, Wang TL: Notch3 gene amplification in ovarian cancer. Cancer Res 2006, 66: 6312–6318.
Nakayama K, Nakayama N, Jinawath N, Salani R, Kurman RJ, Shih Ie M, Wang TL: Amplicon profiles in ovarian serous carcinomas. Int J Cancer 2007, 120: 2613–2617.
Choi JH, Park JT, Davidson B, Morin PJ, Shih Ie M, Wang TL: Jagged-1 and Notch3 juxtacrine loop regulates ovarian tumor growth and adhesion. Cancer Res 2008, 68: 5716–5723.
Park JT, Shih Ie M, Wang TL: Identification of Pbx1, a potential oncogene, as a Notch3 target gene in ovarian cancer. Cancer Res 2008, 68: 8852–8860.
Chen X, Thiaville MM, Chen L, Stoeck A, Xuan J, Gao M, Shih Ie M, Wang TL: Defining Notch3 target genes in ovarian cancer. Cancer Res 2012, 72: 2294–2303.
Ivan C, Hu W, Bottsford-Miller J, Zand B, Dalton HJ, Liu T, Huang J, Nick AM, Lopez-Berestein G, Coleman RL, Baggerly KA, Sood AK: Epigenetic analysis of the Notch superfamily in high-grade serous ovarian cancer. Gynecol Oncol 2013, 128: 506–511.
Jung SG, Kwon YD, Song JA, Back MJ, Lee SY, Lee C, Hwang YY, An HJ: Prognostic significance of Notch3 gene expression in ovarian serous carcinoma. Cancer Sci 2010, 101: 1977–1983.
Park JT, Chen X, Trope CG, Davidson B, Shih Ie M, Wang TL: Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am J Path 2010, 177: 1087–1094.
Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, Ishikawa M, Iida K, Nakayama S, Otsuki Y, Miyazaki K: Notch3 overexpression as potential therapeutic target in advanced stage chemoresistant ovarian cancer. Am J Clin Pathol 2012, 138: 535–544.
Hopfer O, Zwahlen D, Fey MF, Aebi S: The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer 2005, 93: 709–718.
Rose SL, Kunnimalaiyaan M, Drenzek J, Seiler N: Notch 1 signaling is active in ovarian cancer. Gynecol Oncol 2010, 117: 130–133.
Wang M, Wang J, Wang L, Wu L, Xin X: Notch1 expression correlates with tumor differentiation status in ovarian carcinoma. Med Oncol 2010, 27: 1329–1335.
Kluk MJ, Ashworth T, Wang H, Knoechel B, Mason EF, Morgan EA, Dorfman D, Pinkus G, Weigert O, Hornick JL, Chirieac LR, Hirsch M, Oh DJ, South AP, Leigh IM, Pourreyron C, Cassidy AJ, Deangelo DJ, Weinstock DM, Krop IE, Dillon D, Brock JE, Lazar AJ, Peto M, Cho RJ, Stoeck A, Haines BB, Sathayanrayanan S, Rodig S, Aster JC: Gauging Notch1 Activation in Cancer Using Immunohistochemistry. PloS one 2013, 8: e67306.
Oktem G, Sanci M, Bilir A, Yildirim Y, Kececi SD, Ayla S, Inan S: Cancer stem cell and embryonic development-associated molecules contribute to prognostic significance in ovarian cancer. Int J Gyn Cancer 2012, 22: 23–29.
Jordan CT, Guzman ML, Noble M: Cancer stem cells. N Engl J Med 2006, 355: 1253–1261.
O'Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T, Pu C, Wang Z, Qiao L, Duan W: Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett 2014, 344: 180–187.
Shah MM, Zerlin M, Li BY, Herzog TJ, Kitajewski JK, Wright JD: The role of Notch and gamma-secretase inhibition in an ovarian cancer model. Anticancer Res 2013, 33: 801–808.
Foster R, Buckanovich RJ, Rueda BR: Ovarian cancer stem cells: working towards the root of stemness. Cancer Lett 2013, 338: 147–157.
Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, Zhang K, Conner M, Landen CN: Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res 2012, 18: 869–881.
Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, Luk L, Titley I, Carden CP, Box G, Hudson DL, Kaye SB, Brown R: Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther 2011, 10: 325–335.
Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP: Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68: 4311–4320.
Vathipadiekal V, Saxena D, Mok SC, Hauschka PV, Ozbun L, Birrer MJ: Identification of a potential ovarian cancer stem cell gene expression profile from advanced stage papillary serous ovarian cancer. PloS one 2012, 7: e29079.
Espinoza I, Miele L: Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett 2013, 341: 41–45.
Singh A, Settleman J: EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010, 29: 4741–4751.
Ahmed N, Abubaker K, Findlay J, Quinn M: Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr Cancer Drug Tar 2010, 10: 268–278.
Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F: Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007, 31: 277–283.
Marchini S, Fruscio R, Clivio L, Beltrame L, Porcu L, Nerini IF, Cavalieri D, Chiorino G, Cattoretti G, Mangioni C, Milani R, Torri V, Romualdi C, Zambelli A, Romano M, Signorelli M, di Giandomenico S, D'Incalci M: Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer 2013, 49: 520–530.
Gupta N, Xu Z, El-Sehemy A, Steed H, Fu Y: Notch3 induces epithelial-mesenchymal transition and attenuates carboplatin-induced apoptosis in ovarian cancer cells. Gynecol Oncol 2013, 130: 200–206.
Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, Biswas S, Turley H, Heikamp E, Hainfellner JA, Harris AL: Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res 2007, 67: 11244–11253.
Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL: Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res 2005, 65: 8690–8697.
Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH: The Notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137: 1124–1135.
Lu C, Bonome T, Li Y, Kamat AA, Han LY, Schmandt R, Coleman RL, Gershenson DM, Jaffe RB, Birrer MJ, Sood AK: Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res 2007, 67: 1757–1768.
Hu W, Lu C, Dong HH, Huang J, Shen DY, Stone RL, Nick AM, Shahzad MM, Mora E, Jennings NB, Lee SJ, Roh JW, Matsuo K, Nishimura M, Goodman BW, Jaffe RB, Langley RR, Deavers MT, Lopez-Berestein G, Coleman RL, Sood AK: Biological roles of the Delta family Notch ligand Dll4 in tumor and endothelial cells in ovarian cancer. Cancer Res 2011, 71: 6030–6039.
Espinoza I, Miele L: Notch inhibitors for cancer treatment. Pharmacol Ther 2013, 139: 95–110.
Takebe N, Nguyen D, Yang SX: Targeting Notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther 2014, 141: 140–149.
Olsauskas-Kuprys R, Zlobin A, Osipo C: Gamma secretase inhibitors of Notch signaling. OncoTargets and therapy 2013, 6: 943–955.
Lewis HD, Leveridge M, Strack PR, Haldon CD, O'Neil J, Kim H, Madin A, Hannam JC, Look AT, Kohl N, Draetta G, Harrison T, Kerby JA, Shearman MS, Beher D: Apoptosis in T cell acute lymphoblastic leukemia cells after cell cycle arrest induced by pharmacological inhibition of Notch signaling. Chem Biol 2007, 14: 209–219.
Chen J, Kesari S, Rooney C, Strack PR, Shen H, Wu L, Griffin JD: Inhibition of Notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 2010, 1: 822–835.
Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, de Wilde RF, De Oliveira E, Zhang Q, Puig O, Matsui W, Hidalgo M, Maitra A, Rajeshkumar NV: The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther 2012, 11: 1999–2009.
Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, Schwartz GK: Gamma-secretase inhibitors abrogate oxaliplatin-induced activation of the Notch1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res 2009, 69: 573–582.
Konishi J, Kawaguchi KS, Vo H, Haruki N, Gonzalez A, Carbone DP, Dang TP: Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res 2007, 67: 8051–8057.
Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, Stathis A, Hirte HW, Razak AR, Reedijk M, Chen Z, Cohen B, Zhang WJ, Wang L, Ivy SP, Moore MJ, Oza AM, Siu LL, McWhirter E: A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs 2014, 32: 243–249.
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, Lorusso P: Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 2012, 30: 2307–2313.
Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT, Froehlich AM, Paranilam J, Hayes DF, Wicha MS, Chang JC: Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res 2013, 19: 1512–1524.
Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari A, Shapiro GI, Boylan JF, Xu ZX, Wang K, Koehler A, Song J, Middleton SA, Deutsch J, Demario M, Kurzrock R, Wheler JJ: Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol 2012, 30: 2348–2353.
Wang M, Ma X, Wang J, Wang L, Wang Y: Pretreatment with the gamma-secretase inhibitor DAPT sensitizes drug-resistant ovarian cancer cells to cisplatin by downregulation of Notch signaling. Int J Oncol 2014, 44: 1401–1409.
McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, Chin KT, Partridge JC, Poole BB, Cheng KH, Daggett J Jr, Cullen K, Kantoff E, Hasselbatt K, Berkowitz J, Muto MG, Berkowitz RS, Aster JC, Matulonis UA, Dinulescu DM: Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A 2012, 109: E2939–2948.
Groeneweg JW, DiGloria CM, Yuan J, Richardson WS, Growdon WB, Sathyanarayanan S, Foster R, Rueda BR: Inhibition of Notch signaling in combination with Paclitaxel reduces platinum-resistant ovarian tumor growth. Front Oncol 2014, 4: 171.
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar A, Demuth T, Kun LE, Boyett JM, Gilbertson RJ: Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 2011, 29: 3529–3534.
Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, Zhang T, Kamel-Reid S, Ivy SP, Hotte SJ, Razak AA, Chen EX, Brana I, Wizemann M, Wang L, Siu LL, Bedard PL: A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs 2013, 31: 1182–1191.
Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, Cohen B, Zhang WJ, Clarke B, Zhang T, Kamel-Reid S, Chen H, Ivy SP, Razak AR, Oza AM, Chen EX, Hirte HW, McGarrity A, Wang L, Siu LL, Hotte SJ: A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer 2013, 109: 943–949.
Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, Almhanna K, Kim R, Valone T, Jump H, Sullivan D: A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer 2012, 48: 997–1003.
De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, Blatchford PJ, Quackenbush K, Messersmith W: A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs 2014,.
Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, Bonal DM, Charytonowicz E, Gladoun N, de la Iglesia-Vicente J, Petrylak DP, Benson MC, Silva JM, Cordon-Cardo C: Suppression of acquired docetaxel resistance in prostate cancer through depletion of Notch1 and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22: 373–388.
Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, Miele L: ErbB-2 inhibition activates Notch1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene 2008, 27: 5019–5032.
Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS: Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther 2011, 10: 9–15.
Steg AD, Katre AA, Goodman B, Han HD, Nick AM, Stone RL, Coleman RL, Alvarez RD, Lopez-Berestein G, Sood AK, Landen CN: Targeting the Notch ligand JAGGED1 in both tumor cells and stroma in ovarian cancer. Clin Cancer Res 2011, 17: 5674–5685.
Drenzek JG, Seiler NL, Jaskula-Sztul R, Rausch MM, Rose SL: Xanthohumol decreases Notch1 expression and cell growth by cell cycle arrest and induction of apoptosis in epithelial ovarian cancer cell lines. Gynecol Oncol 2011, 122: 396–401.
Zhang X, Samadi AK, Roby KF, Timmermann B, Cohen MS: Inhibition of cell growth and induction of apoptosis in ovarian carcinoma cell lines CaOV3 and SKOV3 by natural withanolide withaferin A. Gynecol Oncol 2012, 124: 606–612.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Groeneweg, J.W., Foster, R., Growdon, W.B. et al. Notch signaling in serous ovarian cancer. J Ovarian Res 7, 95 (2014). https://doi.org/10.1186/s13048-014-0095-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-014-0095-1