
Abstract
AXL is a member of the TAM (TYRO3, AXL, and MERTK) receptor tyrosine kinases family (RTKs), and its abnormal expression has been linked to clinicopathological features and poor prognosis of cancer patients. There is mounting evidence supporting AXL's role in the occurrence and progression of cancer, as well as drug resistance and treatment tolerance. Recent studies revealed that reducing AXL expression can weaken cancer cells' drug resistance, indicating that AXL may be a promising target for anti-cancer drug treatment. This review aims to summarize the AXL's structure, the mechanisms regulating and activating it, and its expression pattern, especially in drug-resistant cancers. Additionally, we will discuss the diverse functions of AXL in mediating cancer drug resistance and the potential of AXL inhibitors in cancer treatment.
Similar content being viewed by others


Background
Cancer remains a leading cause of death worldwide, with its incidence and mortality burden continuing to rise rapidly [1]. Despite significant progress in research and the development of cancer treatment strategies such as targeted therapy and immunotherapy, drug resistance remains a major obstacle to effective therapeutic interventions against cancer [2, 3]. The drug resistance can be intrinsic or acquired and reflects the result of multiple genetic and epigenetic alterations in cancer cells [4, 5]. Therefore, urgent measures must be taken to identify new therapeutic strategies that can target intrinsic and acquired resistance mechanisms.
AXL is a member of the TAM (TYRO3, AXL, and MERTK) receptor tyrosine kinases (RTKs) family [6]. By binding to its primary ligand, the growth arrest-specific protein 6 (GAS6), AXL participates in various signal transduction cascades and plays a critical role in various biological processes including cell proliferation, survival, migration, efferocytosis, angiogenesis, platelet aggregation and fibrosis, and regulation of natural killer (NK) cell development [7, 8]. Multiple lines of evidence indicate that AXL is also involved in cancer progression and treatment tolerance. For example, in lung cancers, AXL interacts with epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 3 (ERBB3, also known as HER3) to maintain the activation status of downstream signal pathway, which confers intrinsic resistance to osimertinib in non-small cell lung cancer (NSCLC) cells [9]. AXL also promotes the transcription level of MYC, which leads to the imbalance of purine metabolism and accelerates the emergence of drug-resistant mutations in NSCLC [10]. In colorectal cancers, AXL induces the expression of Twist family BHLH transcription factor 1 (TWIST1) and mediates resistance to polo-like kinase 1 (PLK1) inhibitor [11]. Overexpression of AXL has also been found in a variety of other cancers, including clear cell renal cell carcinoma (ccRCC) [12], hepatocellular carcinoma (HCC) [13], and cholangiocarcinoma (CCA) [14], which is associated with poor prognosis of these cancer patients [12,13,14]. These findings suggest that AXL could be a useful therapeutic target to address the issue of cancer drug resistance.
This review aims to describe the structure of AXL and its expression regulation, summarize the expression pattern of AXL in cancers, and further discuss the role of AXL plays in cancer occurrence development, particularly in anti-cancer drug resistance. Additionally, we will explore the potential of AXL as a therapeutic target to overcome tumor chemoresistance.
Structure and activation of AXL
The AXL gene is located on chromosome 19q13.2, it includes 20 exons and encodes an 894-amino acid polypeptide with multiple domains, which can be divided into three parts (Fig. 1A) [15]. Exons 1–10 mainly encode two fibronectin type III (FNIII) domains and two immunoglobulin (IG)-like domains constituting the extracellular part, which is involved in binding with ligands (Fig. 1B and C). Exon 11 encodes extracellular proteolytic cleavage sites and a transmembrane domain, and exons 12–20 encode intracellular domain with tyrosine kinase activity [15, 16].

The structure of AXL and GAS6. A The AXL protein comprises an intracellular domain, single helix transmembrane region, two fibronectin type III (FNIII) domains, and two immunoglobulin (IG)-like domains. On the other hand, GAS6 consists of a γ-carboxyglutamic acid (GLA) domain, a loop region, four epidermal growth factor (EGF)-like repeats, and two globular laminin G like (LG) domains. In Figure B and C, we can observe the interaction between AXL and GAS6, front both the front and top sides respectively, as visualized in the Protein Data Bank (PDB) with the identifier 2C5D
The main ligand of AXL [17] is GAS6, which consists of a γ-carboxyglutamic acid (GLA) domain at N-terminus, a loop region, four EGF-like repeats in the middle, and two globular laminin G like (LG) domains at C-terminus [18]. When AXL binds to GAS6, the complex dimerizes with another GAS6-AXL complex to form a 2:2 homodimerized complex with no direct AXL/AXL or GAS6/GAS6 contact, followed by trans-autophosphorylation of the tyrosine residues in the intracellular domain of AXL [19,20,21]. AXL phosphorylation is required for recruitment of corresponding adaptor molecules and effector proteins and ultimately to activate downstream signal pathways [19,20,21]. Six phosphorylation sites have been found in the AXL kinase domain, of which three (Tyr698, Tyr702 and Tyr703) are considered to be related to autophosphorylation and AXL activation, while the other three (Tyr779, Tyr821 and Tyr866) are involved in the docking and signal transduction of adaptor proteins [22].
In addition to GAS6, protein S (PROS1) has also been identified as a ligand of AXL. By phosphorylating AXL and activating downstream NF-κB signal pathway, PROS1 promotes Glioblastoma (GBM) tumor growth [23]. Besides AXL’s ligands, other TAM family members [24] or non-TAM proteins also affect AXL activation. For instance, co-immunoprecipitation experiments suggest that the AXL and TYRO3 receptors are closely associated, which significantly enhance GAS6 mediated AXL phosphorylation [24, 25].
Furthermore, AXL can be activated by interacting with several other non-TAM family member proteins via GAS6 independent mechanisms. For instance, it has been found that AXL heterodimerize with ERBB receptor family members, platelet-derived growth factor receptor (PDGFR), and mesenchymal to epithelial transition factor (MET) and is activated without GAS6 participation [26,27,28]. In addition, activated EGFR phosphorylates AXL Tyr779, leading to ligand-independent AXL activity and activation of more diversified down-stream signaling pathways than those triggered by EGFR alone [26, 29]. Similarly, the activated HER2 forms a complex with AXL and activates AXL in a GAS6-independent manner, which accelerates epithelial-mesenchymal transition (EMT) and metastasis of breast cancer cells [30]. AXL is required for vascular endothelial growth factor-A (VEGF-A) dependent activation of PI3K (phosphoinositide 3-kinase)/AKT (protein kinase B) in endothelial cells. Under the stimulation of VEGF-A, VEGF receptor-2 (VEGFR-2) activates the Src family kinase (SFK), and then promotes the GAS6-independent activation of AXL, which is necessary for vascular permeability and corneal neovascularization [31].
Regulation of AXL
In recent years, AXL has emerged as a key player in various biological processes, including immune regulation, cellular signaling, and cancer progression [7, 8]. Consequently, the regulation of AXL has become an area of intense research interest. Several mechanisms have been identified that regulate AXL expression and function, including transcriptional, post-transcriptional, and post-translational regulation (Fig. 2).

The regulation of AXL. A Expression of AXL is regulated by various transcription factors. B AXL undergoes post-transcriptional regulation. C The protein level of AXL is regulated in the post-translation stage
Transcriptional regulation of AXL is achieved through the binding of various transcription factors to the AXL promoter region, such as TEA domain (TEAD), activation protein-1 (AP-1), and hypoxia-inducible transcription factor-1α (HIF-1α) [32,33,34]. TEAD combines with AXL promoter to enhance its promoter activity, in turn mediating the resistance of colorectal cancer to 5-FU [34]. AP-1 can bind to AP-1 motif of Axl promoter [32]. Blocking JNK (c-Jun N-terminal kinase)/AP-1 inhibits AXL transcription and attenuates drug resistance to PI3Kα therapy in esophagus cancer and head and neck squamous cell carcinoma [35]. Additionally, epigenetic modifications have been shown to modulate AXL expression. Some studies have found that methylation in the GC rich region of AXL promoter can restrict AXL gene expression [36].
Post-transcriptional regulation of AXL involves non-coding RNAs. Various micro-RNAs (miRNAs), such as miR-34a, miR-432, and miR-202-5p, can negatively regulate AXL expression by binding to the 3'UTR of the AXL mRNA and promoting its degradation [37,38,39]. LncTASR, a long noncoding RNA (lncRNA), can directly bind to 5′ UTR of AXL mRNA to stabilize AXL mRNA [40]. Additionally, alternative splicing of the AXL transcript can result in the production of different AXL isoforms, which may have distinct functions and regulation [41].
Finally, post-translational regulation plays a critical role in maintaining protein stability and activity. For instance, by mediating AXL sialylation, ST3 β-Galactoside α-2,3-Sialyltransferase 1 (ST3GAL1) can increase the affinity between AXL and GAS6, and induces activation of AXL [42]. Heat shock protein 90 (HSP90), a molecular chaperone, can correctly fold proteins and be required to stabilize AXL [43], while other molecules, such as carboxy terminus of HSP70 interacting protein (CHIP) and E3 ubiquitin ligase CBL-b (casitas B lymphoma-b), can ubiquitinate AXL protein and induce its proteasome degradation [44,45,46]. Additionally, AXL can be cleaved by the A disintegrin and metalloproteinases (ADAM) 10 and ADAM17 to generate soluble AXL (sAXL), which inhibit AXL function and could be a promising biomarker for predicting cancer progression [47].
In summary, the regulation of AXL is a complex process that involves various mechanisms, including transcriptional, post-transcriptional, and post-translational regulation. A better understanding of these mechanisms is crucial for the development of novel therapeutic strategies targeting AXL in cancer and other diseases.
Expression of AXL in cancers
The receptor tyrosine kinase AXL has been shown to be highly expressed in several major types of cancers and is closely associated with tumor progression [48]. In particular, upregulated AXL mRNA expression has been observed in ccRCC, where it is associated with worse overall survival (OS) and can serve as an independent predictor of prognosis in ccRCC patients [12]. Moreover, high AXL expression has been reported in CCA compared to the adjacent normal tissue, the patients with high AXL levels have a higher risk of developing metastasis and a shorter OS [14]. Similarly, in HCC patients, high AXL expression could serve as a biomarker of higher recurrence and lower OS after hepatectomy [13]. Notably, higher AXL expression is a potent independent predictor of poor progression-free survival (PFS) or OS in patients with HPV-negative tumors treated by surgery alone [49] and patients with lung adenocarcinoma [50].
Furthermore, increased AXL mRNA and/or protein has been observed in other cancers, such as papillary thyroid carcinoma [51] and pancreatic ductal adenocarcinoma (PDAC) [52]. The enzymatic processing of AXL leads to the production of sAXL, and plasma sAXL is significantly increased in HCC and PDAC, making it a candidate biomarker for early diagnosis of these cancers [52,53,54,55], thus highlighting the potential value of sAXL in cancer diagnosis and prognosis prediction.
In recent years, studies have shown that high AXL expression is closely associated with treatment-resistant cancers (Table 1). For instance, high AXL expression has been found in NSCLC patients with low response to EGFR tyrosine kinase inhibitor (TKI) [9], as well as in erlotinib or osimertinib resistant cancer cell lines [56, 57]. Similarly, in patients with ovarian cancer or endometrial cancer, high AXL expression is associated with poor chemoresponse [58, 59] and patients with ccRCC who have high AXL expression shows lower objective response rate to PD-1 inhibition therapy [60]. In patients with colorectal cancer who received anti-EGFR treatments, those with high AXL expression show lower PFS rates than those with low AXL [61]. In addition, cell line expression data also reveal that high AXL expression is found in drug-resistant breast cancer cells and small-cell lung cancer cells (SCLC), but not in drug-sensitive cells [62, 63]. Overall, these findings suggest that AXL may play a critical role in promoting resistance to cancer treatments and further highlight the potential of AXL as a therapeutic target for cancer treatment.
Functions of AXL in drug-resistant cancer
Multiple studies have demonstrated that AXL is involved in various signaling pathways that are critical for cancer initiation and progression (Fig. 3). However, the precise mechanisms underlying AXL-mediated drug resistance in cancer cells remain largely unclear. Here, we will specifically discuss the role AXL in promoting drug tolerance of cancer cells to treatment.

Signal pathways mediated by AXL in the occurrence and development of cancer. AXL play a crucial role in the occurrence and development of cancer through various signal pathways
EMT
EMT is a critical process by which epithelial cells acquire a mesenchymal phenotype, which enables them to invade and metastasize to distant organs. This mechanism has been shown to contribute to drug resistance in cancer cells [64,65,66]. A growing body of evidence suggests that AXL is closely associated with EMT (Fig. 4), and can be used as an EMT marker in several types of cancer, including esophageal squamous cell carcinoma (ESCC) [67]. In oral squamous cell carcinoma (OSCC), AXL upregulates Snail expression and promotes EMT via AKT/GSK-3β (Glycogen Synthase Kinase 3β)/β-catenin signaling pathway [68]. AXL also induces the upregulation of ZEB1 (zinc finger E-box binding homeobox 1) transcription and mediates the drug resistance of breast cancer to doxorubicin through the same pathway [69]. In colorectal cancer, increased AXL contributes to the upregulation of TWIST1, which is directly related to EMT and mediates resistance to PLK1 inhibitors [11]. Previous studies have confirmed that TGF-β (transforming growth factor-β)/SMAD3 (SMAD family member 3) participates in EMT of lung adenocarcinoma cells [70]. In HCC, the interaction between AXL and 14–3-3ζ leads to phosphorylation of Ser213 in SMAD3, inducing the upregulation of TGF-β target genes such as Snail and autocrine TGF-β secretion [71].

AXL-mediated epithelial–mesenchymal transition (EMT) in drug-resistant cancer. AXL is known to activate EMT transcription factors through three pathways to promote EMT transformation and drug resistance of cancer cells. These pathways include AKT/GSK-3β/β-catenin, TGF-β/Smad3, and PI3K/AKT/HIF-1α
On the other hand, inhibition of GAS6/AXL axis reduces Snail and N-cadherin but upregulates E-cadherin, inhibiting EMT in esophageal cancer cells [72]. Similarly, miR-625-3p can directly target AXL and reverse TGF-β1 induced EMT, enhancing sensitivity to gefitinib in NSCLC [73]. Hypoxia has been shown to increased activity of HIFs and promote tumor progression by inducing EMT [74]. Interference with AXL leads to downregulation of HIF-1α, which, in turn, reduces EMT induced by hypoxia and enhances the immunotherapeutic responses in HER2 breast cancer [75].
DNA damage and DNA damage response (DDR)
Stimulation such as ultraviolet light can cause DNA replication stress (RS), leading to stalled replication forks and DNA damage that introduces genomic instability [76]. Genomic instability is a key hallmark of cancer and is closely linked to drug-resistance [77]. Cancer cells respond to DNA damage through various ways, collectively known as DDR (Fig. 5). These responses can be summarized into four aspects: activation of various repair pathways, metabolic reprogramming, blocking cell cycle process, and inducing cell death in the cases of irreparable damage [78,79,80]. It has been well established that aberrant DDR contributes to cancer progression and resistance to DNA-damaging drugs.

AXL affect DNA damage and DNA damage response (DDR) in drug-resistant cancer. DNA damage and repair are dynamic processes that require a delicate balance. AXL plays a crucial role in maintaining this balance by not only inhibiting DNA damage but also participating in multiple processes of DNA repair, such as DNA damage response (DDR), metabolic reprogramming, cell cycle arrest, and apoptosis
Several studies have revealed that AXL is associated with DNA damage. AXL has been found to attenuate reactive oxygen species (ROS) production and inhibit DNA double-strand breaks (DSB) [10], on the other hand, inhibition of AXL leads to enhanced RS and increased DNA damage [59]. Additionally, when combined with other drugs, AXL can interfere with replication fork progression. Although carboplatin and GAS6 inhibitor AVB500 alone shows no effect on replication forks, the combination of the two significantly hinder the progress of replication forks in ovarian cancer cells [59]. Similarly, use of AXL inhibitor BGB324 with ATR inhibitors can cause collapse of replication fork in NSCLC [81]. It is worth noting that the combination of PARP inhibitor and AVB-500 increases DNA damage and genomic instability by increasing replication fork speed rather than stalling the fork. In addition, to affecting DNA replication process, AXL is also involved in multiple repair pathways of DDR [59]. Inhibition of GAS6/AXL axis reduces RAD51 foci and increase 53BP1 foci, inhibiting homologous recombination (HR) and increasing the sensitivity of ovarian cancer to carboplatin [59]. Translesion synthesis (TLS) is a basic pathway for repairing DNA damage caused by replication arrest, but it also serves as the main source of cell mutation [82]. Mono-ubiquitination of proliferating cell nuclear antigen (PCNA) induced by RAD18 is critical for TLS [83]. A recent study found that AXL neddylates and activates RAD18 to enhance TLS and accelerate the emergence of T790M in resistant NSCLC cells [10].
Furthermore, AXL is involved in upregulation and activation of MYC, leading to an imbalance in purine metabolism and increased adaptive mutability [10]. C-ABL and p73 play important roles in the process of cellular apoptosis caused by DNA damage [84], and both are interfered with by AXL to enhance cisplatin resistance in esophageal cancer [85] through impeding nuclear aggregation of c-ABL and impairing p73 protein stability. The effect of AXL on cell cycle progression is still under debate. One study found that AXL inhibition activates ATR/CHK1 (checkpoint kinase 1) and sensitizes NSCLC cells to ATR inhibitors. The combination of AXL and ATR inhibitors can prematurely activate cyclin dependent kinase 1 (CDC2) and induce mitotic catastrophes [81]. However, other have reported AXL overexpression activates the ERK/p90RSK and mTOR pathways, thereby activating CHK1 to promote cell survival and mediate primary and acquired resistance to WEE1 inhibition in SCLC [63]. In summary, AXL has been demonstrated to mediate drug resistance by influencing DNA damage and repair, but its intrinsic mechanism for DDR remains to be specifically explored.
Immunosuppression
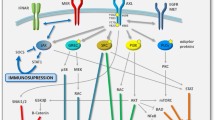
Although significant progress has been made in immunotherapy by activating the immune system to eliminate tumors in recent years, drug resistance remains a major obstacle to its application in clinical practice [86]. AXL plays an essential role in shaping the process of tumor immune tolerance (Fig. 6) [87]. AXL endows cancer cells with resistance to treatment through activation of PI3K/AKT pathway and up-regulation of programmed cell death ligand 1 (PD-L1) transcription in head and neck cancer cells, thereby inhibiting the immune killing effect of the body [88].

AXL-mediated immunosuppression in drug-resistant cancer. AXL plays a key role in mediating immunosuppression through both intrinsic shaping of cancer cells and extrinsic modification of microenvironment surrounding tumors
In the case of GBM, immune-related cells in tumor microenvironment, such as tumor- associated microglia and macrophages, secrete PROS1 to promote the growth of glioma cells, which phosphorylates and activates AXL in glioma stem cells, thus inhibition of AXL could significantly improve the efficacy of Navurizumab [23]. Additionally, tumor-related macrophages secrete 14–3-3ζ, which interacts and phosphorylates AXL to activate downstream pathways to promote the tolerance of PDAC cells to chemotherapy [89]. AXL could affect cytotoxic immune response against tumors by reducing the expression of intercellular adhesion molecule 1 (ICAM1) and UL16 binding protein 1 (ULBP1) in mesenchymal human lung cancer cells. Both of these contribute to the immune resistance to NK and cytotoxic T lymphocytes (CTL) cells [90]. Since ULBP1 plays a role in the recognition of target cells [91] and the combination of ICAM1 and lymphocyte function-associated antigen 1 (LFA-1) enhances immune cells to kill the target cells [92], targeting AXL is beneficial to the formation of anti-tumor microenvironment and enhances the treatment response. Indeed, depletion of AXL facilitates expression of MHC-I, infiltration of dendritic cells and CD8 + T cells, and T-cell-mediated immune response [93]. Particularly, inhibiting AXL in dendritic cells can induce the secretion of type I interferon, promote the expansion of CD8 + T cells, and sensitize NSCLC carrying serine/threonine kinase 11 (STK11/LKB1) mutant to pabolizumab [94]. Recent study in leukemia cells has found GAS6/AXL axis is required to skew macrophages toward a tumor-promoting tissue repair phenotype to establish a suppressive environment to prevent immune attacks [95]. It has been proposed that AXL inhibition can be achieved by blocking TBK1 (TANK binding kinase 1)/NF-κB pathway, a key signal pathway of immune cells, changes the composition of chemokines and cytokines in tumor microenvironment, making tumor sensitive to treatment [96].
Activation of oncogenic bypass pathway
Ample evidence suggests AXL mediates resistance to multiple anti-cancer drugs. In NSCLC with EGFR mutation, the activation of several signal pathways, such as MAPK/ERK and PI3K/AKT, is considered to be one of the major mechanisms responsible for acquiring drug resistance against EGFR TKIs [97]. In the case of resistance to osimertinib, multiple pathways seem to be involved, including the MAPK/ERK pathway, SRC and its downstream AKT signal pathway, and EGFR-induced signal transduction triggered by AXL [9, 56, 98,99,100]. In breast cancers, the heterodimerization of AXL and HER2 leads to the acquired resistance to anti-HER2 drug trastuzumab by activating AKT and ERK pathways [62].
Other functions
AXL can contribute to drug tolerance, and its inhibition has been shown to reduce drug resistance in several types of cancer. For instance, inhibition of AXL reduces the phosphorylation of M2 isoform of pyruvate kinase (PKM2) at Y105, which decreases glycolysis and enhances chemosensitivity of human ovarian cancer cells to cisplatin [101]. In endometrial cancer cells, inhibition of AXL down-regulates various glycolytic metabolites, leading to increased sensitivity to paclitaxel [58]. AXL has also been found to activate Akt/β-catenin pathway which up-regulates the transcription level of c-MYC and promotes resistance of esophageal adenocarcinoma to epirubicin [102]. Furthermore, AXL can mediate the resistance of head and neck cancer to cetuximab through two mechanisms: (1) Activation of HER3 via up-regulation of HER3 ligand neuromodulator 1 (NRG1) [103]; and (2) Activation of c-ABL kinase via Tyr821 of AXL [104].
Targeting AXL to surmount drug resistance in cancer therapy
There is ample evidence supporting the important role of AXL in drug resistance across various types of cancers. Therefore, targeting AXL is a promising strategy for addressing drug resistance. Drugs that inhibit AXL can be grouped based on their mechanisms of action, including small molecule selective inhibitors (such as BGB324 and TP-0903), antibody–drug conjugates (such as BA3011), anti-AXL Fc fusion protein AVB-S6-500, and multitargeted inhibitors (such as ONO-7475, Merestinib and Sitravatinib) [105]. Experimental data from both in vivo and in vitro studies suggest that carboplatin/paclitaxel combined with AVB-S6-500 is more effective than chemotherapy alone [59]. Additionally, the combination of nivolumab and BGB324 prolongs the survival period of mice with GBM [23], and the combination of TP0903 and WEE1 inhibitor can overcome the resistance of SCLC to WEE1 inhibitor [63]. Table 2 listed AXL inhibitors that have entered clinical trials, some of which may help to overcome drug resistance and enhance treatment sensitivity when combined with other therapies.
In addition to the AXL-targeted drugs mentioned above, many monoclonal antibodies specific to AXL have been shown to inhibit the growth of cancer cells, including YW327.6S2, 20G7-D9, Mab173 and DAXL-88 [106]. However, most of these drugs are still in preclinical trial stage and there is no available data regarding the efficacy [106]. Anti-AXL chimeric antigen receptor (CAR)-T-cell therapy is a new precise targeted immunotherapy for cancer, which is currently under clinical trials (NCT05128786 and NCT03393936). Due to its significant effect on AXL-positive osteomyeloid leukemia cells and TNBC cell models [107, 108], it could be a promising regime for cancer treatment. Another strategy targeting AXL is nucleic acid aptamers, a short stretch of nucleotide that can bind to specific target molecules with high affinity, low toxicity and are easy to synthesize [109]. The aptamer GL21.T fulfils these criteria, preliminary data show it binds AXL with high affinity, blocks AXL-dependent signal transduction pathway, and inhibits tumor migration and invasion [110]. However, more evidence is needed to support the application of these drugs in cancer treatment.
Discussion and future perspectives
Drug resistance remains a significant challenge in cancer treatment, but recent studies suggest that AXL may be a promising target to address this issue. In addition to GAS6-dependent activation, AXL can be activated by interacting with various partners, such as PROS1 and EGFR, which are overexpressed in many cancers, particularly those that are drug-resistant. Because AXL plays a role in cancer resistance through multiple mechanisms, AXL-target drugs could be an effective strategy to alleviate drug resistance by inhibiting EMT transformation, interfering with DNA damage and DDR, inhibiting anti-tumor immune microenvironment, attenuating reactivation of oncogenic bypass, metabolic disorder and so on. Numerous ongoing clinical trials are targeting AXL alone or in combination with other drugs, demonstrating significant clinical therapeutic effect. However, several issues need to be considered when using combination therapy with AXL inhibitors. AXL activity is potent and extensive in many biological processes, and the side effects resulting from AXL targeting treatment should be carefully evaluated in clinical application. Since combination therapy does not always generate curative effect, it may inhibit immunity and metabolic remodeling at some point, and this needs to be further clarified to provide clear guidance for combined immunosuppressive therapy.
With the development of nanomedicine, the drug delivery system based on nanocarriers (NDDS) shows a good application prospect in cancer treatment [111]. NDDS can not only improve the solubility of chemotherapy drugs and deliver higher doses of drugs, but also reduce the toxicity of systemic chemotherapy to normal tissues [112, 113]. Based on this, the use of nanocarriers to deliver AXL inhibitors may be an effective strategy to reduce their toxic side effects and enhance their efficacy. However, there are still many challenges that need to be addressed before nanotechnology can achieve widespread clinical application.
In short, targeting AXL is a promising new strategy to delay or even eliminate the development of drug resistance due to its extensive biological effects and functional diversity. As AXL-targeted drug improve and the underlying mechanism of AXL-drug resistance is better understood, AXL inhibition is expected to provide promising strategies for the treatment of cancer patients.
Availability of data and materials
Not applicable.
Abbreviations
- RTKs:
-
Receptor tyrosine kinases
- GAS6:
-
Growth arrest-specific protein 6
- NK:
-
Natural killer
- EGFR:
-
Epidermal growth factor receptor
- HER3:
-
Human epidermal growth factor receptor 3
- NSCLC:
-
Non-small cell lung cancer
- TWIST1:
-
Twist family BHLH transcription factor 1
- PLK1:
-
Polo-like kinase 1
- ccRCC:
-
Clear cell renal cell carcinoma
- HCC:
-
Hepatocellular carcinoma
- CCA:
-
Cholangiocarcinoma
- FNIII:
-
Fibronectin type III
- IG:
-
Immunoglobulin
- GLA:
-
γ-Carboxyglutamic acid
- LG:
-
Laminin G
- PROS1:
-
Protein S
- GBM:
-
Glioblastoma
- PDGFR:
-
Platelet-derived growth factor receptor
- MET:
-
Mesenchymal to epithelial transition factor
- EMT:
-
Epithelial-mesenchymal transition
- VEGF-A:
-
Vascular endothelial growth factor-A
- PI3K:
-
Phosphoinositide 3-kinase
- AKT:
-
Protein kinase B
- VEGFR-2:
-
VEGF receptor-2
- SFK:
-
Src family kinase
- TEAD:
-
TEA domain
- AP-1:
-
Activation protein-1
- HIF-1α:
-
Hypoxia-inducible transcription factor-1α
- JNK:
-
C-Jun N-terminal kinase
- miRNAs:
-
Micro-RNAs
- lncRNA:
-
Long noncoding RNA
- ST3GAL1:
-
ST3 β-Galactoside α-2,3-Sialyltransferase 1
- HSP90:
-
Heat shock protein 90
- CHIP:
-
Carboxy terminus of HSP70 interacting protein
- CBL-b:
-
Casitas B lymphoma-b
- ADAM:
-
A disintegrin and metalloproteinases
- sAXL:
-
Soluble AXL
- OS:
-
Overall survival
- PDAC:
-
Pancreatic ductal adenocarcinoma
- TKI:
-
Tyrosine kinase inhibitors
- SCLC:
-
Small-cell lung cancer cells
- ESCC:
-
Esophageal squamous cell carcinoma
- OSCC:
-
Oral squamous cell carcinoma
- GSK-3β:
-
Glycogen synthase kinase 3β
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- TGF-β:
-
Transforming growth factor-β
- SMAD3:
-
SMAD family member 3
- DDR:
-
DNA damage response
- RS:
-
Replication stress
- ROS:
-
Reactive oxygen species
- DSB:
-
Double-strand breaks
- HR:
-
Homologous recombination
- TLS:
-
Translesion synthesis
- PCNA:
-
Proliferating cell nuclear antigen
- CHK1:
-
Checkpoint kinase 1
- CDC2:
-
Cyclin dependent kinase 1
- ERK:
-
Extracellular signal-regulated kinase
- p90RSK:
-
90 KDa ribosomal S6 kinase
- WEE1:
-
WEE1 G2 checkpoint kinase
- PD-L1:
-
Programmed cell death ligand 1
- ICAM1:
-
Intercellular adhesion molecule 1
- ULBP1:
-
UL16 binding protein 1
- CTL:
-
Cytotoxic T lymphocytes
- LFA-1:
-
Lymphocyte function-associated antigen 1
- STK11/LKB1:
-
Serine/threonine kinase 11
- TBK1:
-
TANK binding kinase 1
- NF-κB:
-
Nuclear factor-κB
- MAPK:
-
Mitogen-activated protein kinase
- PKM2:
-
M2 isoform of pyruvate kinase
- NRG1:
-
Neuromodulator 1
- CAR:
-
Chimeric antigen receptor
- MZF-1:
-
Myeloid zinc finger 1
- EZH2:
-
Enhancer of zeste homolog 2
- NFI:
-
Nuclear factor I
- SP1/SP3:
-
Specificity protein 1/3
- hnRNP-L:
-
Heterogeneous nuclear ribonucleoprotein-L
- ZNF224:
-
Zinc finger protein 224
- PTBP1:
-
Polypyrimidine tract binding protein 1
- METTL3:
-
Methyltransferase 3
- JAK:
-
Janus kinase, STAT1: signal transducer and activator of transcription 1
- SOCS1:
-
Suppressor of cytokine signaling 1
- mTOR:
-
Mechanistic target of rapamycin kinase
- Ang-2:
-
Angiogenin-2
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309.
Chatterjee N, Bivona TG. Polytherapy and Targeted Cancer Drug Resistance. Trends Cancer. 2019;5(3):170–82.
Cabanos HF, Hata AN. Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers (Basel). 2021;13(11):2666.
Ward RA, Fawell S, Floc’h N, Flemington V, McKerrecher D, Smith PD. Challenges and Opportunities in Cancer Drug Resistance. Chem Rev. 2021;121(6):3297–351.
Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S, Ambros PF, et al. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991;6(11):2113–20.
Du W, Brekken RA. Does Axl have potential as a therapeutic target in pancreatic cancer? Expert Opin Ther Targets. 2018;22(11):955–66.
Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11): a009076.
Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10(1):259.
Noronha A, Belugali Nataraj N, Lee JS, Zhitomirsky B, Oren Y, Oster S, et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022;12(11):2666–83.
Solanes-Casado S, Cebrian A, Rodriguez-Remirez M, Mahillo I, Garcia-Garcia L, Rio-Vilarino A, et al. Overcoming PLK1 inhibitor resistance by targeting mevalonate pathway to impair AXL-TWIST axis in colorectal cancer. Biomed Pharmacother. 2021;144: 112347.
Wang Y, Tian Y, Liu S, Wang Z, Xing Q. Prognostic value and immunological role of AXL gene in clear cell renal cell carcinoma associated with identifying LncRNA/RBP/AXL mRNA networks. Cancer Cell Int. 2021;21(1):625.
Hsu CC, Hsieh PM, Chen YS, Lo GH, Lin HY, Dai CY, et al. Axl and autophagy LC3 expression in tumors is strongly associated with clinical prognosis of hepatocellular carcinoma patients after curative resection. Cancer Med. 2019;8(7):3453–63.
Khamko R, Wasenang W, Daduang J, Settasatian C, Limpaiboon T. Combined OPCML and AXL Expression as a Prognostic Marker and OPCML Enhances AXL Inhibitor in Cholangiocarcinoma. In Vivo. 2022;36(3):1168–77.
O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11(10):5016–31.
Lee CH, Chun T. Anti-Inflammatory Role of TAM Family of Receptor Tyrosine Kinases Via Modulating Macrophage Function. Mol Cells. 2019;42(1):1–7.
Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80(4):661–70.
Lew ED, Oh J, Burrola PG, Lax I, Zagorska A, Traves PG, et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife. 2014;3:9953.
Di Stasi R, De Rosa L, D’Andrea LD. Therapeutic aspects of the Axl/Gas6 molecular system. Drug Discov Today. 2020;25(12):2130–48.
Auyez A, Sayan AE, Kriajevska M, Tulchinsky E. AXL Receptor in Cancer Metastasis and Drug Resistance: When Normal Functions Go Askew. Cancers (Basel). 2021;13(19):4864.
Wium M, Ajayi-Smith AF, Paccez JD, Zerbini LF. The Role of the Receptor Tyrosine Kinase Axl in Carcinogenesis and Development of Therapeutic Resistance: An Overview of Molecular Mechanisms and Future Applications. Cancers (Basel). 2021;13(7):1521.
Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. 1997;14(22):2619–31.
Sadahiro H, Kang KD, Gibson JT, Minata M, Yu H, Shi J, et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res. 2018;78(11):3002–13.
Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178(9):5635–42.
Brown JE, Krodel M, Pazos M, Lai C, Prieto AL. Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS ONE. 2012;7(5): e36800.
Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6(287):ra66.
Vouri M, Croucher DR, Kennedy SP, An Q, Pilkington GJ, Hafizi S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis. 2016;5(10): e266.
Antony J, Tan TZ, Kelly Z, Low J, Choolani M, Recchi C, et al. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci Signal. 2016;9(448):ra97.
Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–60.
Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018;23(5):1476–90.
Ruan GX, Kazlauskas A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012;31(7):1692–703.
Mudduluru G, Leupold JH, Stroebel P, Allgayer H. PMA up-regulates the transcription of Axl by AP-1 transcription factor binding to TRE sequences via the MAPK cascade in leukaemia cells. Biol Cell. 2010;103(1):21–33.
Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111(37):13373–8.
Pobbati AV, Hong W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics. 2020;10(8):3622–35.
Badarni M, Prasad M, Balaban N, Zorea J, Yegodayev KM, Joshua BZ, et al. Repression of AXL expression by AP-1/JNK blockage overcomes resistance to PI3Ka therapy. JCI Insight. 2019;5(8):e125341.
Mudduluru G, Allgayer H. The human receptor tyrosine kinase Axl gene–promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci Rep. 2008;28(3):161–76.
Lim D, Cho JG, Yun E, Lee A, Ryu HY, Lee YJ, et al. MicroRNA 34a-AXL Axis Regulates Vasculogenic Mimicry Formation in Breast Cancer Cells. Genes (Basel). 2020;12(1):9.
Wu J, Zhou Z. MicroRNA-432 Acts as a Prognostic Biomarker and an Inhibitor of Cell Proliferation, Migration, and Invasion in Breast Cancer. Clin Breast Cancer. 2021;21(4):e462–70.
Zhang J, Du C, Zhang L, Wang Y, Zhang Y, Li J. lncRNA GSEC Promotes the Progression of Triple Negative Breast Cancer (TNBC) by Targeting the miR-202-5p/AXL Axis. Onco Targets Ther. 2021;14:2747–59.
Shi H, Sun Y, He M, Yang X, Hamada M, Fukunaga T, et al. Targeting the TR4 nuclear receptor-mediated lncTASR/AXL signaling with tretinoin increases the sunitinib sensitivity to better suppress the RCC progression. Oncogene. 2020;39(3):530–45.
Shen L, Lei S, Zhang B, Li S, Huang L, Czachor A, et al. Skipping of exon 10 in Axl pre-mRNA regulated by PTBP1 mediates invasion and metastasis process of liver cancer cells. Theranostics. 2020;10(13):5719–35.
Pietrobono S, Anichini G, Sala C, Manetti F, Almada LL, Pepe S, et al. ST3GAL1 is a target of the SOX2-GLI1 transcriptional complex and promotes melanoma metastasis through AXL. Nat Commun. 2020;11(1):5865.
Chen Y, Zhang Y, Chen S, Liu W, Lin Y, Zhang H, et al. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) sensitize melanoma cells to MEK inhibition and inhibit metastasis and relapse by inducing degradation of AXL. Pigment Cell Melanoma Res. 2022;35(2):238–51.
Yang H, Liang SQ, Xu D, Yang Z, Marti TM, Gao Y, et al. HSP90/AXL/eIF4E-regulated unfolded protein response as an acquired vulnerability in drug-resistant KRAS-mutant lung cancer. Oncogenesis. 2019;8(9):45.
Sun LW, Kao SH, Yang SF, Jhang SW, Lin YC, Chen CM, et al. Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis. Cells. 2021;10(11):2919.
Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508–12.
Miller MA, Oudin MJ, Sullivan RJ, Wang SJ, Meyer AS, Im H, et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016;6(4):382–99.
Zhang S, Xu XS, Yang JX, Guo JH, Chao TF, Tong Y. The prognostic role of Gas6/Axl axis in solid malignancies: a meta-analysis and literature review. Onco Targets Ther. 2018;11:509–19.
Busch CJ, Hagel C, Becker B, Oetting A, Mockelmann N, Droste C, et al. Tissue Microarray Analyses Suggest Axl as a Predictive Biomarker in HPV-Negative Head and Neck Cancer. Cancers (Basel). 2022;14(7):1829.
de Miguel-Perez D, Bayarri-Lara CI, Ortega FG, Russo A, Moyano Rodriguez MJ, Alvarez-Cubero MJ, et al. Post-Surgery Circulating Tumor Cells and AXL Overexpression as New Poor Prognostic Biomarkers in Resected Lung Adenocarcinoma. Cancers (Basel). 2019;11(11):1750.
Wei M, Wang Y, Liu Y, Li D, He X. AXL, along with PROS1, is overexpressed in papillary thyroid carcinoma and regulates its biological behaviour. World J Surg Oncol. 2022;20(1):334.
Martinez-Bosch N, Cristobal H, Iglesias M, Gironella M, Barranco L, Visa L, et al. Soluble AXL is a novel blood marker for early detection of pancreatic ductal adenocarcinoma and differential diagnosis from chronic pancreatitis. EBioMedicine. 2022;75: 103797.
Reichl P, Fang M, Starlinger P, Staufer K, Nenutil R, Muller P, et al. Multicenter analysis of soluble Axl reveals diagnostic value for very early stage hepatocellular carcinoma. Int J Cancer. 2015;137(2):385–94.
Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116(4):415–23.
Song X, Wu A, Ding Z, Liang S, Zhang C. Soluble Axl Is a Novel Diagnostic Biomarker of Hepatocellular Carcinoma in Chinese Patients with Chronic Hepatitis B Virus Infection. Cancer Res Treat. 2020;52(3):789–97.
Murakami Y, Kusakabe D, Watari K, Kawahara A, Azuma K, Akiba J, et al. AXL/CDCP1/SRC axis confers acquired resistance to osimertinib in lung cancer. Sci Rep. 2022;12(1):8983.
Sun Q, Lu Z, Zhang Y, Xue D, Xia H, She J, et al. Integrin beta3 Promotes Resistance to EGFR-TKI in Non-Small-Cell Lung Cancer by Upregulating AXL through the YAP Pathway. Cells. 2022;11(13):2078.
Bruce SF, Cho K, Noia H, Lomonosova E, Stock EC, Oplt A, et al. GAS6-AXL Inhibition by AVB-500 Overcomes Resistance to Paclitaxel in Endometrial Cancer by Decreasing Tumor Cell Glycolysis. Mol Cancer Ther. 2022;21(8):1348–59.
Mullen MM, Lomonosova E, Toboni MD, Oplt A, Cybulla E, Blachut B, et al. GAS6/AXL Inhibition Enhances Ovarian Cancer Sensitivity to Chemotherapy and PARP Inhibition through Increased DNA Damage and Enhanced Replication Stress. Mol Cancer Res. 2022;20(2):265–79.
Terry S, Dalban C, Rioux-Leclercq N, Adam J, Meylan M, Buart S, et al. Association of AXL and PD-L1 Expression with Clinical Outcomes in Patients with Advanced Renal Cell Carcinoma Treated with PD-1 Blockade. Clin Cancer Res. 2021;27(24):6749–60.
Cardone C, Blauensteiner B, Moreno-Viedma V, Martini G, Simeon V, Vitiello PP, et al. AXL is a predictor of poor survival and of resistance to anti-EGFR therapy in RAS wild-type metastatic colorectal cancer. Eur J Cancer. 2020;138:1–10.
Adam-Artigues A, Arenas EJ, Martinez-Sabadell A, Braso-Maristany F, Cervera R, Tormo E, et al. Targeting HER2-AXL heterodimerization to overcome resistance to HER2 blockade in breast cancer. Sci Adv. 2022;8(20):eabk2746.
Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J, et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res. 2017;23(20):6239–53.
Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5(5):390–401.
Tulchinsky E, Demidov O, Kriajevska M, Barlev NA, Imyanitov E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim Biophys Acta Rev Cancer. 2019;1871(1):29–39.
Luond F, Sugiyama N, Bill R, Bornes L, Hager C, Tang F, et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev Cell. 2021;56(23):3203–21 e11.
Zhang G, Kong X, Wang M, Zhao H, Han S, Hu R, et al. AXL is a marker for epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Oncol Lett. 2018;15(2):1900–6.
Li YY, Tao YW, Gao S, Li P, Zheng JM, Zhang SE, et al. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EBioMedicine. 2018;36:209–20.
Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y, et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics. 2016;6(8):1205–19.
Kitamura K, Seike M, Okano T, Matsuda K, Miyanaga A, Mizutani H, et al. MiR-134/487b/655 cluster regulates TGF-beta-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther. 2014;13(2):444–53.
Reichl P, Dengler M, van Zijl F, Huber H, Fuhrlinger G, Reichel C, et al. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology. 2015;61(3):930–41.
Kong L, Lu X, Chen X, Wu Y, Zhang Y, Shi H, et al. Qigesan inhibits esophageal cancer cell invasion and migration by inhibiting Gas6/Axl-induced epithelial-mesenchymal transition. Aging (Albany NY). 2020;12(10):9714–25.
Du W, Sun L, Liu T, Zhu J, Zeng Y, Zhang Y, et al. The miR-625-3p/AXL axis induces non-T790M acquired resistance to EGFR-TKI via activation of the TGF-beta/Smad pathway and EMT in EGFR-mutant non-small cell lung cancer. Oncol Rep. 2020;44(1):185–95.
Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer. 2016;2(12):758–70.
Goyette MA, Elkholi IE, Apcher C, Kuasne H, Rothlin CV, Muller WJ, et al. Targeting Axl favors an antitumorigenic microenvironment that enhances immunotherapy responses by decreasing Hif-1alpha levels. Proc Natl Acad Sci U S A. 2021;118(29):e2023868118.
Saxena S, Zou L. Hallmarks of DNA replication stress. Mol Cell. 2022;82(12):2298–314.
Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31–46.
Turgeon MO, Perry NJS, Poulogiannis G. DNA Damage, Repair, and Cancer Metabolism. Front Oncol. 2018;8:15.
Gourley C, Balmana J, Ledermann JA, Serra V, Dent R, Loibl S, et al. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J Clin Oncol. 2019;37(25):2257–69.
Huang RX, Zhou PK. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 2020;5(1):60.
Ramkumar K, Stewart CA, Cargill KR, Della Corte CM, Wang Q, Shen L, et al. AXL Inhibition Induces DNA Damage and Replication Stress in Non-Small Cell Lung Cancer Cells and Promotes Sensitivity to ATR Inhibitors. Mol Cancer Res. 2021;19(3):485–97.
Zhang S, Zhou T, Wang Z, Yi F, Li C, Guo W, et al. Post-Translational Modifications of PCNA in Control of DNA Synthesis and DNA Damage Tolerance-the Implications in Carcinogenesis. Int J Biol Sci. 2021;17(14):4047–59.
Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425(6954):188–91.
Rozenberg JM, Zvereva S, Dalina A, Blatov I, Zubarev I, Luppov D, et al. Dual Role of p73 in Cancer Microenvironment and DNA Damage Response. Cells. 2021;10(12):3516.
Hong J, Peng D, Chen Z, Sehdev V, Belkhiri A. ABL regulation by AXL promotes cisplatin resistance in esophageal cancer. Cancer Res. 2013;73(1):331–40.
Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol. 2021;16:223–49.
Engelsen AST, Lotsberg ML, Abou Khouzam R, Thiery JP, Lorens JB, Chouaib S, et al. Dissecting the Role of AXL in Cancer Immune Escape and Resistance to Immune Checkpoint Inhibition. Front Immunol. 2022;13: 869676.
Skinner HD, Giri U, Yang LP, Kumar M, Liu Y, Story MD, et al. Integrative Analysis Identifies a Novel AXL-PI3 Kinase-PD-L1 Signaling Axis Associated with Radiation Resistance in Head and Neck Cancer. Clin Cancer Res. 2017;23(11):2713–22.
D’Errico G, Alonso-Nocelo M, Vallespinos M, Hermann PC, Alcala S, Garcia CP, et al. Tumor-associated macrophage-secreted 14-3-3zeta signals via AXL to promote pancreatic cancer chemoresistance. Oncogene. 2019;38(27):5469–85.
Terry S, Abdou A, Engelsen AST, Buart S, Dessen P, Corgnac S, et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol Res. 2019;7(11):1789–802.
Schmiedel D, Mandelboim O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front Immunol. 2018;9:2040.
Reina M, Espel E. Role of LFA-1 and ICAM-1 in cancer. Cancers (Basel). 2017;9(11):153.
Aguilera TA, Rafat M, Castellini L, Shehade H, Kariolis MS, Hui AB, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun. 2016;7:13898.
Li H, Liu Z, Liu L, Zhang H, Han C, Girard L, et al. AXL targeting restores PD-1 blockade sensitivity of STK11/LKB1 mutant NSCLC through expansion of TCF1(+) CD8 T cells. Cell Rep Med. 2022;3(3): 100554.
Tirado-Gonzalez I, Descot A, Soetopo D, Nevmerzhitskaya A, Schaffer A, Kur IM, et al. AXL Inhibition in Macrophages Stimulates Host-versus-Leukemia Immunity and Eradicates Naive and Treatment-Resistant Leukemia. Cancer Discov. 2021;11(11):2924–43.
Ludwig KF, Du W, Sorrelle NB, Wnuk-Lipinska K, Topalovski M, Toombs JE, et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018;78(1):246–55.
Tian X, Gu T, Lee MH, Dong Z. Challenge and countermeasures for EGFR targeted therapy in non-small cell lung cancer. Biochim Biophys Acta Rev Cancer. 2022;1877(1): 188645.
Liu YN, Tsai MF, Wu SG, Chang TH, Tsai TH, Gow CH, et al. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int J Cancer. 2019;145(6):1609–24.
Lotsberg ML, Wnuk-Lipinska K, Terry S, Tan TZ, Lu N, Trachsel-Moncho L, et al. AXL Targeting Abrogates Autophagic Flux and Induces Immunogenic Cell Death in Drug-Resistant Cancer Cells. J Thorac Oncol. 2020;15(6):973–99.
Dong M, Xiao Q, Hu J, Cheng F, Zhang P, Zong W, et al. Targeting LRIG2 overcomes resistance to EGFR inhibitor in glioblastoma by modulating GAS6/AXL/SRC signaling. Cancer Gene Ther. 2020;27(12):878–97.
Tian M, Chen XS, Li LY, Wu HZ, Zeng D, Wang XL, et al. Inhibition of AXL enhances chemosensitivity of human ovarian cancer cells to cisplatin via decreasing glycolysis. Acta Pharmacol Sin. 2021;42(7):1180–9.
Hong J, Maacha S, Belkhiri A. Transcriptional upregulation of c-MYC by AXL confers epirubicin resistance in esophageal adenocarcinoma. Mol Oncol. 2018;12(12):2191–208.
Iida M, McDaniel NK, Kostecki KL, Welke NB, Kranjac CA, Liu P, et al. AXL regulates neuregulin1 expression leading to cetuximab resistance in head and neck cancer. BMC Cancer. 2022;22(1):447.
McDaniel NK, Iida M, Nickel KP, Longhurst CA, Fischbach SR, Rodems TS, et al. AXL Mediates Cetuximab and Radiation Resistance Through Tyrosine 821 and the c-ABL Kinase Pathway in Head and Neck Cancer. Clin Cancer Res. 2020;26(16):4349–59.
Tanaka M, Siemann DW. Therapeutic targeting of the Gas6/Axl signaling pathway in cancer. Int J Mol Sci. 2021;22(18):9953.
Sang YB, Kim JH, Kim CG, Hong MH, Kim HR, Cho BC, et al. The Development of AXL Inhibitors in Lung Cancer: Recent Progress and Challenges. Front Oncol. 2022;12: 811247.
Cho JH, Okuma A, Al-Rubaye D, Intisar E, Junghans RP, Wong WW. Engineering Axl specific CAR and SynNotch receptor for cancer therapy. Sci Rep. 2018;8(1):3846.
Wei J, Sun H, Zhang A, Wu X, Li Y, Liu J, et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell Immunol. 2018;331:49–58.
Hwang JA, Hur JY, Kim Y, Im JH, Jin SH, Ryu SH, et al. Efficacy of newly discovered DNA aptamers targeting AXL in a lung cancer cell with acquired resistance to Erlotinib. Transl Cancer Res. 2021;10(2):1025–33.
Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, Insabato L, et al. Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther. 2012;20(12):2291–303.
Majumder J, Taratula O, Minko T. Nanocarrier-based systems for targeted and site specific therapeutic delivery. Adv Drug Deliv Rev. 2019;144:57–77.
Al-Zoubi MS, Al-Zoubi RM. Nanomedicine tactics in cancer treatment: Challenge and hope. Crit Rev Oncol Hematol. 2022;174: 103677.
Gowd V, Ahmad A, Tarique M, Suhail M, Zughaibi TA, Tabrez S, et al. Advancement of cancer immunotherapy using nanoparticles-based nanomedicine. Semin Cancer Biol. 2022;86(Pt 2):624–44.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Nature Science Foundation of China (grant number: 81773218, 82102805 and 82272722) and the Natural Science Foundation of Hunan Province (2020JJ4122, 2021JJ40890, 2023JJ30788).
Author information
Authors and Affiliations
Contributions
SQF and QYW designed the research. YXT drafted the manuscript. SQF and HJZ improved the structure. QYW and YXT revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tang, Y., Zang, H., Wen, Q. et al. AXL in cancer: a modulator of drug resistance and therapeutic target. J Exp Clin Cancer Res 42, 148 (2023). https://doi.org/10.1186/s13046-023-02726-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-023-02726-w