
Abstract
The conventional method used to obtain a tumor biopsy for hepatocellular carcinoma (HCC) is invasive and does not evaluate dynamic cancer progression or assess tumor heterogeneity. It is thus imperative to create a novel non-invasive diagnostic technique for improvement in cancer screening, diagnosis, treatment selection, response assessment, and predicting prognosis for HCC. Circulating tumor DNA (ctDNA) is a non-invasive liquid biopsy method that reveals cancer-specific genetic and epigenetic aberrations. Owing to the development of technology in next-generation sequencing and PCR-based assays, the detection and quantification of ctDNA have greatly improved. In this publication, we provide an overview of current technologies used to detect ctDNA, the ctDNA markers utilized, and recent advances regarding the multiple clinical applications in the field of precision medicine for HCC.
Similar content being viewed by others
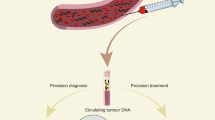

Background
Hepatocellular carcinoma (HCC) is a malignant tumor with high morbidity and mortality worldwide, ranking sixth in incidence and fourth in cancer-related mortality [1, 2]. Many patients are at an advanced stage at the time of diagnosis, thus losing the possibility for curative surgery. Patients with early-stage HCC (BCLC stage A) who undergo radical surgical resection or ablation still have a 50–70% recurrence rate [3]. Thus, early diagnosis and precise and timely implementation of therapeutic agents are key steps by which to improve prognosis.
The early disseminated recurrence of HCC is mainly evaluated by serum alpha-fetoprotein (AFP) levels, imaging studies, and tissue biopsies. Currently, screening of patients with HCC mainly relies on the serum AFP and ultrasound of the liver, the combination of which has a sensitivity of only 63% [4]. In addition, imaging and tumor biopsy have limited diagnostic potential and sensitivity. For example, ultrasound is an operator-dependent modality [5] and some lesions are difficult to access. Moreover, the molecular pathogenesis is highly heterogeneous and complex in HCC, and the information provided from a single biopsy always fails to reflect the heterogeneity. Therefore, it is imperative to create an efficient method to detect early HCC and to guide precise management of the cancer.
The analytes of liquid biopsy, such as circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), microRNA (miRNA), extracellular vesicles (EVs), proteomics, metabolomics, and transcriptomics have shown the potential to overcome these limitations. Each of those analytes targeted by the liquid biopsy has advantages and limitations in addressing clinical needs (Table 1). Specifically, ctDNA is a cornerstone of a liquid biopsy. The term ctDNA refers to the approximately 150–200 base fractions of total circulating-free DNA (cfDNA), which originates from tumor cells (Fig. 1) [6]. There are multiple mechanisms that have been postulated for the release of ctDNA by apoptosis or necrosis of tumor cells or products from macrophages that have phagocytized necrotic tumor cells [7].

Illustration of common liquid biopsy markers:circulating tumor DNA (ctDNA), microRNA (miRNA), and extracellular vesicles (EVs)
A number of studies have focused on ctDNA as a novel biomarker for early diagnosis, surveillance for recurrence or progression, and prognostication in several common malignancies [8,9,10,11]. In our review, we have outlined the current status of ctDNA detection, marker selection, and emphasize the strong role of ctDNA in precision medicine for HCC.
Current methodologies for detection of ctDNA
Ultrasensitive technology for detection of ctDNA is needed considering that ctDNA is highly fragmented by nature and diluted among overall cfDNA in patients, especially for patients with early-stage cancer who have a light tumor burden, presenting < 0.1% according to previous studies [12,13,14].
Pre-analytical analysis
Appropriate sample preparation can significantly lower the false-negative rate related to a low amount of ctDNA input for numerous assays. Conventional approaches involve complicated sample preparation steps, including sample collection, matrix selection, conservation, thawing, sample processing, and ctDNA extraction. The interval between venipuncture and sample processing should be short, while a specialized blood collection tube containing a preservative, such as Streck (Streck, Omaha, NE) or PAXgene (PreAnalytiX GmbH, Hombrechtikon Switzerland) blood collection tubes, make samples stable [15,16,17]. Furthermore, a storage temperature up to − 80 °C is required and < 3 freeze and thaw cycles are recommended [18]. During sample processing, a two-step high-speed centrifugation is always performed [16]. Protocols that are created for ctDNA extraction based on spin column, magnetic bead, and phase isolation, vary with plasma volumes and extraction methods. There are commercial kits, such as the QIAamp Circulating Nucleic Acid™ kit (Qiagen, Germany) have been applied [19].
New technologies for the isolation of ctDNA from background contaminates have improved the potency of pre-analytic procedures, especially microfluidics and nanotechnology. For example, Sonnenberg et al. [20] proposed a dielectrophoresis-based microarray device that separates cfDNA into the microelectrodes embedded in high-field regions and performs detection of the concentrated cfDNA on-chip by fluorescence. Lee et al. [21] also reported nanochip- and nanowire-based assays, capturing the ctDNA by switching the oxidation state of the conducting polymer followed by release. Therefore, the research approaches allow for better cfDNA yields, efficient processing, and less loss and damage during extraction.
Analytic approaches
Currently, ctDNA analysis is generally performed using PCR-based methods, including real-time PCR, droplet digital PCR (ddPCR), beads, emulsion, amplification, and magnetics (BEAMing), as well as sequencing-based technologies.
A range of real-time PCR-based assays have been designed for the detection of targeted SNV in ctDNA [22,23,24]. Although the traditional assays are easy to operate and cost-effective, most of the traditional assays have low sensitivity and a limited number of targeted loci can be analyzed. Advances in co-amplification at lower denaturation temperature (COLD)-PCR [25], bidirectional pyrophosphorolysis-activated polymerization (bi-PAP) [26], and Intplex [27,28,29] were developed for analysis of low-abundance mutations.
Digital PCR was defined as one of the standard reference methodologies for the analysis of ctDNA with high sensitivity and the ability to quantify results. The basic workflow schematic is depicted in Fig. 2. The dPCR assay allows the identification of rare target-mutated genes by compartmentalization and amplification. The method consists of limited dilution, separations of a single sequence into each microcompartment, individual amplification, and immunofluorescence staining for specific sequences. Droplet digital PCR (ddPCR) involves millions of monodisperse droplets generated by microfluidic emulsification to create PCR microreactors that can perform millions of reactions in parallel [30, 31]. Another method with high-resolution detection (beads, emulsion, amplification, and magnetics [BEAMing]) can detect mutations as rare as 0.01% [32]. In the process, the single magnetic bead tethered with a starting DNA template is partitioned into each water-in-oil microemulsion, where thousands of amplification reactions are subsequently performed. Then, the beads are purified and hybridized with allele-specific fluorophore probes to discriminate mutant genes from wild types [33,34,35].

Schematic workflow of droplet-based digital PCR (dPCR) and next-generation sequencing (NGS). dPCR consists of sample isolation, limited dilution, separations of a single sequence into each microcompartment, individual amplification, and immunofluorescence staining for specific sequence. NGS method profiles target genes by fracturing the DNA molecules into small sequences and proceeds with massive parallel sequencing
In contrast to dPCR methodology screening for pre-defined variants, NGS-based technologies make entire genome sequencing feasible and allow for detection of non-hotspot relevant mutation sequences. With the advent of NGS, every unknown mutation and emergent cancer-related genetic alterations during the tumor evolution period can be profiled by fracturing the DNA molecules into small sequences and massive parallel sequencing of multigenes [36, 37]. Frequently-applied assays, such as tagged-amplicon deep sequencing (Tam-Seq) [37], a safe-sequencing system (Safe-SeqS) [38], cancer personalized profiling by deep sequencing (CAPP-Seq) [39], and ion torrent [40], permitted sequencing of multiple targets. As for non-target variants, NGS technology, such as whole-exome sequencing (WES) [41], whole-genome sequencing (WGS) [42], and Methyl-Seq [43, 44], are also available for genome-wide sequencing. Nevertheless, NGS-based genome-wide ctDNA analyses have a higher requirement for ctDNA concentration and a sensitivity of 1–5%, making NGS-based genome-wide ctDNA analyses unsuitable for monitoring residual disease before disease relapse [45].
In addition to NGS-based untargeted sequencing, personalized analysis of rearranged ends (PARE) is a PCR-based approach allowing for untargeted identification of cancer-specific rearrangements in ctDNA, while digital karyotyping provides untargeted information of chromosomally-changed genomes or new genomic regions [16, 46].
The aforementioned technologies are limited by complicated sample preparation and interference from bioenvironmental components. Thus, many advanced technologies have been developed for ultrasensitive detection of multiplex minor variants without those limitations. Plasmonic nanoparticles are used in surface-enhanced Raman scattering (SERS) nanosensors for signal-amplification and mutations are identified based on specific Raman spectroscopy [47, 48]. For matrix-assisted laser desorption/ ionization time of flight mass spectrometry (MALDI-TOF-MS), biotin-labeled extended products are captured and eluted, then dispensed onto bioarrays for spectrum profiles [49]. Relatively, the electrochemical biosensors are more widespread and easier to fabricate, time- and cost-effective, rapidly responsive, and portable. The device incorporates immobilized DNA as a molecular recognition element on the electrode surface. The introduction of nanostructured materials as an interfacial film enables improved recognition capability and increased signal output intensity [50, 51]. A summary and comparison of all these technologies are shown in Table 2. Despite all advantages, these devices are not widely used in clinics. Perhaps there is still a gap in the translation from laboratory prototypes to clinical devices or there is a reluctance from users to this new and unfamiliar technology.
Available guidelines
Various pre-analytic factors, such as time interval and temperature of biofluids before purification, storage temperature, collection tubes, relevant stabilization reagents, and extraction protocols, can result in variable DNA yields, sequence bias, sample contaminations, and DNA degradation [52]. During the subsequent analysis, amplification bias, sequencing artefacts, and adoption of different laboratory techniques can all influence the final results. Thus, establishing a standard operating procedure (SOP) and strict quality control are of great significance in increasing the validity and comparability of ctDNA analysis results.
Many efforts were paid to construct a unified SOP. For ctDNA isolation, the European Committee for Standardization (CEN), as part of the Standardization of generic Pre-analytical procedures for In-vitro DIAgnostics for Personalized Medicine (SPIDIA) program, have proposed recommended guidelines for sample preparation (ISO 20186-3:2019 document [https://standards.cen.eu/]). The European Consortium Cancer ID (https://www.cancer-id.eu) and the United States Working Groups Blood Profiling Atlas in Cancer (BloodPAC [https://www.bloodpac.org/]) published best-practice protocols when implementing liquid biopsies [53]. Additionally, before NGS-based technologies are applied, full validation, cyclic testing, and external quality assessment should be done according to ISO 15189 [54].
Molecular profiling of ctDNA for HCC
Hot-spot mutated genes in ctDNA
Understanding the molecular features of the pathogenesis underlying HCC can shed light on the development of targeted agents in HCC. The majority of approved agents are angiogenesis inhibitors and targeting multiple tyrosine kinase receptors, such as VEGFR and PDGFR [55]. Sorafenib was the first molecular medicine approved, while lenvatinib, regorafenib, and cabozantinib have also received approval recently. Active agents blocking immune checkpoint programmed cell death protein 1 (PD1) or its ligands (PDL1) have also been approved as anchor drugs, including ramucirumab, nivolumab, and pembrolizumab [56]. Despite this, therapeutic applications of target drugs derived from genomic alterations are still slow to be adopted and worthy of further investigation at the genomic level.
The tumorigenesis and development of HCC involve many complex genes and signaling pathways [57]. To date, the description of the genomic landscape of HCC patients at an early stage is mainly derived from the excised surgical tissues, and the recurrent genomic alterations are TERT, TP53, CDKN2A, CTNNB1, AXIN1, ARID1A, ARID2, MLL2, NFE2L2, and KEAP1 [58]. The analysis of mutation detection in ctDNA found that 27 of 48 pre-operative samples of patients in an early stage had at least one mutation in TP53, CTNNB1, and TERT [59]. It has been reported that TERT promoter (51%), TP53 (32%), CTNNB1 (17%), PTEN (8%), AXIN1 (6%), ARID2 (6%), KMT2D (6%), and TSC2 (6%) were prevalent in ctDNA analysis of 121 advanced HCC patients according to targeted ultra-deep sequencing [60]. Notably, mutations in TP53 and CTNNB1 were excluded. Using ddPCR targeting detection, at least one of the recurrent mutated loci situated in TP53 c.747G > T (p.R249S), CTNNB1 c.121A > G (p.T41A), CTNNB1 c.133 T > C (p.S45A), and TERT c.-124C > T have been detected in the peripheral blood of HCC patients, without being detected in normal HCC tissues or mononuclear cells of blood samples [61]. Studying the biological mechanisms of the aforementioned mutated genes in tumorgenesis and progression (Fig. 3) is conducive to screening of targeted populations and selection of therapeutic drugs, thus providing broad prospects for the clinical application of precision medicine (Table 3).

High frequency genetic markers of hepatocellular carcinoma and the key pathways
TP53
The TP53 gene is closely related to the p53 signaling pathway and aberrations are involved in many biological regulation processes, generating increased proliferation, active epithelial-to-mesenchymal transition, and increased angiogenesis. There are > 120 various alterations and monitoring the TP53 codon 249 mutation is particularly significant for clinical practice [74]. A recent study showed that the diagnostic ability of TP53 c.747G > T (p.R249S) detected in ctDNA led to positive outcomes in > 20% HCC patients, in contrast to only 3–4% of patients with pancreatic and gastric cancer, but was not detected in any of the healthy controls [75]. The presence of R249S in ctDNA reveals patients more likely to have high AFP values or high HBV virus loads and increased quantities of hepato-carcinogenic risk factors, in addition to a poor prognosis [76, 77]. Anti-angiogenesis drugs, such as bevacizumab or Wee1 inhibitors, can be used as inhibitors [62,63,64].
TERT
A mutation in the promoter region of the TERT gene always occurs early during HCC oncogenesis and is regarded as a driver gene for HCC carcinogenesis [78]. The expression of mutational TERT genes results in telomeres extending compensates for eroded telomeric ends and allows for epithelial cell immortalization [79]. Frequent occurrences of TERT promoter mutations located at − 124 and − 146 bp relative to the start codon in various cancers, especially alterations in -124C > T, clearly boost transcriptional activity in HCC cell lines [80]. Furthermore, patients with this type of mutation in ctDNA are more prone to have vascular invasion (p = 0.005) and are positively correlated with an advanced TNM stage (p < 0.0001), large intrahepatic tumor size (p = 0.05), high des-gamma carboxyprothrombin value (p = 0.005), and increased mortality [81, 82]. Telomerase-targeting compounds, like GRN163L, BIBR1532, or compounds that interfere with RNA, can decrease telomere length, which is expected to be applied in the following treatment, but still needs clinical evaluation [65, 66].
CTNNB1 and AXIN
CTNNB1 and AXIN are the key genes involved in the WNT/β-catenin pathway [83, 84]. Mutated CTNNB1 produces mutated β-catenin, which can escape phosphorylation and degradation. Negative regulation of mutated AXIN1or APC prevents the destruction complex from functioning, thus accelerating accumulation of β-catenin [85, 86]. The overaccumulation β-catenin will promote tumorigenesis or cancer progression. Analysis of CTNNB1 mutations (c.121A > G, c.133 T > C) had a frequency of 17% in ctDNA, while the positive rate was 6% for AXIN1 [60]. The expression of those genes can function as a compound tumor promoter involved in the progression of HCC based on an analysis of HCC tissue samples, which is consistent with the findings of a targeted sequence analysis of ctDNA [87, 88]. The recently reported small molecule blockade that aimed at attacking WNT ligands or receptors, such as LGK874 and OMP-54F28, preventing β-catenin degradation, such as NSAIDs, or inhibiting β-catenin from interacting with nuclear transcription, such as vitamin D and CWP232291, to block the WNT signaling pathway, are still in phase I or II clinical trials [67,68,69].
CDKN2A
Inactivation of the cyclin-dependent kinase inhibitor, CDKN2A, emerges in 7% of advanced HCC patients based on digital ctDNA sequencing and leads to overexpression of CDK4/6 [89]. With mutated CDKN2A, the upregulated CDK4/6 accelerates the G1/S phase transition in the cell cycle through thee CDK4/6-Rb-myc pathway and eventually promotes cell proliferation [70]. Additionally, patients with CDKN2A silencing correlate with an advanced stage and aggressive biological behavior [90]. Thus, CDK4/6 inhibitors leading to cell cycle arrest and cell death induction, such as palbociclib, ribociclib, and abemaciclib, can provide an effective target treatment for HCC patients with CDKN2A loss of function [70].
ARID1A and ARID2
ARID1A and ARID2 are crucial components of the switch/sucrose non-fermentable (SWI/SNF) complex, an adenosine triphosphate-dependent complex participating in gene transcription stimulation or suppression via chromosomal remodeling [91]. Inactivated mutations of ARID1A or ARID2 frequent present in many HCC patients and are clinically associated with cancer development [92]. Although ARID1A is found in 14.3% of the target HCC population and 6% for ARID2 through ctDNA analysis [60, 71]. Inhibitors targeting the mutated SWI/SNF complex warrant further investigation.
Altered methylations in ctDNA
Greater than 98% of methylation reactions occur on the cytosine of 5′-cytosine-phosphate-guanine-3′ (CpG) dinucleotide and catalyzed by DNA methyltransferase [93]. Previous studies showed that focal hypermethylation changes can drive inactivation of key tumor suppressor genes, dysregulation of regulatory regions that control cell cycle and growth, or reduced response to therapy. Hypomethylation of some gene sequences also occurs during the HCC-promoting process [94]. Abnormal epigenetic aberration of DNA methylation often occurs before tumor formation or development and can be considered as early tumor biomarkers for diagnosis or the identification tool to discriminate people at high risk of developing cancer [95]. Additionally, cancer type-specific methylation signatures displayed in different samples can help to identify the cancer tissue of origin, for tumor cells originating from different tissue types may share similar genotypes but exhibit a unique methylation profile [96, 97].
Hypermethylated changes
Villanueva et al. [98] reported that the DNA methylation aberrant landscape of HCC is depicted by the prevalence of RASSF1A, APC, NEFH, IGF2, SEPT9, and EFNB2. In an analysis of cfDNA, hypermethylation of p15, p16, APC, SPINT2, SFRP1, TFPI2, GSTP1, and RASSF1A were shown to be closely related to HCC initiation and progression [72]. The value of SEPT9 promoter methylation detected in cfDNA was also emphasized by Oussalah et al. [73] for it can discriminate HCC from cirrhotic patients with an area under the receiver operating characteristic curve (AUC) of 0.944. Additionally, Lu et al. [99] demonstrated that hypermethylation of RASSF1A, COX2, and APC genes detected in ctDNA can identify those HCC patients with negative AFP levels and is associated with greater susceptibility to tumor recurrence and poor survival prognosis. Recently, methylation status in ctDNA detected by a panel of several methylation sites has been considered to be a promising tool. For example, a panel of 10 DNA methylation markers was constructed and validated with high diagnostic sensitivity and specificity, in addition to a close relationship to tumor burden and clinical outcomes (p < 0.001) [100]. It has also been shown that a panel of 6 methylated DNA markers tested in a phase I pilot study and validated in a phase II clinical cohort study had a sensitivity of 95% and a specificity of 92% when HCC was detected among high-risk controls (AUC of 0.94) [101].
Hypomethylated changes
DNA hypomethylation may be involved in HCC through many mechanisms, including destabilization of chromosomes, repression free of imprinted genes, aberrant epigenetic expression, and activation of retrotransposition [102]. It is reported that ctDNA assay of the hypomethylation level nearby HBV integration sites can serve as an early detection tool for HCC [103]. Other genes, such as CTCFL promoters and UBE2Q1, the hypomethylation status of which in ctDNA are also believed to be relevant to the diagnostic and monitoring period for HCC patients [104, 105].
To conclude, assessment of ctDNA derived from peripheral blood samples may facilitate the diagnosis, staging, and surveillance of HCC, and offers signaling pathway inhibitors or targets for precision therapy based on the specific mutation identified.
Diagnostic value of ctDNA for HCC patients
Early diagnosis of HCC
Before the genetic and epigenetic landscape of HCC was well defined, many studies investigated quantitative changes in cfDNA levels to achieve the goal for early detection of HCC [106, 107]. The cfDNA levels, however, can also increase with exercise, inflammation, surgery, or tissue injury in healthy individuals [14], leading to an application limitation.
Cancer-related sequences detected by liquid biopsy are often well-known mutations that have already shown clinical relevance, which limits the application of mutated genes in early tumor diagnosis [59, 108]. Cohen et al. (29) combined mutations in ctDNA and circulating proteins for several types of common cancers for early detection of tumors (Cancer SEEK). For HCC, the sensitivity was nearly 95% and it detected nearly 100% of HCC patients in an early stage (stage I) among a cohort of 44 patients with liver cancer [75].
In contrast to mutational sequences in ctDNA, the genome-wide distribution of numerous, densely clustered DNA methylation aberrations significantly impact robust cancer detection and high sensitivity in cancer diagnostics. Aberrant methylation in the promoter region is always involved in the initiation of HCC [109]. Based on previous findings, ctDNA positivity precedes imaging findings and prior to positive AFP values [99, 110]. Analysis of methylation alterations in ctDNA has been reported to accurately distinguish early-stage HCC patients (BCLC stage 0/A) from non-HCC and high-risk patients with a history of HBV infection or liver cirrhosis [111]. Chen et al. [112] also described an assay interrogating cancer-specific methylation signals in ctDNA, and exhibted the potency of early diagnosis among 5 types of tumor types (liver cancer contained), outperforming conventional diagnosis by up to 4 years. These results suggest the feasibility of ctDNA as an early-onset biomarker for HCC detection.
Wong et al. [113] first reported the positive rate of methylation in p15 and p16 is 48% and the rate of p15/p16 detection can be as high as 92%. RASSF1A was also confirmed to have a valuable role in the early diagnosis of HCC by Mohamed et al. [114], with a sensitivity of 90%. Moreover, in their study, RASSF1A can also discriminate HCC patients from healthy patients with a predictive accuracy of 77.5% based on logistic regression analysis, and it can also differentiate HCC and hepatitis C patients with an area under the receiver operating characteristic curve (AUC) value of 0.733 nmol/L and predictive accuracy of 72.5%. In another study conducted by Xu et al. [100], a 10-methylation marker panel was constructed as a diagnostic prediction model, with a sensitivity of 85.7% and a specificity of 94.3% for HCC patients in the training cohort, and a sensitivity of 83.3% and a specificity of 90.5% in the validation cohort. Thus, the combined detection of methylation status among multiple genes can effectively improve the diagnostic efficacy. Xu et al. [100] also pointed out that a combined prognostic score can differentiate high-risk liver disease and HCC. Similarly, A 32-gene diagnostic model was developed by Cai et al. [111], which had superior performance in distinguishing early HCC or small tumors (≤ 2 cm) from non-HCC compared to AFP (AUC = 88.4; 95% CI: 85.8–91.1%). The model can discriminate HCC from chronic HBV or cirrhosis.
Etiologic diagnosis for HCC
In addition, several investigations have shown that multiple carcinogens for HCC, such as chronic HBV or HCV virus infection, alcohol abuse, NAFLD/NASH, and aflatoxin B1, may have different somatic mutations [115]. Based on an analysis of the correlation between tumor tissues and their carcinogens, mutations in the TERT promoter are prevalent in HCV-induced HCC, as well as the CTNNB1, ARID2, and GPC3 sequences [116]. Specific mutations in the HLA region, KIF1B, STAT4, GRIK1, ErbB2, TP53, and PTEN are mainly found in HCC caused by HBV [117], and the genomic aberration of HCC samples in the region of TP53 and GPCR subfamily members (ADGRB1, ADGRB2, and ADGRB3) are closely related to aflatoxin B1 [118]. In addition, changes in DNA sequences in patients with alcohol-related HCC have recurrent mutations in CTNNB1, TERT, ARID1A, SMARCA2, and PNPLA3 I148M [119]. Furthermore, patients with NAFLD/NASH are more prone to exhibit mutations in rare germline hTERT, and genes involved in calcium signaling, such as Sav, YAP, and TAZ [120].
It has been suggested that detection of specific genomic aberrations in ctDNA will distinguish HCC types and facilitate individualized treatment of HCC patients; however, retrospective studies have demonstrated that patients with TERT promoter mutations in ctDNA are closely related to HCV infection [121] and ERBB2 alterations within ctDNA are more likely to be identified with characteristics of HBV infection [74], thus confirming the possibility of identifying HCC subtypes by ctDNA.
Monitoring response to therapy by ctDNA
Response to targeted therapy
Thus far, no identical genomic profile has been detected, suggesting that analyzing the mutational genomic landscape of a patient can enable customized treatment. For example, Ikeda et al. [89] evaluated 14 patients using a commercial NGS panel and showed that advanced HCC patients with PTEN-inactivating and MET-activating mutations can benefit from therapy with sirolimus and cabozantinib, which are inhibitors of the relevant pathways. Patients with CDKN2A-inactivating and CTNNB1-activating mutations who received palbocilib (a CDK4/6 inhibitor) and celecoxib (a Cox-2/Wnt inhibitor) subsequently had decreased AFP levels.
In addition, the gene sequence of ctDNA may change as the pressure of treatment changes. Theoretically, ctDNA shares the same tumor genetic information with primary tumor cells from which they originated and represent a real-time biomarker due to the rapid clearance [14]. In a recent study, ctDNA of a patient treated with capecitabine was profiled at multiple time points and displayed a decrease in initial ARID1A and BRCA2 mutational alleles during systemic treatment and emergence of TP53 aberration after disease progression [89]. Making this speculation more powerful, Alunni-Fabbroni et al. [122] reported that the majority of genomic variants (68%) were discovered after the beginning of sorafenib treatment, the first-line targeted therapy for advanced HCC patients [123], indicating that treatment alone may affect the selection of gene cloning. Additionally, ctDNA can also serve as an eligible tool for evaluating the treatment efficiency of refametinib monotherapy and refametinib plus sorafenib combined therapy in advanced HCC patients with a mutational RAS allele [124]. Above all, it is speculated that we can monitor disease progression of HCC patients and make timely response treatment measures by analyzing the ctDNA genomic profile serially.
The molecular ctDNA may also serve as a biomarker for predicting drug resistance. DNA methylation alterations in cell lines can actuate EMT-mediated resistance to sorafenib in HCC patients at an advanced stage [125]. This non-invasive method of obtaining genomic drug resistance information avoids the difficulty of re-obtaining and analyzing biopsy tissues of advanced HCC patients.
There are some biologic and technique limitations that need to be addressed. Due to the complexity of the signal interaction network, the tumor microenvironment, and diverse genetic backgrounds, HCC has high tumoral heterogeneity [126]. Thus, a single biomarker may be inadequate for personalized medicine selection. Moreover, the low incidence of the potential predictive biomarkers, as shown in Table 3, makes it difficult to drive further clinical trials. The task appears to be more daunting for the presence of comorbid cirrhosis in most patients with HCC because drug-related toxicity would be another limitation. A consensus of standard operating procedures to ensure accuracy of ctDNA test has not been achieved. Current available methodologies are time-consuming and costly, and most have insufficient sensitivity and cannot cover the entire genomic loci [16].
Response to immunotherapy
Immunotherapy, which can be represented by immune checkpoint blockade (ICB), has transformed clinical practice in cancer treatment. At present, ramucirumab is recommended as second-line medication after sorafenib for advanced HCC patients with serum AFP levels ≥400 ng/mL [127]. And a large phase III study (IMbrave 150) reported that atezolizumab plus bevacizumab improves prognostic outcomes superior to sorafenib [128]. On 29 May 2020, the combination of atezolizumab and bevacizumab for the treatment of unresectable or metastatic HCC patients was approved by the Food and Drug Administration [129]. Despite the initial successes achieved with ICB systemic therapy, patients suitable for immunotherapy need to be identified using molecular assays.
Previous studies have proposed that ctDNA can be used to measure the tumor mutation burden, referring to the total number of alterations per mega-base in a specific exon region of tumor genomic sequences and identify tumor patients who have a high likelihood of response to immunotherapy [130, 131]. This response can be detected 38 days earlier than the radiographic response [132]. Moreover, ctDNA can differentiate the true progress from pseudo-progression caused by inflammation from ICB therapy [133] and alterations in some specific genes may be related to immune-related adverse events [134]. Relevant studies have mainly focused on melanoma, non-small cell lung cancer, and gastric cancer [134,135,136]. There is still a gap in clinical research of ctDNA in ICB therapy for HCC patients because mutational DNA molecules in the HCC population have not been pre-defined and those aberrations which exist in HCC can also be detected in benign hepatic diseases [137]. These deficiencies can be overcome by establishing a panel consisting of HCC-associated genetic aberrations for sequencing assay.
Response to surgical therapy
Hepatic resection, liver transplantation, and local ablation have been suggested to be the standard curative treatment method for early-stage HCC patients [138, 139]. Nevertheless, the postoperative recurrence rate remains at a high level of > 60% HCC patients within 5 years [140]. A considerable number of post-operative patients may have occult micrometastases or minimal residual disease (MRD) without clinical or radiologic signs; however, ctDNA can serve as a biomarker to detect MRD.
In a recent study, 34 HCC patients underwent surgical resection followed by other adjuvant therapies during the follow-up period in China underwent ctDNA detection using NGS-based technology [110]. The study indicated that ctDNA identified 10 of 17 patients had a recurrence within 1 year, prior to serum protein biomarker detection. One patient with consistent ctDNA-positivity had a recurrence on day 610, suggesting that he had MRD for a relatively long period. Further discussion of their study suggested that patients with ctDNA-positivity were thought to be at high risk for recurrence and metastasis (log-rank, p < 0.001) by Kaplan–Meier analysis. Another study relied on ddPCR technology to identify four hot-spot mutants (TP53-rs28934572, TRET-rs1242535815, CTNNB1-rs121913412, and CTNNB1-rs121913401) and arrived at a similar conclusion that specific aberrations displayed in ctDNA can be defined as an independent risk factor of HCC patients for post-operative recurrence [88]. Moreover, ctDNA can track longitudinal changes of different mutants and therapeutic responses in real-time monitoring. For example, Cai et al. [141] reported a patient monitored with a somatic mutation (HCKp.V174M). The alteration was detected after the first TACE treatment, and then became undetectable after the second hepatic surgery and sharply increased during the second recurrence. In summary, the somatic mutation frequency of ctDNA is capable of detecting a recurrence in advance, unlike traditional imaging tests and protein biomarkers.
Predictive value of ctDNA for prognosis
ctDNA levels have been reported to be closely correlated with tumor burden, cell proliferation, and Edmondson grade [142]. It has been reported that patients with a high level of ctDNA are more likely to have metastases and worse survival outcomes [122]. Nevertheless, the practicability of this research with only 13 patients as entities in the study cohort requires further verification.
In contrast, targeted ctDNA analysis of intra-tumoral heterogeneity enables prediction of survival outcomes among HCC patients. TERT promoter mutations were the recurrent point mutations and aberrant alteration of TERT C228T has been shown to be associated with increased mortality when detected in ctDNA [81, 121]. Alterations in other driver genes, such as TP53 and CTNNB1, also have a negative performance for prognosis [108, 143]. MLH is a pivotal gene for mismatch repair during DNA replication and the defections impact genomic instability and cancer development [144]. The presence of the alteration in MLH1 chr3:37025749 T > A exhibited a worse survival rate [145].
Recently, Li et al. [146] also reported that promotor methylation of insulin-like growth factor binding protein 7 (IGFBP7) in the cfDNA is an independent risk factor for 155 patients undergoing surgical resection, indicating that continuous detection of tumor-specific driver gene mutations and methylation in ctDNA can be unrestricted by genetic heterogeneity and achieve the prediction goal of HCC.
Limitations and future perspective
Although the potential application of ctDNA is promising for monitoring the occurrence, development, and prognosis of HCC, there is still controversy regarding the clinical utility.
First, the expression of non-DNA based alterations, including hormone receptors or other proteins, cannot be identified by ctDNA analysis, which also plays a significant role in the diagnosis and treatment strategies of tumors. Thus, the histologic information carried by ctDNA is incomplete. Second, analysis of clinical relevance of ctDNA concentrated on systemic therapy and hepatectomy, lacking investigations in transarterial chemoembolization (TACE), selective internal radiation therapy (SIRT), locational ablations and immunotherapy, leading to insufficient evidence supported the clinical utility. Nevertheless, a considerable number of clinical trials registered with clinicaltrials.gov and some selected patents, as presented in Tables S1 and S2, respectively, highlight the potency of ctDNA in HCC management. Third, a universal tool with high sensitivity and specificity to ensure the accuracy of research results is urgently needed for clinicians. Although the detection of ctDNA mutations and methylation have been successfully applied in advanced common cancers, 15% of patients with metastatic cancer may not have sufficient ctDNA levels to allow for mutational profiling from plasma [147]. It should also be mentioned that most data are from Asian countries and with restricted samples. Thus, these data have limited generalizability.
To select the most appropriate mutation profiling specimens guiding clinical-decision making, it is necessary to perform a comparative study of CTC-derived DNA (CTC-DNA), ctDNA, and tumor tissue DNA (tumor DNA; Table 4). Sundaresan et al. [148, 149] believe the overall performance of ctDNA is superior to CTCs, but there is still a 20–30% blank that needs to be covered by combination analysis of ctDNA and CTC-DNA in non-small-cell lung cancer, a finding also corroborated in colorectal cancer [150], thoracic cancer [151], metastatic prostate cancer [152], and HCC [153].
On the other hand, some researchers argue that matching genomic biomarkers with systematic therapies is not sufficient. First, greater than half of the patients do not have actionable alterations. Second, there is a wide range of non-genetic factors relevant to oncogenesis and progression, and some mutations may result in different responses to the same drugs in different cancers. Moreover, the identification of splice variants rely on mRNA analysis instead of genomic NGS. The immunoassays are also closely related to RNA-based analysis [154]. Furthermore, with advances in technologies, it is possible to conduct RNA analysis of a single CTC [155]. Jan et al. [156] demonstrated that tissue -based RNA profiling can be transferred to CTC-RNA expression analysis and the expression can provide guidance on treatment selection. Some researchers have also reported an increased match rate by incorporation of transcriptomic data [157, 158]. Thus, it is hypothesized that combining ctDNA and CTC-RNA data may improve the predictability of the treatment response. Although there is no definitive answer to this important question, it emphasizes the desired direction of research in liquid biopsy (multi-parametric co-analysis) to facilitate the development of precision oncology.
Last, the currently used methodologies require full preparation of biological material and expensive specialized laboratory equipment, increasing the difficulty in popularization.
Conclusion
In conclusion, ctDNA is a transformative biomarker aimed at precision monitoring of HCC patients during the overall course of treatment. Superior performance in initial diagnosis, optimal selection of relevant targeted therapy or immunotherapy, and a timely decision of the need to transform therapeutic strategies are of great significance for improving the survival outcome of HCC patients and development of precision oncology for HCC. Because the choice of ctDNA markers has not yet reached agreement and detection technology is time consuming and expensive, ctDNA analysis should be further explored when applied to patients with HCC.
Availability of data and materials
Not applicable.
Change history
13 July 2021
A Correction to this paper has been published: https://doi.org/10.1186/s13046-021-02032-3
Abbreviations
- CTNNB1:
-
Cadherin-associated protein beta 1
- AXIN:
-
Axis inhibition
- AFP:
-
Alpha-fetoprotein
- TERT:
-
Telomerase reverse transcriptase
- TP53:
-
Tumor protein p53
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- ARID1A:
-
AT-rich interaction domain 1A
- MLL2:
-
Mixed-lineage leukemia 2
- NFE2L2:
-
Nuclear factor, erythroid 2 like 2
- KEAP1:
-
Kelch-like ECH-associated protein 1
- KMT2D:
-
Lysine methyltransferase 2D
- TSC2:
-
TSC complex subunit 2
- RASSF1A:
-
Ras association domain family member 1
- NEFH:
-
Neurofilament heavy
- IGF2:
-
Insulin-like growth factor 2
- SEPT9:
-
SEPTIN9
- TFPI2:
-
Tissue factor pathway inhibitor 2
- GSTP1:
-
Glutathione S-transferase pi 1
- SMARCA2 – SWI/SNF related:
-
matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 2
- PNPLA3:
-
Patatin-like phospholipase domain containing 3
- ERBB2:
-
Erb-b2 receptor tyrosine kinase 2
- PTEN:
-
Phosphatase and tensin homolog
- BRCA2:
-
Breast cancer 2
- RAS:
-
Resistance to audiogenic seizures
- IGFBP7:
-
Insulin-like growth factor binding protein 7
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- VEGFR:
-
Vascular endothelial growth factor receptor
- PDGFR:
-
Platelet-derived growth factor
- RASSF1A:
-
Ras association (RalGDS/AF-6) domain family member 1
- COX2:
-
cytochrome c oxidase subunit II
- CTCFL:
-
CCCTC-binding factor like
- UBE2Q1:
-
ubiquitin conjugating enzyme E2 Q1
- BCLC stage:
-
Barcelona Clinic Liver Cancer stage
References
Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C, et al. The burden of primary liver Cancer and underlying etiologies from 1990 to 2015 at the global, regional, and National Level: results from the global burden of disease study 2015. JAMA Oncol. 2017;3(12):1683–91. https://doi.org/10.1001/jamaoncol.2017.3055.
Feng J, Li J, Wu L, Yu Q, Ji J, Wu J, et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39(1):126. https://doi.org/10.1186/s13046-020-01629-4.
EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908–43. https://doi.org/10.1016/j.jhep.2011.12.001.
Tzartzeva K, Obi J, Rich NE, Parikh ND, Marrero JA, Yopp A, et al. Surveillance Imaging and Alpha Fetoprotein for Early Detection of Hepatocellular Carcinoma in Patients With Cirrhosis: A Meta-analysis. Gastroenterology. 2018;154:1706–18 e1701.
Wong RJ, Ahmed A, Gish RG. Elevated alpha-fetoprotein: differential diagnosis - hepatocellular carcinoma and other disorders. Clin Liver Dis. 2015;19(2):309–23. https://doi.org/10.1016/j.cld.2015.01.005.
Corcoran RB, Chabner BA. Application of cell-free DNA analysis to Cancer treatment. N Engl J Med. 2018;379(18):1754–65. https://doi.org/10.1056/NEJMra1706174.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–38. https://doi.org/10.1038/nrc.2017.7.
Chae YK, Oh MS. Detection of minimal residual disease using ctDNA in lung Cancer: current evidence and future directions. J Thorac Oncol. 2019;14(1):16–24. https://doi.org/10.1016/j.jtho.2018.09.022.
Clatot F. Review ctDNA and breast Cancer. Recent Results Cancer Res. 2020;215:231–52. https://doi.org/10.1007/978-3-030-26439-0_12.
Kruger S, Heinemann V, Ross C, Diehl F, Nagel D, Ormanns S, et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann Oncol. 2018;29(12):2348–55. https://doi.org/10.1093/annonc/mdy417.
Yang JD, Liu MC, Kisiel JB. Circulating tumor DNA and hepatocellular carcinoma. Semin Liver Dis. 2019;39(4):452–62. https://doi.org/10.1055/s-0039-1688503.
Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–86. https://doi.org/10.1200/JCO.2012.45.2011.
Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014;4(6):650–61. https://doi.org/10.1158/2159-8290.CD-13-1014.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90. https://doi.org/10.1038/nm.1789.
Franczak C, Filhine-Tresarrieu P, Gilson P, Merlin JL, Au L, Harlé A. Technical considerations for circulating tumor DNA detection in oncology. Expert Rev Mol Diagn. 2019;19(2):121–35. https://doi.org/10.1080/14737159.2019.1568873.
Gorgannezhad L, Umer M, Islam MN, Nguyen NT, Shiddiky MJA. Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip. 2018;18(8):1174–96. https://doi.org/10.1039/C8LC00100F.
Gobbini E, Swalduz A, Levra MG, Ortiz-Cuaran S, Toffart AC, Pérol M, et al. Implementing ctDNA analysis in the clinic: challenges and opportunities in non-small cell lung cancer. Cancers (Basel). 2020;12(11). https://doi.org/10.3390/cancers12113112.
Zavridou M, Mastoraki S, Strati A, Tzanikou E, Chimonidou M, Lianidou E. Evaluation of preanalytical conditions and implementation of quality control steps for reliable gene expression and DNA methylation analyses in liquid biopsies. Clin Chem. 2018;64(10):1522–33. https://doi.org/10.1373/clinchem.2018.292318.
Diefenbach RJ, Lee JH, Kefford RF, Rizos H. Evaluation of commercial kits for purification of circulating free DNA. Cancer Genet. 2018;228-229:21–7. https://doi.org/10.1016/j.cancergen.2018.08.005.
Sonnenberg A, Marciniak JY, Rassenti L, Ghia EM, Skowronski EA, Manouchehri S, et al. Rapid electrokinetic isolation of cancer-related circulating cell-free DNA directly from blood. Clin Chem. 2014;60(3):500–9. https://doi.org/10.1373/clinchem.2013.214874.
Lee H, Jeon S, Seo JS, Goh SH, Han JY, Cho Y. A novel strategy for highly efficient isolation and analysis of circulating tumor-specific cell-free DNA from lung cancer patients using a reusable conducting polymer nanostructure. Biomaterials. 2016;101:251–7. https://doi.org/10.1016/j.biomaterials.2016.06.003.
Gilson P. Enrichment and analysis of ctDNA. Recent Results Cancer Res. 2020;215:181–211. https://doi.org/10.1007/978-3-030-26439-0_10.
Soda N, Rehm BHA, Sonar P, Nguyen NT, Shiddiky MJA. Advanced liquid biopsy technologies for circulating biomarker detection. J Mater Chem B. 2019;7(43):6670–704. https://doi.org/10.1039/C9TB01490J.
Keppens C, Palma JF, Das PM, Scudder S, Wen W, Normanno N, et al. Detection of EGFR variants in plasma: a multilaboratory comparison of a real-time PCR EGFR mutation test in Europe. J Mol Diagn. 2018;20(4):483–94. https://doi.org/10.1016/j.jmoldx.2018.03.006.
Mauger F, How-Kit A, Tost J. COLD-PCR technologies in the area of personalized medicine: methodology and applications. Mol Diagn Ther. 2017;21(3):269–83. https://doi.org/10.1007/s40291-016-0254-8.
Madic J, Piperno-Neumann S, Servois V, Rampanou A, Milder M, Trouiller B, et al. Pyrophosphorolysis-activated polymerization detects circulating tumor DNA in metastatic uveal melanoma. Clin Cancer Res. 2012;18(14):3934–41. https://doi.org/10.1158/1078-0432.CCR-12-0309.
Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20(4):430–5. https://doi.org/10.1038/nm.3511.
Thierry AR. A targeted Q-PCR-based method for point mutation testing by analyzing circulating DNA for cancer management care. Methods Mol Biol. 2016;1392:1–16. https://doi.org/10.1007/978-1-4939-3360-0_1.
Kerachian MA, Azghandi M, Javadmanesh A, Ghaffarzadegan K, Mozaffari-Jovin S. Selective capture of plasma cell-free tumor DNA on magnetic beads: a sensitive and versatile tool for liquid biopsy. Cell Oncol (Dordr). 2020;43(5):949–56. https://doi.org/10.1007/s13402-020-00536-2.
Perkins G, Lu H, Garlan F, Taly V. Droplet-based digital PCR: application in Cancer research. Adv Clin Chem. 2017;79:43–91. https://doi.org/10.1016/bs.acc.2016.10.001.
Zhang Y, Xu Y, Zhong W, Zhao J, Chen M, Zhang L, et al. Total DNA input is a crucial determinant of the sensitivity of plasma cell-free DNA EGFR mutation detection using droplet digital PCR. Oncotarget. 2017;8(4):5861–73. https://doi.org/10.18632/oncotarget.14390.
Diehl F, Schmidt K, Durkee KH, Moore KJ, Goodman SN, Shuber AP, et al. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology. 2008;135(2):489–98. https://doi.org/10.1053/j.gastro.2008.05.039.
Ilié M, Hofman P. Pros: can tissue biopsy be replaced by liquid biopsy? Transl Lung Cancer Res. 2016;5(4):420–3. https://doi.org/10.21037/tlcr.2016.08.06.
O'Leary B, Hrebien S, Beaney M, Fribbens C, Garcia-Murillas I, Jiang J, et al. Comparison of BEAMing and droplet digital PCR for circulating tumor DNA analysis. Clin Chem. 2019;65(11):1405–13. https://doi.org/10.1373/clinchem.2019.305805.
Rowe SP, Luber B, Makell M, Brothers P, Santmyer J, Schollenberger MD, et al. From validity to clinical utility: the influence of circulating tumor DNA on melanoma patient management in a real-world setting. Mol Oncol. 2018;12(10):1661–72. https://doi.org/10.1002/1878-0261.12373.
Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54. https://doi.org/10.1038/nm.3519.
Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra168.
Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26(8):1715–22. https://doi.org/10.1093/annonc/mdv177.
Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–55. https://doi.org/10.1038/nbt.3520.
Namløs HM, Boye K, Mishkin SJ, Barøy T, Lorenz S, Bjerkehagen B, et al. Noninvasive detection of ctDNA reveals intratumor heterogeneity and is associated with tumor burden in gastrointestinal stromal tumor. Mol Cancer Ther. 2018;17(11):2473–80. https://doi.org/10.1158/1535-7163.MCT-18-0174.
Manier S, Park J, Capelletti M, Bustoros M, Freeman SS, Ha G, et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat Commun. 2018;9(1):1691. https://doi.org/10.1038/s41467-018-04001-5.
Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet. 2016;48(10):1273–8. https://doi.org/10.1038/ng.3648.
Garrett-Bakelman FE, Sheridan CK, Kacmarczyk TJ, Ishii J, Betel D, Alonso A, et al. Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J Vis Exp. 2015:e52246. https://doi.org/10.3791/52246.
Sun Z, Vaisvila R, Hussong LM, Yan B, Baum C, Saleh L, et al. Nondestructive enzymatic deamination enables single-molecule long-read amplicon sequencing for the determination of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Genome Res. 2021;31(2):291–300. https://doi.org/10.1101/gr.265306.120.
Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16(7):409–24. https://doi.org/10.1038/s41571-019-0187-3.
Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14.
Zhang Y, Mi X, Tan X, Xiang R. Recent progress on liquid biopsy analysis using surface-enhanced Raman spectroscopy. Theranostics. 2019;9(2):491–525. https://doi.org/10.7150/thno.29875.
Lyu N, Rajendran VK, Diefenbach RJ, Charles K, Clarke SJ, Engel A, et al. Multiplex detection of ctDNA mutations in plasma of colorectal cancer patients by PCR/SERS assay. Nanotheranostics. 2020;4(4):224–32. https://doi.org/10.7150/ntno.48905.
Lamy PJ, van der Leest P, Lozano N, Becht C, Duboeuf F, Groen HJM, et al. Mass spectrometry as a highly sensitive method for specific circulating tumor DNA analysis in NSCLC: a comparison study. Cancers (Basel). 2020;12(10). https://doi.org/10.3390/cancers12103002.
Campuzano S, Serafín V, Gamella M, Pedrero M, Yáñez-Sedeño P, Pingarrón JM. Opportunities, challenges, and prospects in electrochemical biosensing of circulating tumor DNA and its specific features. Sensors (Basel). 2019;19(17). https://doi.org/10.3390/s19173762.
Rodda AE, Parker BJ, Spencer A, Corrie SR. Extending circulating tumor DNA analysis to ultralow abundance mutations: techniques and challenges. ACS Sens. 2018;3(3):540–60. https://doi.org/10.1021/acssensors.7b00953.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36(16):1631–41. https://doi.org/10.1200/JCO.2017.76.8671.
Ungerer V, Bronkhorst AJ, Holdenrieder S. Preanalytical variables that affect the outcome of cell-free DNA measurements. Crit Rev Clin Lab Sci. 2020;57(7):484–507. https://doi.org/10.1080/10408363.2020.1750558.
Heeke S, Hofman V, Long-Mira E, Lespinet V, Lalvée S, Bordone O, et al. Use of the ion PGM and the GeneReader NGS systems in daily routine practice for advanced lung adenocarcinoma patients: a practical point of view reporting a comparative study and assessment of 90 patients. Cancers (Basel). 2018;10(4). https://doi.org/10.3390/cancers10040088.
Hilmi M, Neuzillet C, Calderaro J, Lafdil F, Pawlotsky JM, Rousseau B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: current knowledge and future research directions. J Immunother Cancer. 2019;7(1):333. https://doi.org/10.1186/s40425-019-0824-5.
Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: looking outside the box. J Hepatol. 2020;72(2):342–52. https://doi.org/10.1016/j.jhep.2019.09.010.
Wu L, Li J, Liu T, Li S, Feng J, Yu Q, et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019;8(10):4806–20. https://doi.org/10.1002/cam4.2388.
Nault JC, Martin Y, Caruso S, Hirsch TZ, Bayard Q, Calderaro J, et al. Clinical impact of genomic diversity from early to advanced hepatocellular carcinoma. Hepatology. 2020;71(1):164–82. https://doi.org/10.1002/hep.30811.
Huang A, Zhang X, Zhou SL, Cao Y, Huang XW, Fan J, et al. Detecting circulating tumor DNA in hepatocellular carcinoma patients using droplet digital PCR is feasible and reflects Intratumoral heterogeneity. J Cancer. 2016;7(13):1907–14. https://doi.org/10.7150/jca.15823.
von Felden J, Craig AJ, Garcia-Lezana T, Labgaa I, Haber PK, D'Avola D, et al. Mutations in circulating tumor DNA predict primary resistance to systemic therapies in advanced hepatocellular carcinoma. Oncogene. 2020;40:140–51. https://doi.org/10.1038/s41388-020-01519-1.
Mocan T, Simão AL, Castro RE, Rodrigues CMP, Słomka A, Wang B, et al. Liquid biopsies in hepatocellular carcinoma: are we winning? J Clin Med. 2020;9(5). https://doi.org/10.3390/jcm9051541.
Wheler JJ, Janku F, Naing A, Li Y, Stephen B, Zinner R, et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: implications for targeted therapeutics. Mol Cancer Ther. 2016;15(10):2475–85. https://doi.org/10.1158/1535-7163.MCT-16-0196.
Janku F, Kaseb AO, Tsimberidou AM, Wolff RA, Kurzrock R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget. 2014;5(10):3012–22. https://doi.org/10.18632/oncotarget.1687.
Kogiso T, Nagahara H, Hashimoto E, Ariizumi S, Yamamoto M, Shiratori K. Efficient induction of apoptosis by wee1 kinase inhibition in hepatocellular carcinoma cells. PLoS One. 2014;9(6):e100495. https://doi.org/10.1371/journal.pone.0100495.
Berardinelli F, Coluzzi E, Sgura A, Antoccia A. Targeting telomerase and telomeres to enhance ionizing radiation effects in in vitro and in vivo cancer models. Mutat Res. 2017;773:204–19. https://doi.org/10.1016/j.mrrev.2017.02.004.
Tang M, Li Y, Zhang Y, Chen Y, Huang W, Wang D, et al. Disease mutant analysis identifies a new function of DAXX in telomerase regulation and telomere maintenance. J Cell Sci. 2015;128(2):331–41. https://doi.org/10.1242/jcs.159467.
Ozcan M, Altay O, Lam S, Turkez H, Aksoy Y, Nielsen J, et al. Improvement in the current therapies for hepatocellular carcinoma using a systems medicine approach. Adv Biosyst. 2020;4:e2000030.
He S, Tang S. WNT/β-catenin signaling in the development of liver cancers. Biomed Pharmacother. 2020;132:110851. https://doi.org/10.1016/j.biopha.2020.110851.
Khemlina G, Ikeda S, Kurzrock R. The biology of hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16(1):149. https://doi.org/10.1186/s12943-017-0712-x.
Digiacomo G, Fumarola C, La Monica S, Bonelli MA, Cretella D, Alfieri R, et al. Simultaneous combination of the CDK4/6 inhibitor palbociclib with regorafenib induces enhanced anti-tumor effects in hepatocarcinoma cell lines. Front Oncol. 2020;10:563249. https://doi.org/10.3389/fonc.2020.563249.
Huang A, Zhao X, Yang XR, Li FQ, Zhou XL, Wu K, et al. Circumventing intratumoral heterogeneity to identify potential therapeutic targets in hepatocellular carcinoma. J Hepatol. 2017;67(2):293–301. https://doi.org/10.1016/j.jhep.2017.03.005.
Mezzalira S, De Mattia E, Guardascione M, Dalle Fratte C, Cecchin E, Toffoli G. Circulating-free DNA analysis in hepatocellular carcinoma: a promising strategy to improve Patients' Management and therapy outcomes. Int J Mol Sci. 2019;20(21). https://doi.org/10.3390/ijms20215498.
Oussalah A, Rischer S, Bensenane M, Conroy G, Filhine-Tresarrieu P, Debard R, et al. Plasma mSEPT9: a novel circulating cell-free DNA-based epigenetic biomarker to diagnose hepatocellular carcinoma. EBioMedicine. 2018;30:138–47. https://doi.org/10.1016/j.ebiom.2018.03.029.
Kaseb AO, Sánchez NS, Sen S, Kelley RK, Tan B, Bocobo AG, et al. Molecular profiling of hepatocellular carcinoma using circulating cell-free DNA. Clin Cancer Res. 2019;25(20):6107–18. https://doi.org/10.1158/1078-0432.CCR-18-3341.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–30. https://doi.org/10.1126/science.aar3247.
Marchio A, Amougou Atsama M, Béré A, Komas NP, Noah Noah D, Atangana PJA, et al. Droplet digital PCR detects high rate of TP53 R249S mutants in cell-free DNA of middle African patients with hepatocellular carcinoma. Clin Exp Med. 2018;18(3):421–31. https://doi.org/10.1007/s10238-018-0502-9.
Shen T, Li SF, Wang JL, Zhang T, Zhang S, Chen HT, et al. TP53 R249S mutation detected in circulating tumour DNA is associated with prognosis of hepatocellular carcinoma patients with or without hepatectomy. Liver Int. 2020;40(11):2834–47. https://doi.org/10.1111/liv.14581.
Nault JC, Zucman-Rossi J. TERT promoter mutations in primary liver tumors. Clin Res Hepatol Gastroenterol. 2016;40(1):9–14. https://doi.org/10.1016/j.clinre.2015.07.006.
In der Stroth L, Tharehalli U, Günes C, Lechel A. Telomeres and telomerase in the development of liver cancer. Cancers (Basel). 2020;12. https://doi.org/10.3390/cancers12082048.
Kim YJ, Yoo JE, Jeon Y, Chong JU, Choi GH, Song DG, et al. Suppression of PROX1-mediated TERT expression in hepatitis B viral hepatocellular carcinoma. Int J Cancer. 2018;143(12):3155–68. https://doi.org/10.1002/ijc.31731.
Oversoe SK, Clement MS, Pedersen MH, Weber B, Aagaard NK, Villadsen GE, et al. TERT promoter mutated circulating tumor DNA as a biomarker for prognosis in hepatocellular carcinoma. Scand J Gastroenterol. 2020;55:1–8. https://doi.org/10.1080/00365521.2020.1837928.
Hirai M, Kinugasa H, Nouso K, Yamamoto S, Terasawa H, Onishi Y, et al. Prediction of the prognosis of advanced hepatocellular carcinoma by TERT promoter mutations in circulating tumor DNA. J Gastroenterol Hepatol. 2020. https://doi.org/10.1111/jgh.15227.
Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–39 e1224.
Dai W, Xu L, Yu X, Zhang G, Guo H, Liu H, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72(5):909–23. https://doi.org/10.1016/j.jhep.2019.12.015.
Monga SP. β-Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology. 2015;148(7):1294–310. https://doi.org/10.1053/j.gastro.2015.02.056.
Wang W, Smits R, Hao H, He C. Wnt/β-catenin signaling in liver cancers. Cancers (Basel). 2019;11. https://doi.org/10.3390/cancers11070926.
Kim E, Lisby A, Ma C, Lo N, Ehmer U, Hayer KE, et al. Promotion of growth factor signaling as a critical function of β-catenin during HCC progression. Nat Commun. 2019;10(1):1909. https://doi.org/10.1038/s41467-019-09780-z.
Wang J, Huang A, Wang YP, Yin Y, Fu PY, Zhang X, et al. Circulating tumor DNA correlates with microvascular invasion and predicts tumor recurrence of hepatocellular carcinoma. Ann Transl Med. 2020;8(5):237. https://doi.org/10.21037/atm.2019.12.154.
Ikeda S, Tsigelny IF, Skjevik ÅA, Kono Y, Mendler M, Kuo A, et al. Next-generation sequencing of circulating tumor DNA reveals frequent alterations in advanced hepatocellular carcinoma. Oncologist. 2018;23(5):586–93. https://doi.org/10.1634/theoncologist.2017-0479.
Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–11. https://doi.org/10.1038/ng.3252.
Zhou Y, Xu Q, Tao L, Chen Y, Shu Y, Wu Z, et al. Enhanced SMARCD1, a subunit of the SWI/SNF complex, promotes liver cancer growth through the mTOR pathway. Clin Sci (Lond). 2020;134(12):1457–72. https://doi.org/10.1042/CS20200244.
Hu B, Lin JZ, Yang XB, Sang XT. The roles of mutated SWI/SNF complexes in the initiation and development of hepatocellular carcinoma and its regulatory effect on the immune system: a review. Cell Prolif. 2020;53:e12791.
Hlady RA, Zhao X, Pan X, Yang JD, Ahmed F, Antwi SO, et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics. 2019;9(24):7239–50. https://doi.org/10.7150/thno.35573.
Herman JG, Farooq M. Noninvasive diagnostics for early detection of lung Cancer: challenges and potential with a focus on changes in DNA methylation. Cancer Epidemiol Biomark Prev. 2020;29:2416–22. https://doi.org/10.1158/1055-9965.epi-20-0704.
Holčáková J. Effect of DNA methylation on the development of Cancer. Klin Onkol. 2018;31(Suppl 2):41–5. https://doi.org/10.14735/amko20182S41.
Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol. 2016;17(10):1386–95. https://doi.org/10.1016/S1470-2045(16)30297-2.
Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392(10149):777–86. https://doi.org/10.1016/S0140-6736(18)31268-6.
Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Méndez-González J, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. 2015;61(6):1945–56. https://doi.org/10.1002/hep.27732.
Lu CY, Chen SY, Peng HL, Kan PY, Chang WC, Yen CJ. Cell-free methylation markers with diagnostic and prognostic potential in hepatocellular carcinoma. Oncotarget. 2017;8(4):6406–18. https://doi.org/10.18632/oncotarget.14115.
Xu RH, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155–61. https://doi.org/10.1038/nmat4997.
Kisiel JB, Dukek BA, RVSRK, Ghoz HM, Yab TC, Berger CK, et al. Hepatocellular carcinoma detection by plasma methylated DNA: discovery, phase I pilot, and phase II clinical validation. Hepatology. 2019;69(3):1180–92. https://doi.org/10.1002/hep.30244.
Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25(2):196–209. https://doi.org/10.1016/j.ccr.2014.01.003.
Zhang H, Dong P, Guo S, Tao C, Chen W, Zhao W, et al. Hypomethylation in HBV integration regions aids non-invasive surveillance to hepatocellular carcinoma by low-pass genome-wide bisulfite sequencing. BMC Med. 2020;18(1):200. https://doi.org/10.1186/s12916-020-01667-x.
Chen MM, Zhao RC, Chen KF, Huang Y, Liu ZJ, Wei YG, et al. Hypomethylation of CTCFL promoters as a noninvasive biomarker in plasma from patients with hepatocellular carcinoma. Neoplasma. 2020;67(04):909–15. https://doi.org/10.4149/neo_2020_190819N789.
Hu N, Fan XP, Fan YC, Chen LY, Qiao CY, Han LY, et al. Hypomethylated ubiquitin-conjugating enzyme2 Q1 (UBE2Q1) gene promoter in the serum is a promising biomarker for hepatitis B virus-associated hepatocellular carcinoma. Tohoku J Exp Med. 2017;242(2):93–100. https://doi.org/10.1620/tjem.242.93.
Yan L, Chen Y, Zhou J, Zhao H, Zhang H, Wang G. Diagnostic value of circulating cell-free DNA levels for hepatocellular carcinoma. Int J Infect Dis. 2018;67:92–7. https://doi.org/10.1016/j.ijid.2017.12.002.
Oh CR, Kong SY, Im HS, Kim HJ, Kim MK, Yoon KA, et al. Genome-wide copy number alteration and VEGFA amplification of circulating cell-free DNA as a biomarker in advanced hepatocellular carcinoma patients treated with Sorafenib. BMC Cancer. 2019;19(1):292. https://doi.org/10.1186/s12885-019-5483-x.
Liao W, Yang H, Xu H, Wang Y, Ge P, Ren J, et al. Noninvasive detection of tumor-associated mutations from circulating cell-free DNA in hepatocellular carcinoma patients by targeted deep sequencing. Oncotarget. 2016;7(26):40481–90. https://doi.org/10.18632/oncotarget.9629.
Hlady RA, Zhou D, Puszyk W, Roberts LR, Liu C, Robertson KD. Initiation of aberrant DNA methylation patterns and heterogeneity in precancerous lesions of human hepatocellular cancer. Epigenetics. 2017;12(3):215–25. https://doi.org/10.1080/15592294.2016.1277297.
Cai Z, Chen G, Zeng Y, Dong X, Li Z, Huang Y, et al. Comprehensive liquid profiling of circulating tumor DNA and protein biomarkers in long-term follow-up patients with hepatocellular carcinoma. Clin Cancer Res. 2019;25(17):5284–94. https://doi.org/10.1158/1078-0432.CCR-18-3477.
Cai J, Chen L, Zhang Z, Zhang X, Lu X, Liu W, et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut. 2019;68(12):2195–205. https://doi.org/10.1136/gutjnl-2019-318882.
Chen X, Gole J, Gore A, He Q, Lu M, Min J, et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun. 2020;11(1):3475. https://doi.org/10.1038/s41467-020-17316-z.
Wong IH, Lo YM, Yeo W, Lau WY, Johnson PJ. Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clin Cancer Res. 2000;6(9):3516–21.
Mohamed NA, Swify EM, Amin NF, Soliman MM, Tag-Eldin LM, Elsherbiny NM. Is serum level of methylated RASSF1A valuable in diagnosing hepatocellular carcinoma in patients with chronic viral hepatitis C? Arab J Gastroenterol. 2012;13(3):111–5. https://doi.org/10.1016/j.ajg.2012.06.009.
Feng J, Dai W, Mao Y, Wu L, Li J, Chen K, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):24. https://doi.org/10.1186/s13046-020-1528-x.
Tornesello ML, Buonaguro L, Izzo F, Buonaguro FM. Molecular alterations in hepatocellular carcinoma associated with hepatitis B and hepatitis C infections. Oncotarget. 2016;7(18):25087–102. https://doi.org/10.18632/oncotarget.7837.
Mathew S, Abdel-Hafiz H, Raza A, Fatima K, Qadri I. Host nucleotide polymorphism in hepatitis B virus-associated hepatocellular carcinoma. World J Hepatol. 2016;8(10):485–98. https://doi.org/10.4254/wjh.v8.i10.485.
Zhang W, He H, Zang M, Wu Q, Zhao H, Lu LL, et al. Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterology. 2017;153:249–62 e242.
Nahon P, Nault JC. Constitutional and functional genetics of human alcohol-related hepatocellular carcinoma. Liver Int. 2017;37(11):1591–601. https://doi.org/10.1111/liv.13419.
Li X, Wang H, Li T, Wang L, Wu X, Liu J, et al. Circulating tumor DNA/circulating tumor cells and the applicability in different causes induced hepatocellular carcinoma. Curr Probl Cancer. 2020;44(2):100516. https://doi.org/10.1016/j.currproblcancer.2019.100516.
Jiao J, Watt GP, Stevenson HL, Calderone TL, Fisher-Hoch SP, Ye Y, et al. Telomerase reverse transcriptase mutations in plasma DNA in patients with hepatocellular carcinoma or cirrhosis: prevalence and risk factors. Hepatol Commun. 2018;2(6):718–31. https://doi.org/10.1002/hep4.1187.
Alunni-Fabbroni M, Rönsch K, Huber T, Cyran CC, Seidensticker M, Mayerle J, et al. Circulating DNA as prognostic biomarker in patients with advanced hepatocellular carcinoma: a translational exploratory study from the SORAMIC trial. J Transl Med. 2019;17(1):328. https://doi.org/10.1186/s12967-019-2079-9.
Feng J, Wu L, Ji J, Chen K, Yu Q, Zhang J, et al. PKM2 is the target of proanthocyanidin B2 during the inhibition of hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):204. https://doi.org/10.1186/s13046-019-1194-z.
Lim HY, Merle P, Weiss KH, Yau T, Ross P, Mazzaferro V, et al. Phase II studies with Refametinib or Refametinib plus Sorafenib in patients with RAS-mutated hepatocellular carcinoma. Clin Cancer Res. 2018;24(19):4650–61. https://doi.org/10.1158/1078-0432.CCR-17-3588.
Galle E, Thienpont B, Cappuyns S, Venken T, Busschaert P, Van Haele M, et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin Epigenetics. 2020;12(1):27. https://doi.org/10.1186/s13148-020-0821-z.
Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: challenges and opportunities. Cancer Lett. 2019;460:1–9. https://doi.org/10.1016/j.canlet.2019.114428.
Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–96. https://doi.org/10.1016/S1470-2045(18)30937-9.
Iseda N, Itoh S, Tomiyama T, Morinaga A, Wang H, Shimagaki T, et al. Immune response on outcomes in hepatocellular carcinoma. Gan To Kagaku Ryoho. 2020;47(9):1303–6.
Casak SJ, Donoghue M, Fashoyin-Aje L, Jiang X, Rodriguez L, Shen YL, et al. FDA approval summary: Atezolizumab plus bevacizumab for the treatment of patients with advanced unresectable or metastatic hepatocellular carcinoma. Clin Cancer Res. 2020;27:1836–41. https://doi.org/10.1158/1078-0432.ccr-20-3407.
Singal G, Miller PG, Agarwala V, Li G, Kaushik G, Backenroth D, et al. Association of Patient Characteristics and Tumor Genomics with Clinical Outcomes among Patients with non-Small Cell Lung Cancer Using a Clinicogenomic database. Jama. 2019;321(14):1391–9. https://doi.org/10.1001/jama.2019.3241.
Jensen TJ, Goodman AM, Kato S, Ellison CK, Daniels GA, Kim L, et al. Genome-wide sequencing of cell-free DNA identifies copy-number alterations that can be used for monitoring response to immunotherapy in Cancer patients. Mol Cancer Ther. 2019;18(2):448–58. https://doi.org/10.1158/1535-7163.MCT-18-0535.
Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early assessment of lung Cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res. 2018;24(8):1872–80. https://doi.org/10.1158/1078-0432.CCR-17-1341.
Ashida A, Sakaizawa K, Uhara H, Okuyama R. Circulating tumour DNA for monitoring treatment response to anti-PD-1 immunotherapy in melanoma patients. Acta Derm Venereol. 2017;97(10):1212–8. https://doi.org/10.2340/00015555-2748.
Jin Y, Chen DL, Wang F, Yang CP, Chen XX, You JQ, et al. The predicting role of circulating tumor DNA landscape in gastric cancer patients treated with immune checkpoint inhibitors. Mol Cancer. 2020;19(1):154. https://doi.org/10.1186/s12943-020-01274-7.
Herbreteau G, Langlais A, Greillier L, Audigier-Valette C, Uwer L, Hureaux J, et al. Circulating tumor DNA as a prognostic determinant in small cell lung cancer patients receiving atezolizumab. J Clin Med. 2020;9(12). https://doi.org/10.3390/jcm9123861.
Marsavela G, Lee J, Calapre L, Wong SQ, Pereira MR, McEvoy AC, et al. Circulating tumor DNA predicts outcome from first-, but not second-line treatment and identifies melanoma patients who may benefit from combination immunotherapy. Clin Cancer Res. 2020;26(22):5926–33. https://doi.org/10.1158/1078-0432.CCR-20-2251.
Weng J, Atyah M, Zhou C, Ren N. Prospects and challenges of circulating tumor DNA in precision medicine of hepatocellular carcinoma. Clin Exp Med. 2020;20(3):329–37. https://doi.org/10.1007/s10238-020-00620-9.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–14. https://doi.org/10.1016/S0140-6736(18)30010-2.
Vogel A, Cervantes A, Chau I, Daniele B, Llovet JM, Meyer T, et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):871–3. https://doi.org/10.1093/annonc/mdy510.
Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–2. https://doi.org/10.1002/hep.24199.
Cai ZX, Chen G, Zeng YY, Dong XQ, Lin MJ, Huang XH, et al. Circulating tumor DNA profiling reveals clonal evolution and real-time disease progression in advanced hepatocellular carcinoma. Int J Cancer. 2017;141(5):977–85. https://doi.org/10.1002/ijc.30798.
Ng CKY, Di Costanzo GG, Tosti N, Paradiso V, Coto-Llerena M, Roscigno G, et al. Genetic profiling using plasma-derived cell-free DNA in therapy-naïve hepatocellular carcinoma patients: a pilot study. Ann Oncol. 2018;29(5):1286–91. https://doi.org/10.1093/annonc/mdy083.
An Y, Guan Y, Xu Y, Han Y, Wu C, Bao C, et al. The diagnostic and prognostic usage of circulating tumor DNA in operable hepatocellular carcinoma. Am J Transl Res. 2019;11:6462–74.
Ma G, Ge Y, Gu D, Du M, Chu H, Chen J, et al. Functional annotation of colorectal cancer susceptibility loci identifies MLH1 rs1800734 associated with MSI patients. Gut. 2016;65(7):1227–8. https://doi.org/10.1136/gutjnl-2016-311543.
Kim SS, Eun JW, Choi JH, Woo HG, Cho HJ, Ahn HR, et al. MLH1 single-nucleotide variant in circulating tumor DNA predicts overall survival of patients with hepatocellular carcinoma. Sci Rep. 2020;10(1):17862. https://doi.org/10.1038/s41598-020-74494-y.
Li F, Qiao CY, Gao S, Fan YC, Chen LY, Wang K. Circulating cell-free DNA of methylated insulin-like growth factor-binding protein 7 predicts a poor prognosis in hepatitis B virus-associated hepatocellular carcinoma after hepatectomy. Free Radic Res. 2018;52(4):455–64. https://doi.org/10.1080/10715762.2018.1443448.
Yang YC, Wang D, Jin L, Yao HW, Zhang JH, Wang J, et al. Circulating tumor DNA detectable in early- and late-stage colorectal cancer patients. Biosci Rep. 2018;38(4). https://doi.org/10.1042/BSR20180322.
Sundaresan TK, Sequist LV, Heymach JV, Riely GJ, Jänne PA, Koch WH, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res. 2016;22(5):1103–10. https://doi.org/10.1158/1078-0432.CCR-15-1031.
Liu HE, Vuppalapaty M, Wilkerson C, Renier C, Chiu M, Lemaire C, et al. Detection of EGFR mutations in cfDNA and CTCs, and comparison to tumor tissue in non-small-cell-lung-cancer (NSCLC) patients. Front Oncol. 2020;10:572895. https://doi.org/10.3389/fonc.2020.572895.
Takeda K, Yamada T, Takahashi G, Iwai T, Ueda K, Kuriyama S, et al. Analysis of colorectal cancer-related mutations by liquid biopsy: utility of circulating cell-free DNA and circulating tumor cells. Cancer Sci. 2019;110(11):3497–509. https://doi.org/10.1111/cas.14186.
Freidin MB, Freydina DV, Leung M, Montero Fernandez A, Nicholson AG, Lim E. Circulating tumor DNA outperforms circulating tumor cells for KRAS mutation detection in thoracic malignancies. Clin Chem. 2015;61(10):1299–304. https://doi.org/10.1373/clinchem.2015.242453.
Hodara E, Morrison G, Cunha A, Zainfeld D, Xu T, Xu Y, et al. Multiparametric liquid biopsy analysis in metastatic prostate cancer. JCI Insight. 2019;4(5). https://doi.org/10.1172/jci.insight.125529.
Kelley RK, Magbanua MJ, Butler TM, Collisson EA, Hwang J, Sidiropoulos N, et al. Circulating tumor cells in hepatocellular carcinoma: a pilot study of detection, enumeration, and next-generation sequencing in cases and controls. BMC Cancer. 2015;15(1):206. https://doi.org/10.1186/s12885-015-1195-z.
Lin VTG, Yang ES. The pros and cons of incorporating transcriptomics in the age of precision oncology. J Natl Cancer Inst. 2019;111(10):1016–22. https://doi.org/10.1093/jnci/djz114.
Huang X, Liu S, Wu L, Jiang M, Hou Y. High throughput single cell RNA sequencing, bioinformatics analysis and applications. Adv Exp Med Biol. 2018;1068:33–43. https://doi.org/10.1007/978-981-13-0502-3_4.
Jan YJ, Yoon J, Chen JF, Teng PC, Yao N, Cheng S, et al. A circulating tumor cell-RNA assay for assessment of androgen receptor signaling inhibitor sensitivity in metastatic castration-resistant prostate cancer. Theranostics. 2019;9(10):2812–26. https://doi.org/10.7150/thno.34485.
Rodon J, Soria JC, Berger R, Miller WH, Rubin E, Kugel A, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25(5):751–8. https://doi.org/10.1038/s41591-019-0424-4.
Tuxen IV, Rohrberg KS, Oestrup O, Ahlborn LB, Schmidt AY, Spanggaard I, et al. Copenhagen prospective personalized oncology (CoPPO)-clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res. 2019;25(4):1239–47. https://doi.org/10.1158/1078-0432.CCR-18-1780.
Acknowledgements
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Funding
This study was supported by the following agencies: (1) the National Natural Science Foundation of China (grant number: 82002539); (2) the Natural Science Foundation of Shanghai (grant number: 19ZR1447700); (3) the Health System Innovation Project of Shanghai Putuo Science and Technology Commission (grant numbers: PTKWWS201801 and PTKWWS201903); (4) WBN Hepatology Research Fund of China Hepatitis Prevention and Treatment Foundation (grant number: CFHPC2019031); and (5) The Yangfan Plan of Shanghai Science and Technology Commission (grant number: 20YF1443300).
Author information
Authors and Affiliations
Contributions
YL and YZ contributed equally to this work and share first authorship. Conceptualization, YL, YZ, LW, QY, and JJ; writing—original draft preparation, YL; writing—review and editing, JW and WD; visualization, YL; supervision, CG; funding acquisition, JF, JW and CG. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the authors identified an error in the author affiliations. Chuanyong Guo’s first affiliation was missing; Chuanyong Guo’s affiliations are therefore: 1Department of Gastroenterology, Putuo People's Hospital, Tongji University School of Medicine, number 1291, Jiangning road, Putuo, Shanghai, 200060, China. 2Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Number 301, Middle Yanchang road, Jing’an, Shanghai 200072, China
Supplementary Information
Additional file 1: Supplementary Table S1.
Current trials registered with clinicaltrials.gov exploring ctDNA in hepatocellular carcinoma.
Additional file 2: Supplementary Table S2.
Recent patents of ctDNA analysis in hepatocellular carcinoma.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Zheng, Y., Wu, L. et al. Current status of ctDNA in precision oncology for hepatocellular carcinoma. J Exp Clin Cancer Res 40, 140 (2021). https://doi.org/10.1186/s13046-021-01940-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-01940-8