
Abstract
Background
Multiple myeloma (MM) cell motility is a critical step during MM dissemination throughout the body, but how it is regulated remains largely unknown. As hypercalcemia is an important clinical feature of MM, high calcium (Ca2+) and altered Ca2+ signaling could be a key contributing factor to the pathological process.
Methods
Bioinformatics analyses were employed to assess the clinical significance of Ca2+ influx channels in clinical specimens of smoldering and symptomatic MM. Functional and regulatory roles of influx channels and downstream signaling in MM cell migration and invasion were conducted and experimental MM dissemination was examined in a xenograft mouse model using in vivo live imaging and engraftment analysis.
Results
Inhibition of TRPM7, ORAI1, and STIM1 influx channels, which are highly expressed in MM patients, and subsequent blockage of Ca2+ influx by CRISPR/Cas9 and small molecule inhibitors, effectively inhibit MM cell migration and invasion, and attenuate the experimental MM dissemination. Mechanistic studies reveal a nutrient sensor O-GlcNAcylation as a downstream regulator of Ca2+ influx that specifically targets cell adhesion molecules. Hyper-O-GlcNAcylation following the inhibition of Ca2+ influx channels induces integrin α4 and integrin β7 downregulation via ubiquitin-proteasomal degradation and represses the aggressive MM phenotype.
Conclusions
Our findings unveil a novel regulatory mechanism of MM cell motility via Ca2+ influx/O-GlcNAcylation axis that directly targets integrin α4 and integrin β7, providing mechanistic insights into the pathogenesis and progression of MM and demonstrating potential predictive biomarkers and therapeutic targets for advanced MM.
Similar content being viewed by others


Background
Multiple myeloma (MM) is a prototypical clonal B-cell malignancy characterized by an excessive growth and proliferation of terminally differentiated plasma cells (PCs) in the bone marrow (BM). Recent evidence suggests that MM arises from the BM and disseminates throughout the body. Through a process that is similar to metastasis of solid tumors, the disseminated MM cells can settle at different sites including the liver, lung, brain, and other soft tissues [1,2,3]. Advanced MM may be found in up to 30% of patients and is strongly correlated with poor prognosis with an overall survival of less than 6 months [4, 5]. It is widely accepted that BM microenvironment provides support for MM cell growth and survival as well as for the acquisition of aggressive phenotypes. Understanding how MM cells interact and respond to changes in the microenvironment is therefore crucial to the design of more effective anticancer therapies for MM.
One of the critical steps during MM dissemination is increased cell migration and invasion. Such increase is necessary for the intravasation of MM cells from the BM into nearby blood vessels, and for their subsequent extravasation into distant tissues [6, 7]. During the early stage of MM, these cells are exposed to a high level of Ca2+ in the BM niche due to increased bone resorption by the osteoclasts and consequently Ca2+ overload [8, 9]. Clinically, a high level of Ca2+ is generally regarded as the most frequent metabolic complication in MM patients [10]. Physiologically, Ca2+ is a ubiquitous signaling molecule regulating various cellular processes, including cell proliferation, cell death and cell motility. However, the roles of Ca2+ signaling, and specifically Ca2+ influx channels, in cell motility regulation and dissemination of MM are not well understood.
The transient receptor potential melastatin-subfamily member 7 (TRPM7) and the store-operated calcium (SOC) channels comprising of calcium release-activated calcium channel protein 1 (ORAI1) and stromal interaction molecule 1 (STIM1) are the major pathways of Ca2+ entry and intracellular signaling in both cancer and most excitable cells [11,12,13]. TRPM7 is a non-selective cation channel with notable permeability to Ca2+ and Mg2+, though it specifically facilitates Ca2+ influx in various cancer cells [14,15,16]. Additionally, TRPM7 has been shown to regulate B cell development and antigen recognition [17, 18], suggesting its role in MM pathogenesis. ORAI1 is located in the plasma membrane and regulates Ca2+ entry in collaboration with the endoplasmic reticulum Ca2+ sensor STIM1, which has been implicated in the metastasis of several solid tumors, including breast, cervical, and kidney [19,20,21]. Using bioinformatics database, we observed that TRPM7, ORAI1 and STIM1 mRNA expression are upregulated in patient-derived MM cells as compared to normal plasma cells (NPCs). We therefore further investigated the functional roles of these Ca2+ influx channels in MM cell migration and invasion and in the dissemination of MM cells in a mouse model.
An aberrant metabolism is an established hallmark of cancer [22, 23], and O-GlcNAcylation, a post-translational modification (PTM) in the hexosamine biosynthesis pathway, is a critical sensor of metabolic changes. We observed that cellular O-GlcNAcylation is dependent on changes in Ca2+ influx, regardless of channel type. In this study, we aimed to address the following: (a) whether O-GlcNAcylation plays a role in MM cell motility and dissemination; (b) whether Ca2+ influx regulates MM cell motility via O-GlcNAcylation, and (c) what are the downstream targets of Ca2+ influx/O-GlcNAcylation and the associated regulatory mechanisms.
Materials and methods
Reagents
2-APB, SKF96365, and thiamet G were obtained from Tocris Bioscience (Bristol, UK). PugNAc and antibodies for TRPM7, ORAI1, STIM1, OGA, O-GlcNAc and ITGB7 were obtained from Abcam (Cambridge, UK). Antibody for ubiquitin was from Santa Cruz Biotechnology (Dallas, TX, USA) and secondary antibodies were from EMD Millipore (Berlington, MA, USA). MG-132 and all other antibodies were from Cell Signaling Technology (Beverly, MA, USA). Other reagents were from Sigma-Aldrich (Dallas, MA, USA).
Cells and culture
Human MM-derived cell lines RPMI8226 and National Cancer Institute (NCI)-H929 were obtained from the American Type Culture Collection (Manassas, VA, USA). Cell lines were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Invitrogen, Waltham, MA, USA) at 37 °C and 5% CO2. Mycoplasma contamination was regularly checked using a commercial test kit (MycoAlert™ PLUS, Lonza, Cologne, Germany) and was found to be absent in all cell lines tested.
Oncomine™ bioinformatics database
mRNA expression analysis of clinical samples was conducted using Oncomine™ public database (https://www.oncomine.org/resource/main.html). Analyzed datasets included Zhan myeloma [24], Zhan myeloma 3 [25], and Agnelli myeloma 3 [26].
Calcium assay
Intracellular Ca2+ measurements were performed using Fura-2 AM (Abcam, Cambridge, UK) as described previously with minor modifications [21]. After specific treatments, 5 × 104 cells were suspended in HBSS buffer containing 4 μM Fura-2 AM and loaded onto a 96-well black plate for 1 h at 37 °C. Following an internal Ca2+ depletion by 2 μM thapsigargin for 10 min, 2 μM extracellular Ca2+ was added and Fura-2 AM signals were measured using a fluorescence microplate reader (Synergy H1, BioTek, Winooski, VT, USA) at 340/510 or 380/510 nm.
CRISPR/Cas9-mediated gene knockdown
All-in-one pLentiCRISPR v2 plasmids carrying a single guide RNA (sgRNA) against specific target gene, spCas9 and puromycin resistance were obtained from GenScript (Piscataway, NJ, USA), while LentiCas9-blasticidin and TRPM7 sgRNA plasmids were a kind gift from Profs. Zhang, Doench, and Root (Addgene #52962 and #76111). The oligos sequences of all sgRNAs were listed in Additional file 1: Supplementary Table S1. Lentiviral particles were generated in HEK293T cells using pCMV.dR8.2 dvpr packaging and pCMV-VSV-G envelope vectors (Addgene #8454 and 8455). Cells were transduced with the virus in the presence of hexadimethrine bromide (8 μg/mL), selected with puromycin (8 μg/mL) or blasticindin (10 μg/mL), and assessed for gene knockdown efficiency by Western blotting.
Overexpression plasmid and transfection
Cells were transfected with ITGA4 or ITGB7 plasmid (Genscript) using Lipofectamine 3000 Transfection Kit (Life Technologies, Carlsbad, CA). The transfected cells were allowed to recover for 48 h and protein level was determined by Western blotting before each experiment.
Cell motility assay
Cell migration was assessed in a 24-well plate Transwell system (Corning, Kennebunk, ME, USA) using a cell culture insert with a pore size of 5.0 μm, while cell invasion was assessed using an insert coated with 0.5 mg/mL Matrigel (BD Bioscience, San Jose, CA, USA) [27, 28]. After starvation for 4 h, cells (1 × 105 cells/well) were loaded onto the insert chamber containing low-serum (1% FBS) medium and complete medium was added to the lower chamber as a chemoattractant. Migrating/invading cells in the lower chamber were collected at 24–48 h, stained with Hoechst 33342, and visualized under an inverted fluorescence microscope (Eclipse Ti-U, Nikon, Tokyo, Japan).
In vivo disseminated MM xenograft model
All animal studies were performed in accordance with the protocol approved by Institutional Animal Care and Use Committee of West Virginia University (WVU) (#1702005551). Cells were labeled with UBC-RFP-T2A-Luciferase dual reporter for live cell tracking. Male NSG (NOD.Cg-Prkdcscid. Il2rgtm1Wjl/SzJ) mice (WVU Transgenic Animal Core Facility, Morgantown, WV, USA) at 6–8 weeks of age were intravenously injected with 1.5 × 107 luciferase-labeled cells and tumor burden was monitored on an IVIS Lumina II in Vivo Imaging system (PerkinElmer, Waltham, MA, USA). Mice were euthanized 4 weeks after the infusion or as recommended by a veterinarian. Organs were collected and imaged ex vivo to evaluate a disseminated lesion. After which, isolated organs were formalin fixed, paraffin embedded, and cut into 5-μm sections. Engraftment was confirmed by immunohistochemistry (IHC) using CD138 antibody (Invitrogen, #36–2900).
Western blotting and immunoprecipitation (IP)
Protein was extracted using commercial protein lysis buffer (Cell Signaling Technology) supplemented with a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). A total protein of 30–50 μg was subjected to SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with 5% fat-free milk and incubated with indicated primary antibodies overnight at 4 °C and with secondary antibodies conjugated with HRP for 2 h at room temperature. The immunoreactive proteins were captured and analyzed with ECL reagent (Millipore, Billerica, MA) using a digital imaging system (ImageQuant LAS, GE Healthcare, Pittsburgh, PA). For IP, cell lysates (100–200 μg) were incubated with anti-integrin α4 and or anti-integrin β7 antibody at 4 °C overnight, followed by incubation on ice with agarose beads for another 2 h to precipitated the protein-antibody complex. After extensive washing with ice-cold IP lysis buffer, samples were resuspended in protein loading buffer, boiled at 95 °C for 5 min, and subjected to immunoblotting.
Statistical analysis
Data are presented as mean ± SD from three or more independent experiments. Unpaired, two-tailed Student t-test or one-way ANOVA with Tukey’s multiple comparison tests were used to determine statistical significance at the significance level of P < 0.05 (GraphPad Prism, San Diego, CA, USA).
Results
TRPM7, ORAI1 and STIM1 levels are upregulated in MM
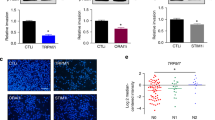
mRNA expression analysis of critical Ca2+ influx channels was first performed in human clinical specimens available on Oncomine™ bioinformatics database. We observed that TRPM7, ORAI1, and STIM1 expression was significantly higher in smoldering MM (SMM), a precancerous form of MM, when compared to NPCs (Fig. 1A–C). An upregulation of STIM1 expression in symptomatic MM was observed in two other datasets (Fig. 1C), while information on TRPM7 and ORAI1 expression in symptomatic MM was not available. These data suggest that aberrant Ca2+ influx channels may be involved in MM pathogenesis and progression and support the previous study demonstrating a close relation between elevated SOC influx channels and clinical outcome of MM patients [29].

Upregulation of TRPM7, ORAI1 and STIM1 in clinical MM specimens and their inhibition by small molecule inhibitors in human MM cells. a-c Differential mRNA expression of TRPM7, ORAI1 and STIM1 in SMM and/or MM tissues in comparison to NPC in clinical datasets available on Oncomine™ bioinformatics database. *P < 0.05, **P < 0.01, ***P < 0.0001 versus NPC; two-tailed Student’s t-test. d, e Effect of SMIs of TRPM7, ORAI1, and STIM1 on intracellular Ca2+ levels. Human MM-derived RPMI8226 and NCI-H929 cells were treated with different concentrations of 2-APB (0–50 μM), AnCoA4 (0–40 μM) and SKF96365 (0–40 μM), and free intracellular Ca2+ levels were measured by a fluorescence plate reader using Fura-2 AM as a probe. Fura-2 signals were measured after a depletion of internal Ca2+ stores at 340/510 and 380/510 nm and reported as relative Fura-2 ratio to non-treated cells (NTX). f, g RPMI8226 and NCI-H929 cells were similarly treated with 2-APB (0–50 μM) (upper), AnCoA4 (0–40 μM) (middle), and SKF96365 (0–40 μM) (lower), and cell migration and invasion were evaluated by Transwell assays at 48 h. The penetrating cells were stained by Hoechst 33342 dye, quantified, and reported as percentage to NTX cells. Representative micrographs of migrating/invading cells stained with Hoechst 33342 are shown (see also Additional file 2: Fig. S4 for additional micrographs). Scale bar = 100 μm. Data are mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus NTX; two-tailed Student’s t-test
Inhibition of Ca2+ influx channels suppresses MM cell migration and invasion
To investigate the potential role of TRPM7, ORAI1, and STIM1 in MM cell motility, we first used various small-molecule inhibitors (SMIs) of the channels, including 2-APB, AnCoA4, and SKF96365, as schematically depicted in Additional file 2: Fig. S1. To ensure that their effect on cell motility was not due to their adverse effects on cell viability and proliferation, we used sub-cytotoxic concentrations of the SMIs, i.e. up to 50 μM for 2-APB and up to 40 μM for AnCoA4 and SKF96365 in MM RPMI8226 and NCI-H929 cells (see Additional file 2: Fig. S2 for MTT assay and Fig. S3 for cell cycle analysis and Annexin V-PE/7-AAD apoptosis assay) in our study. Free intracellular Ca2+ in response to 2-APB treatment was evaluated using Fura-2 AM as a fluorescent probe. Figure 1D and E shows that 2-APB, AnCoA4, and SKF96365 dose-dependently inhibited Ca2+ influx in both RPMI8226 and NCI-H929 cells, indicating that Ca2+ influx channels are functional in these MM cells and that they are responsive to the SMI treatments. Phenotypically, we observed that the cells treated with 2-APB, AnCoA4, and SKF96365 were less migratory and invasive by 30–95% depending on the dose, as evaluated by Transwell assays (Fig. 1F and G; Additional file 2: Fig. S4). These data suggest that Ca2+ influx channels, particularly TRPM7, ORAI1, and STIM1, are critical for MM cell migration and invasion.
Depletion of TRPM7, ORAI1, and STIM1 suppresses MM cell motility
To specifically determine the role of TRPM7, ORAI1, and STIM1 influx channels in MM cell motility, CRISPR/Cas9 system was used to repress the specific gene expression in MM cells. The cells were transduced with lentiviral particles comprising sgRNA against TRPM7 (sgTRPM7), ORAI1 (sgORAI1), and STIM1 (sgSTIM1) and Cas9, and their effect on protein expression was determined by Western blotting (Fig. 2A–C; Additional file 2: Fig. S5). Figure 2D–F shows that sgTRPM7, sgORAI1, and sgSTIM1-transduced cells exhibited significantly lower migratory and invasive activities than vector control (wild-type, WT) cells. We also verified that the sgTRPM7, sgORAI1 and sgSTIM1 cells exhibited a similar proliferative activity, cell cycle profile, and apoptosis rate as their respective WT control cells (Additional file 2: Fig. S6), thus ruling out their anti-proliferative effect on the observed cell motility. Together, the results from our CRISPR/Cas9 gene depletion and SMI studies strongly indicate that TRPM7, ORAI1, and STIM1 are key regulators of MM cell motility.

Inhibition of TRPM7, ORAI1 and STIM1 reduces cell migration and invasion in MM cells likely in concomitant with a decreased cellular O-GlcNAcylation. a–c Western blot analysis showing TRPM7, ORAI1, and STIM1 knockdown efficiency in sgTRPM7, sgORAI1 and sgSTIM1 RPMI8226 cells, respectively. d–f Cell migration and invasion were evaluated by Transwell assays at 48 h, where the penetrating cells were stained by Hoechst 33342 dye. (upper) Representative micrographs of migrating/invading cells are shown. Scale bar = 100 μm. (lower) Quantitative data showing the percentage of migrating/invading cells relative to WT control cells. Data mean ± SD (n = 4). ***P < 0.001 versus WT; two-tailed Student’s t-test. g RPMI8226 cells were treated with 2-APB (0–50 μM), AnCoA4 (0–40 μM), and SKF96365 (0–40 μM) for 24 h, and cellular O-GlcNAcylation level was evaluated by Western blotting. h Quantitative analysis of O-GlcNAcylation level in treated cells, normalized to β-actin and relative to non-treated cells (NTX). Data are mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus NTX; two-tailed Student’s t-test. i–k Cellular O-GlcNAcylation level was compared between sgTRPM7, sgORAI1 or sgSTIM1 and WT RPMI8226 cells. Data are mean ± SD (n = 4). *P < 0.05, ***P < 0.001 versus NTX; two-tailed Student’s t-test
Ca2+ influx acts upstream of cellular O-GlcNAcylation
Recent studies have demonstrated a plausible crosstalk between Ca2+ influx and cellular O-GlcNAcylation in excitable cells [30]. To test this possibility in MM cells, cellular O-GlcNAc level was manipulated in MM RPMI8226 cells and its effect on Ca2+ influx channels was determined by Western blotting, and vice versa. Additional file 2: Fig. S7 shows that the levels of TRPM7, ORAI1, and STIM1 were relatively unchanged in cells treated with OGA inhibitor PugNAc (10–50 μM) or thiamet G (2.5–10 μM), which caused an increase in O-GlcNAc level. In contrast, cells treated with Ca2+ influx channel inhibitors, including 2-APB (10–50 μM), AnCoA4 (10–40 μM), and SKF96365 (10–40 μM), exhibited a dose-dependent increase in cellular O-GlcNAc level as compared to non-treated control (Fig. 2G and H). The increase in cellular O-GlcNAc level upon inhibition of TRPM7, ORAI1, and STIM1 was validated in sgTRPM7, sgORAI1, and sgSTIM1 cells, which displayed elevated O-GlcNAc levels as compared to WT cells (Fig. 2I–K). These data indicate that O-GlcNAcylation is regulated by Ca2+ influx via TRPM7, ORAI1, and STIM1 channels and that O-GlcNAcylation may regulate MM cell motility.
Elevated O-GlcNAcylation reduces migration and invasion of MM cells
Although O-GlcNAcylation has been associated with metastasis of various solid tumors [31], its role in MM cell migration and invasion has not been demonstrated. In this study, we used OGT and OGA inhibitors, as schematically depicted in Fig. 3A, to evaluate the dose-response effect of O-GlcNAcylation on cell motility. To ascertain the clinical significance of O-GlcNAcylation in MM, we also analyzed OGT and MGEA5 (encoding OGA) expression in MM patients using the Oncomine™ database. We found that MGEA5 expression was significantly higher in MM patients as compared to NPCs, whereas the expression of OGT was not significantly different in the two groups (Fig. 3B). These results suggest a reduced O-GlcNAcylation in MM. Treatment of MM cells with PugNAc (10–50 μM) and thiamet G (2.5–10 μM) decreased cell migration and invasion in a dose-dependent manner as compared to non-treated control, in an inverse relationship with the O-GlcNAc level (Fig. 3C and D; Additional file 2: Fig. S8 and S9). Consistently, genetic inhibition of MGEA5 by CRISPR/Cas9 (sgMGEA5), which caused an increase in O-GlcNAcylation, reduced the migration and invasion of MM cells (Fig. 3E and F; Additional file 2: Fig. S10). It should be noted that inhibition of OGA by both SMIs and CRISPR/Cas9 did not affect cell proliferation, cell cycle or apoptosis (Additional file 2: Fig. S11 and S12), indicating that the observed inhibitory effects were indeed due to impaired cell motility. Similar results were obtained in human MM-derived NCI-H929 cells (Additional file 2: Fig. S13). These findings are in good agreement with the TRPM7, ORAI1, and STIM1 inhibition data showing their effect on hyper-O-GlcNAcylation, and with the clinical data linking hypo-O-GlcNAcylation with aggressive MM.

MM cell motility is regulated by Ca2+ influx/O-GlcNAc axis. a Schematic diagram showing O-GlcNAcylation on protein’s serine/threonine residues and SMIs that modulate OGT, including OSMI-1, and OGA, including PugNAc and thiamet G. b MGEA5 (encoding OGA) and OGT mRNA expression in MM tissues in comparison to NPC in a clinical dataset available on Oncomine™ bioinformatics database. Data mean ± SD. *P < 0.05 versus NPC; two-tailed Student’s t-test. c Human MM-derived RPMI8226 cells were treated with various concentrations of PugNAc (0–50 μM) for 24 h and cellular O-GlcNAcylation was analyzed by Western blotting to confirm its induction. (right) Quantitative analysis of O-GlcNAcylation, normalized to β-actin and relative to the non-treated cells (NTX). Data are mean ± SD (n = 4). **P < 0.01, ***P < 0.001 versus NTX; two-tailed Student’s t-test. d Cell migration and invasion in response to PugNAc (0–50 μM) were evaluated by Transwell assays at 48 h, where the penetrating cells were stained with Hoechst 33342 dye. Scale bar = 100 μm. Data are mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus NTX; two-tailed Student’s t-test. e, f RPMI8226 and NCI-H929 cells were genetically knocked down with sgRNA sequence against MGEA5 (encoding OGA) in the CRISPR/Cas9 system. Western blot analysis showing OGA knockdown efficiency in sgMGEA5 cells. After which, cell migration and invasion were evaluated by Transwell assays at 48 h. (see also Additional file 2: Fig. S10C and D for representative micrographs of migrating/invading cells). Quantitative data showing the percentage of migrating/invading cells relative to WT control cells. Data mean ± SD (n = 4). ***P < 0.001 versus WT; two-tailed Student’s t-test. g–k Rescue experiments were performed in sgTRPM7, sgORAI1, and sgSTIM1 RPMI8226 cells by treating the cells with OGT inhibitor OSMI-1. g, h Cellular O-GlcNAcylation level in sgTRPM7, sgORAI1, and sgSTIM1 cells in the presence or absence of OSMI-1 (3 μM) for 24 h was evaluated by Western blotting. Representative immunoblots (g) and quantitative analysis of O-GlcNAcylation levels (h) indicate that OSMI-1 could reverse the hyper-O-GlcNAcylation in sgTRPM7, sgORAI1, and sgSTIM1 cells. Data mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus non-treated WT control cells or non-treated sgTRPM7, sgORAI1 or sgSTIM1 cells; one-way ANOVA with Tukey’s multiple comparison test. ns, not significant. i–k Cell migration and invasion in sgTRPM7, sgORAI1, and sgSTIM1 cells in the presence or absence of OSMI-1 (3 μM) were evaluated by Transwell assays at 48 h. (see also Additional file 2: Fig. S14A–C for representative micrographs of migrating/invading cells). Quantitative data showing the percentage of migrating/invading cells relative to non-treated WT cells. Data mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus non-treated WT control cells or non-treated sgTRPM7, sgORAI1 or sgSTIM1 cells; one-way ANOVA with Tukey’s multiple comparison test. ns, not significant
Hypo-O-GlcNAcylation rescues impaired cell motility upon inhibition of TRPM7, ORAI1, and STIM1
Based on the findings that inhibition of TRPM7, ORAI1, and STIM1 elevated cellular O-GlcNAcylation level, and that inhibition of such channels and O-GlcNAcylation reduced MM cell migration and invasion, we postulated that TRPM7, ORAI1, and STIM1 may regulate MM cell motility through O-GlcNAcylation. Hence, hypo-O-GlcNAcylation should be able to rescue the effects on cell motility caused by inhibition of the channels. To test this possibility, sgTRPM7, sgORAI1, and sgSTIM1 cells were treated with OGT inhibitor OSMI-1 (3 μM), which reduced O-GlcNAcylation (Fig. 3G and H), and its effects on cell migration and invasion were determined by Transwell assays. Figure 3I–K shows that OSMI-1 rescued the inhibitory effects in sgTRPM7, sgORAI1, and sgSTIM1 cells, substantiating that TRPM7, ORAI1, and STIM1 regulate MM cell motility through O-GlcNAcylation (Additional file 2: Fig. S14). These results demonstrated a novel role of Ca2+ influx/O-GlcNAc via TRPM7, ORAI1, and STIM1 channels in regulating MM cell motility.
Disruption of Ca2+ influx/O-GlcNAc axis suppresses tumor burden in MM xenograft model
Having demonstrated that Ca2+ influx acts upstream of O-GlcNAcylation in regulating MM cell motility, we experimentally verified the involvement of TRPM7, which showed the most pronounced effect on cell motility, and O-GlcNAcylation in MM dissemination in a xenograft mouse model [32, 33]. sgTRPM7, sgMGEA5 and WT cells were genetically labeled with luciferase and intravenously injected into NSG mice via tail vein to induce systemic disease, followed by a weekly bioluminescence imaging for quantitative analysis of tumor progression. Bioluminescence analyses revealed that both sgTRPM7 and sgMGEA5 cells yielded substantially lower whole-body signals at 1, 2, 3 and 4 weeks post-injection comparing to WT cells (Fig. 4A, B, F, and G), suggesting less tumor burden and experimental dissemination of MM cells. For better comparison, the luminescence signals in each group were normalized to their initial signals at week 0 and relative to WT control. Figure 4C and H confirmed a lower myeloma burden in mice bearing sgTRPM7 and sgMGEA5 cells at week 1 to 4 post-injection. To support the finding of MM dissemination, mice were euthanatized at the end of the experiments and various organs, including brain, lung, kidney, bone, heart, spleen, and liver, were collected for ex vivo bioluminescence imaging and histochemistry. Figure 4D and I demonstrated substantially lower tumor signals in the various organs in mice bearing sgTRPM7 and sgMGEA5 cells as compared to those in WT mice. These results are consistent with the IHC finding showing a decreased number of infiltrated MM cells in the various organs, including bone (femur) and liver, and the flow cytometric analysis of human CD138+ cells in the BM of femur bone (Fig. 4E and J; see also Additional file 2: Fig. S15–18). Together, these results support the in vitro findings that Ca2+ influx and hypo-O-GlcNAcylation are critical regulators of MM cell motility and dissemination.

Inhibition of Ca2+ influx channels and hyper-O-GlcNAcylation impairs MM dissemination in vivo. a–e Luciferase-labeled sgTRPM7 or WT RPMI8226 cells were injected into NSG mice via tail vein. A Representative bioluminescence imaging of mice taken at the time of inoculation (week 0) and weekly post-injection (week 1, 2, 3 and 4). b, c Quantification of whole-body luminescence signals normalized to their initial signal at week 0 (b) and relative to WT control cells (c). Data mean ± SD (n = 5). ***P < 0.001 versus WT cells; two-tailed Student’s t-test. d Representative ex vivo imaging from isolated internal organs including brain, lung, kidney, bone, heart, spleen, and liver at week 4 post-injection (see also Additional file 2: Fig. S15 for quantification of ex vivo IVIS images). e Representative IHC micrographs of femur bone and liver sections staining for human CD138 (see also Additional file 2: Fig. S17 for additional IHC micrographs). Scale bar = 100 μm. f–j Luciferase-labeled sgMGEA5 or WT RPMI8226 cells were similarly injected into NSG mice via tail vein and bioluminescence imaging and IHC staining were similarly performed as described in A–E (see also Additional file 2: Fig. S18 for additional IHC micrographs)
Elevated O-GlcNAcylation decreases MM cell motility via suppression of integrin α4 and integrin β7
O-GlcNAcylation has been long known to modify proteins and alter their functions and cellular pathways [34]. To gain a more in-depth understanding of the underlying mechanisms, we used a literature mining approach to identify cell adhesion molecules that are involved in MM progression, including the integrin family of proteins such as integrin β1 (ITGB1), β3 (ITGB3), β7 (ITGB7), α4 (ITGA4), α5 (ITGA5) and αV (ITGAV), E-cadherin (E-cad), N-cadherin (N-cad), and β-catenin (B-cat) [35,36,37,38]. The expression level of these proteins in response to thiamet G (2.5–10 μM) treatment was examined in MM cells by Western blotting. Figure 5A and B shows that thiamet G minimally affected ITGB1, ITGAV, ITGA5, N-cad, and B-cat expression, but significantly downregulated ITGB3, ITGA4, ITGB7, and E-cad in a dose-dependent manner (see also Additional file 2: Fig. S19A). Bioinformatics analysis of the adhesion molecules in MM patients using Oncomine™ database revealed that ITGA4 and ITGB7 were significantly upregulated in MM tissues as compared to NPCs, while ITGB3 and CDH1 expression was not different (Fig. 5C; see also Additional file 2: Fig. S19B), suggesting the role of ITGA4 and ITGB7 in MM cell adhesion. Genetic inhibition of ITGA4 and ITGB7 by CRISPR/Cas9 in MM cells, designated as sgITGA4 and sgITGB7 cells (Fig. 6A and B), also showed a substantial reduction in MM cell migration and invasion when compared to WT control (Fig. 6C and D), while having minimal effect on cell proliferation, cell cycle and apoptosis (Additional file 2: Fig. S20). The change in ITGA4 and ITGB7 expression following O-GlcNAcylation was verified in sgMGEA5 cells (Fig. 5 D and E), thus confirming that ITGA4 and ITGB7 are key regulators of MM cell motility under O-GlcNAcylation.

Hyper-O-GlcNAcylation suppresses MM cell motility via ITGA4 and ITGB7. a Human MM-derived RPMI8226 cells were treated with various concentrations of thiamet G (0–10 μM) and analyzed for cell adhesion proteins, including ITGB1, ITGB3, ITGB7, ITGA4, ITGA5, ITGAV, E-cad, N-cad, and B-cat by Western blotting. b Quantitative analysis of the indicated proteins, normalized to β-actin and relative to the non-treated cells (NTX) (see also Additional file 2: Fig. S19 for an additional quantification). Data mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus NTX; two-tailed Student’s t-test. c ITGA4 and ITGB7 expression in MM tissues in comparison to NPC in a clinical dataset available on Oncomine™ bioinformatics database. Data mean ± SD. *P < 0.05, ***P < 0.001 versus NPC; two-tailed Student’s t-test. d, e OGA-knockdown sgMGEA5 RPMI8226 and NCI-H929 cells were evaluated for ITGA4 and ITGB7 levels by Western blotting to confirm that they are downstream of O-GlcNAcylation. (lower) Quantitative analysis of ITGA4 and ITGB7 levels, normalized to β-actin and relative to WT control cells. Data mean ± SD. ***P < 0.001 versus WT; two-tailed Student’s t-test

Inhibition of ITGA4 and ITGB7 critically suppresses MM cell motility. a, b Human MM-derived RPMI8226 cells were genetically knocked down with sgRNA against ITGA4 and ITGB7 by CRISPR/Cas9. (left) Western blot analysis showing knockdown efficiency in sgITGA4 (a) and sgITGB7 (b) cells. c, d Cell migration and invasion were evaluated in sgITGA4 (c) and sgITGB7 (d) cells by Transwell assays at 48 h, where the penetrating cells were stained with Hoechst 33342 dye. (left) Representative micrographs of migrating/invading cells are shown. Scale bar = 100 μm. (right) Quantitative data showing the percentage of migrating/invading cells relative to WT control cells. Data mean ± SD (n = 4). ***P < 0.001 versus WT; two-tailed Student’s t-test
To further validate that ITGA4 and ITGB7 are downstream of Ca2+ influx under the Ca2+ influx/O-GlcNAc axis, rescue experiments were conducted in which ITGA4 and ITGB7 plasmid were ectopically overexpressed in various Ca2+ influx-inhibited cells, including sgTRPM7, sgORAI1, and sgSTIM1 RPMI8226 cells, and their effects on cell motility and protein level were determined. Figure 7A–C showed the reduced ITGA4 and ITGB7 levels in sgTRPM7, sgORAI1, and sgSTIM1 cells, indicating that Ca2+ influx via TRPM7, ORAI1, and STIM1 channels regulated ITGA4 and ITGB7 abundance. Overexpression of ITGA4 and ITGB7 did restore the reduced migration and invasion in sgTRPM7, sgORAI1, and sgSTIM1 cells (Fig. 7D–F), in agreement with the increased ITGA4 and ITGB7 levels, strengthening that the ITGA4 and ITGB7 levels and their functional contributions were indeed regulated by the Ca2+ influx/O-GlcNAc axis.

ITGA4 and ITGB7 are favorable downstream targets of Ca2+ influx/O-GlcNAc-mediated MM cell motility. a–c Rescue experiments were performed in sgTRPM7 (a), sgORAI1 (b), and sgSTIM1 (c) RPMI8226 cells by transfection of the cells with ITGA4, ITGB7 or control pcDNA plasmid. ITGA4 (upper) and ITGB7 (lower) levels were evaluated by Western blot analysis at 48 h post-transfection prior to experiments and representative blots were shown. d–f Cell migration and invasion in ITGA4 or ITGB7-overexpressing sgTRPM7 (d), sgORAI1 (e), and sgSTIM1 (f) cells by Transwell assays at 48 h, where the penetrating cells were stained with Hoechst 33342 dye. Quantitative data showing the percentage of migrating/invading cells relative to transfected control cells. Data mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus pcDNA-transfected control cells; one-way ANOVA with Tukey’s multiple comparison test. #P < 0.05, ##P < 0.01, ###P < 0.001 versus pcDNA-transfected sgTRPM7, sgORAI1, or sgSTIM1 cells; one-way ANOVA with Tukey’s multiple comparison test. ns, not significant
Notably, we also monitored the pan matrix metalloproteinase (MMP) activity using MMP Activity Fluorometric Assay Kit (Abcam) in sgTRPM7, sgORAI1, sgSTIM1 cells as well as in sgMGEA5 cells and WT control cells since MMPs are known to be involved in cancer cell motility of various cancers and have been reported to promote MM dissemination [39, 40]. However, our results showed no significant difference in total MMP activity in sgTRPM7, sgORAI1, sgSTIM1, and sgMGEA5 cells, indicating that MMPs may not play a significant role in the MM dissemination under the experimental Ca2+ influx/O-GlcNAc conditions (Additional file 2: Fig. S21).
O-GlcNAcylation of ITGA4 and ITGB7 induces ubiquitin-mediated proteasomal degradation
Proteasomal degradation is known to play a major role in cellular proteostasis via the regulation of protein stability and degradation [41]. We tested whether O-GlcNAcylation regulates ITGA4 and ITGB7 stability by proteasomal degradation by first performing a rescue experiment using an established proteasome inhibitor, MG-132 (50 μM). Figure 8A shows that ITGA4 and ITGB7 downregulation by thiamet G was rescued by MG-132 treatment, indicating that O-GlcNAcylation regulates ITGA4 and ITGB7 via a proteasome-dependent pathway. Since O-GlcNAcylation and ubiquitination are PTMs that similarly occur at Ser/Thr residues [42], we next determined the interplay between the two on ITGA4 and ITGB7 proteins using co-immunoprecipitation. Figure 8B reveals physical interaction between ITGA4 and ITGB7 and the association between O-GlcNAcylation and ubiquitination of the two proteins. Elevated ITGA4 and ITGB7 O-GlcNAcylation by thiamet G caused an increase in ITGA4 and ITGB7 ubiquitination. These results indicate that O-GlcNAcylation of ITGA4 and ITGB7 promotes their ubiquitin-proteasomal degradation, leading to reduced protein expression and attenuated MM cell motility.

Hyper-O-GlcNAcylation promotes ubiquitin-mediated proteasomal degradation of ITGA4 and ITGB7. a RPMI8226 cells were pretreated with the proteasome inhibitor MG-132 (50 μM) for 3 h, followed by thiamet G (10 μM) treatment for 6 h, and ITGA4 and ITGB7 levels were analyzed by Western blotting. (lower) Quantitative analysis of ITGA4 (left) and ITGB7 (right) levels, normalized to β-actin and relative to non-treated cells (NTX). Data mean ± SD (n = 4). *P < 0.05, ***P < 0.001 versus NTX cells or thiamet G-treated cells alone; one-way ANOVA with Tukey’s multiple comparison test. ns, not significant. b RPMI8226 cells were pretreated with MG-132 to prevent proteasomal degradation and then treated with thiamet G (10 μM) for 6 h. The cells were then subjected to immunoprecipitation (IP) with anti-ITGA4 and anti-ITGB7 antibodies, and the immune complexes were detected for O-GlcNAcylation or ubiquitination by Western blotting. IP inputs were also evaluated for ITGA4, ITGB7 or β-actin for comparison. c A schematic illustration for the regulation of MM cell motility and dissemination by Ca2+ influx/O-GlcNAc axis. Ca2+ influx channels, including TRPM7, ORAI1, and STIM1, regulate Ca2+ entry in MM cells. Following Ca2+ influx channels inhibition, cellular O-GlcNAcylation is elevated and activates ITGA4 and ITGB7. O-GlcNAcylation of ITGA4 and ITGB7 induces downregulation of the two adhesion proteins via ubiquitin (Ub)-proteasomal degradation, causing a repression of MM cell motility and dissemination
Discussion
This study unveils a novel mechanism of MM cell motility and dissemination regulation via Ca2+ influx and O-GlcNAcylation axis. Depletion of Ca2+ influx channels, including TRPM7, ORAI1, and STIM1, in human-derived MM cells causes hyper-O-GlcNAcylation and subsequent ubiquitin-proteasome mediated degradation of ITGA4 and ITGB7, leading to their decreased expression and consequential reduction in MM cell motility and dissemination (Fig. 8C). These cell adhesion molecules as targets of Ca2+ influx/O-GlcNAcylation axis could have a major clinical implication in MM therapies since ITGA4 and ITGB7 are highly expressed in the majority of MM tissues, but not in NPCs.
Ca2+ is a ubiquitous second messenger that controls many essential cellular processes, such as cell proliferation, differentiation, and death [43]. The involvement of Ca2+ signaling in various oncogenic and metastatic processes initially arose from the observations that breast, lung, and prostate cancers often metastasize to the bone, causing osteolysis and high Ca2+ accumulation in the tumor ecosystem [44]. In contrast to solid tumors, BM is the primary site of MM development and dysregulated Ca2+ environment, i.e., due to bone resorption, and therefore could play a key role in MM dissemination. Oncomine™ bioinformatics studies showed that TRPM7, ORAI1, and STIM1 are highly expressed in SMM and MM patients’ tissues compared with healthy NPCs. In addition, an increased Ca2+ influx is linked to high-grade MM (Additional file 2: Fig. S22), suggesting that the elevated Ca2+ influx channels may serve as prognostic markers of MM progression. To substantiate these observations, we additionally performed comparative gene expression analysis between NPCs and newly diagnosed, aggressive MM that were subsequently treated with three total therapies (TT3) using datasets available on Gene Expression Omnibus (GEO; accession number GSE5900 and GSE2658) (Additional file 2: Fig. S23) [45, 46]. Consistent with the Oncomine™ database analyses, TRPM7 and STIM1 are upregulated in TT3 MM cohort, though we did not observe a significant change in ORAI1 expression. Remarkably, the direct linkage between TRPM7 expression and MM progression has never been reported, although an increased expression of ORAI1 and STIM1 in MM that has previously been described and linked to poor clinical outcomes. For example, ORAI1 and STIM1 were found to express at a high level in BM tissues of stage III MM compared to stage I/II MM [29]. Analysis of MM patients, regardless of stage, also indicated a direct relationship between STIM1 expression and the duration of progression-free survival.
While the importance of Ca2+ influx and its channels in cancer progression has been well recognized, the precise mechanisms of regulation are unclear and likely to depend on cancer type and cellular context. Genetic downregulation of TRPM7 was shown to inhibit hypoxia-induced cell motility in androgen-independent prostate cancer cells. Such inhibition was mediated through proteasomal degradation of HIF-1α, which resulted in stabilization and increased binding of HIF-1α to RACK1, [47] suggesting the role of Ca2+ influx in protein stability and function. In the present study, we demonstrated for the first time the role of Ca2+ influx through TRPM7, STIM1 and ORAI1 channels in controlling protein stability and function via O-GlcNAcylation. We provided compelling evidence that Ca2+ influx through the described Ca2+ channels acts upstream of O-GlcNAcylation in an inverse relationship (Fig. 2) and that such Ca2+ influx regulates MM cell motility through O-GlcNAcylation (Fig. 3).
The roles of O-GlcNAcylation in the pathogenesis and progression of solid tumors have been well documented. Hyper-O-GlcNAcylation or elevated O-GlcNAcylation levels has been observed in various tumors, including breast, colon, pancreas, liver, bladder, gastric and lung, and has been attributed to an upregulated OGT and/or downregulated OGA expression [31]. Hyper-O-GlcNAcylation was found to cause an acquired apoptosis resistance in lung carcinoma through p53 and c-Myc, independent of p53 status, and induce cell migration and invasion through caveolin-1 and c-Myc [48]. However, the involvement of O-GlcNAcylation in the pathogenesis of hematologic malignancies has been far less studied and may be different from that of solid tumors. In chronic lymphocytic leukemia (CLL), indolescent clinical behaviors of CLL cells and favorable clinical outcomes correlate well with the higher level of O-GlcNAcylation [49]. Likewise, hyper-O-GlcNAcylation induced by OGA inhibition was found to sensitize bortezomib-induced apoptosis and reverse bortezomib resistance in mantle cell lymphoma (MCL), suggesting the potential utility of OGA inhibitors such as ketoconazole in adjuvant therapy of MCL [50]. In myeloid malignancies, OGT directly stabilizes ASXL1 by O-GlcNAcylation and drives myeloid differentiation associated with the pathogenesis of myelodysplastic syndrome (MDS) [51]. O-GlcNAcylation may also be involved in the pathogenesis of MM since it is involved in the lymphopoiesis of B cells [52, 53], which terminally differentiate into plasma cells.
As MM cells initially reside in the BM microenvironment and adhere to extracellular matrix and/or BM stromal cells, cell adhesion molecules such as integrins and cadherins are crucial for their BM homing, cell survival, proliferation and drug resistance. Integrins are the major adhesion molecules that are highly expressed in MM cells [35]. We demonstrated in this study that ITGA4 and ITGB7 are a direct target of O-GlcNAcylation that regulates MM cell motility and dissemination downstream of Ca2+ influx signaling. Bioinformatics analyses using Oncomine™ and the GEO datasets strengthen the clinical significance of ITGA4 and ITGB7, as their gene expression tend to be upregulated in the MM tissues when compared to healthy NPCs, although ITGB7 was not statistically significant (p = 0.0788) in one of the datasets (Fig. 5 and Additional file 2: Fig. S23). Hyper-O-GlcNAcylation causes a concomitant decrease in ITGA4 and ITGB7 expression (Fig. 5), which could be reversed by the addition of proteasome inhibitor MG-132 (Fig. 8), indicating that O-GlcNAcylation interferes with proteasomal degradation of ITGA4 and ITGB7. Accumulating evidence also indicates the crosstalk between O-GlcNAcylation and ubiquitination to promote either protein stability or turnover [42]. Hyper-O-GlcNAcylation of ITGA4 and ITGB7 was shown to promote their ubiquitination and subsequent proteasomal degradation. Although, hyper-O-GlcNAcylation was reported to induce ITGB1 activation in Hela cells via focal adhesion complex formation [54], this is the first demonstration of the regulation of ITGA4 and ITGB7 by O-GlcNAcylation via ubiquitin-proteasome mediated degradation pathway. It is worth noting that constitutive activation of ITGB7 has been observed in MM cells but not in non-hematopoietic cells/tissues, making ITGB7 a potential therapeutic target for chimeric antigen receptor (CAR) T cells [33].
Conclusion
In summary, the evidence presented here demonstrated that MM cell motility and dissemination could be functionally modulated by the Ca2+ influx/O-GlcNAcylation regulatory axis that directly targets ITGA4 and ITGB7 (Fig. 8C). Our novel findings on the molecular pathways and interactions provide mechanistic insights into the pathogenesis and progression of MM, and identify potential predictive biomarkers and drug targets for advanced MM. Herein, genetic inhibition of Ca2+ influx channel TRPM7 and OGA, which act upstream of ITGA4 and ITGB7, effectively inhibited experimental MM dissemination in vivo, suggesting the potential clinical applications of Ca2+ influx and O-GlcNAcylation modulators. It is worth noting that TRPM7, STIM1/ORAI1, and OGA are reported to be druggable targets [55,56,57] and an administration of currently available SMIs in vivo, e.g. NS8593 for TRPM7, and PugNAc and thiamet G for OGA, could be used as a promising tool for preclinical assessment [58,59,60]. However, development of potent, selective, and high-affinity SMIs for targeting Ca2+ influx and/or O-GlcNAcylation for cancer therapeutics is challenging and still in progress. Further studies would contribute to investigate the effects of novel SMIs in the in vivo disseminated MM xenograft model, which could be advantageous for the future MM treatment to achieve long-term control of the disease.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. Supplementary information is available.
Abbreviations
- B-cat:
-
β-catenin
- BM:
-
Bone marrow
- Ca2+ :
-
Calcium
- CAR:
-
Chimeric antigen receptor
- CLL:
-
Chronic lymphocytic leukemia
- E-cad:
-
E-cadherin
- ITGA4:
-
Integrin α4
- ITGA5:
-
Integrin α5
- ITGAV:
-
Integrin αV
- ITGB1:
-
Integrin β1
- ITGB3:
-
Integrin β3
- ITGB7:
-
Integrin β7
- MCL:
-
Mantle cell lymphoma
- Mg+ :
-
Magnesium
- MM:
-
Multiple myeloma
- N-cad:
-
N-cadherin
- NPCs:
-
Normal plasma cells
- O-GlcNAc:
-
O-linked β-N-acetylglucosamine
- ORAI1:
-
Calcium release-activated calcium channel protein 1
- PCs:
-
Plasma cells
- PTM:
-
Post-translational modification
- sgRNA:
-
Single guide RNA
- SMIs:
-
Small-molecule inhibitors
- SMM:
-
Smoldering multiple myeloma
- SOC:
-
Store-operated calcium
- STIM1:
-
Stromal interaction molecule 1
- TRPM7:
-
Transient receptor potential melastatin-subfamily member 7
- WT:
-
Wild-type
References
Tiu AC, Potdar R, Arguello-Gerra V, Morginstin M. Multiple liver nodules mimicking metastatic disease as initial presentation of multiple myeloma. Case Rep Hemato. 2018;2018:7954816.
Rai S, Sridevi H, Acharya V, Lobo F, Kini J. Pulmonary plasmacytoma in multiple myeloma: a rare case of extramedullary spread. Egypt J Bronchol. 2015;9:293–5.
Gregorio LM, Soyemi TO. Multiple myeloma presenting as dural plasmacytoma. Radiol Case Rep. 2019;14:1007–13.
Weinstock M, Ghobrial IM. Extramedullary multiple myeloma. Leuk Lymphoma. 2013;54:1135–41.
de Haart SJ, Willems SM, Mutis T, Koudijs MJ, van Blokland MT, Lokhorst HM, et al. Comparison of intramedullary myeloma and corresponding extramedullary soft tissue plasmacytomas using genetic mutational panel analyses. Blood Cancer J. 2016;6:e426.
Ghobrial IM. Myeloma as a model for the process of metastasis: implications for therapy. Blood. 2012;120:20–30.
Bhutani M, Foureau DM, Atrash S, Voorhees PM, Usmani SZ. Extramedullary multiple myeloma. Leukemia. 2020;34:1–20.
Hameed A, Brady JJ, Dowling P, Clynes M, O'Gorman P. Bone disease in multiple myeloma: pathophysiology and management. Cancer Growth Metastasis. 2014;7:33–42.
Roodman GD. Pathogenesis of myeloma bone disease. Leukemia. 2009;23(3):435–41.
Oyajobi BO. Multiple myeloma/hypercalcemia. Arthritis Res Ther. 2007;9 Suppl 1(Suppl 1):S4.
Nielsen N, Lindemann O, Schwab A. TRP channels and STIM/ORAI proteins: sensors and effectors of cancer and stroma cell migration. Br J Pharmacol. 2014;171:5524–40.
Chen YF, Chen YT, Chiu WT, Shen MR. Remodeling of calcium signaling in tumor progression. J Biomed Sci. 2013;20:23.
Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609–18.
Yee NS. Role of TRPM7 in cancer: potential as molecular biomarker and therapeutic target. Pharmaceuticals (Basel). 2017;10:39.
Liu L, Wu N, Wang Y, Zhang X, Xia B, Tang J, et al. TRPM7 promotes the epithelial–mesenchymal transition in ovarian cancer through the calcium-related PI3K/AKT oncogenic signaling. J Exp Clin Cancer Res. 2019;38:106.
Middelbeek J, Kuipers AJ, Henneman L, Visser D, Eidhof I, van Horssen R, et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res. 2012;72:4250–61.
Krishnamoorthy M, Buhari FHM, Zhao T, Brauer PM, Burrows K, Cao EY, et al. The ion channel TRPM7 is required for B cell lymphopoiesis. Sci Signal. 2018;11:eaan2693.
Krishnamoorthy M, Wasim L, Buhari FHM, Zhao T, Mahtani T, Ho J, et al. The channel-kinase TRPM7 regulates antigen gathering and internalization in B cells. Sci Signal. 2018;11:eaah6692.
Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–34.
Chen Y-F, Chiu W-T, Chen Y-T, Lin P-Y, Huang H-J, Chou C-Y, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A. 2011;108:15225–30.
Kim J-H, Lkhagvadorj S, Lee M-R, Hwang K-H, Chung HC, Jung JH, et al. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2014;448:76–82.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
Ferrer Christina M, Lynch Thomas P, Sodi Valerie L, Falcone John N, Schwab Luciana P, Peacock Danielle L, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54:820–31.
Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99:1745–57.
Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007;109:1692–700.
Agnelli L, Mosca L, Fabris S, Lionetti M, Andronache A, Kwee I, et al. A SNP microarray and FISH-based procedure to detect allelic imbalances in multiple myeloma: an integrated genomics approach reveals a wide gene dosage effect. Genes Chromosomes Cancer. 2009;48:603–14.
Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell. 2009;16:309–23.
Azab AK, Quang P, Azab F, Pitsillides C, Thompson B, Chonghaile T, et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood. 2012;119:1468–78.
Wang W, Ren Y, Wang L, Zhao W, Dong X, Pan J, et al. Orai1 and Stim1 mediate the majority of store-operated calcium entry in multiple myeloma and have strong implications for adverse prognosis. Cell Physiol Biochem. 2018;48:2273–85.
Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J Biol Chem. 2012;287:39094–106.
de Queiroz RM, Carvalho É, Dias WB. O-GlcNAcylation: the sweet side of the cancer. Front Oncol. 2014;4:132.
Lwin ST, Edwards CM, Silbermann R. Preclinical animal models of multiple myeloma. Bonekey Rep. 2016;5:772.
Hosen N, Matsunaga Y, Hasegawa K, Matsuno H, Nakamura Y, Makita M, et al. The activated conformation of integrin β7 is a novel multiple myeloma–specific target for CAR T cell therapy. Nat Med. 2017;23:1436–43.
Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–65.
Hosen N. Integrins in multiple myeloma. Inflamm Regen. 2020;40:4.
Olson DL, Burkly LC, Leone DR, Dolinski BM, Lobb RR. Anti-α4 integrin monoclonal antibody inhibits multiple myeloma growth in a murine model. Mol Cancer Ther. 2005;4:91–9.
Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011;117:6202–13.
Gkotzamanidou M, Fulciniti M, Martín Sanchez J, Samur MK, Parmigiani G, Anderson KC, et al. LSD1 impairs the epithelial-mesenchymal transition (EMT) and osteoclastogenesis potency in multiple myeloma and synergistically induces cytotoxicity with HDAC inhibitors. Blood. 2014;124:3410.
Kelly T, Børset M, Abe E, Gaddy-Kurten D, Sanderson RD. Matrix metalloproteinases in multiple myeloma. Leuk Lymphoma. 2000;37:273–81.
Zeissig MN, Zannettino ACW, Vandyke K. Tumour dissemination in multiple myeloma disease progression and relapse: a potential therapeutic target in high-risk myeloma. Cancers (Basel). 2020;12:3643.
Jang HH. Regulation of protein degradation by proteasomes in cancer. J Cancer Prev. 2018;23:153–61.
Ruan H-B, Nie Y, Yang X. Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics. 2013;12:3489–97.
Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium–cancer signalling nexus. Nat Rev Cancer. 2017;17:373–80.
Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72.
Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108:2020–8.
Sun J, Muz B, Alhallak K, Markovic M, Gurley S, Wang Z, et al. Targeting CD47 as a novel immunotherapy for multiple myeloma. Cancers (Basel). 2020;8(12):305.
Yang F, Cai J, Zhan H, Situ J, Li W, Mao Y, et al. Suppression of TRPM7 inhibited hypoxia-induced migration and invasion of androgen-independent prostate cancer cells by enhancing RACK1-mediated degradation of HIF-1α. Oxid Med Cell Longev. 2020;2020:6724810.
Luanpitpong S, Angsutararux P, Samart P, Chanthra N, Chanvorachote P, Issaragrisil S. Hyper-O-GlcNAcylation induces cisplatin resistance via regulation of p53 and c-Myc in human lung carcinoma. Sci Rep. 2017;7:10607.
Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia. 2010;24:1588–98.
Luanpitpong S, Chanthra N, Janan M, Poohadsuan J, Samart P, Y UP, et al. Inhibition of O-GlcNAcase sensitizes apoptosis and reverses bortezomib resistance in mantle cell lymphoma through modification of truncated bid. Mol Cancer Ther. 2018;17:484–96.
Inoue D, Fujino T, Sheridan P, Zhang Y-Z, Nagase R, Horikawa S, et al. A novel ASXL1–OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia. 2018;32:1327–37.
Wu J-L, Chiang M-F, Hsu P-H, Tsai D-Y, Hung K-H, Wang Y-H, et al. O-GlcNAcylation is required for B cell homeostasis and antibody responses. Nat Commun. 2017;8:1854.
Wu J-L, Wu H-Y, Tsai D-Y, Chiang M-F, Chen Y-J, Gao S, et al. Temporal regulation of Lsp1 O-GlcNAcylation and phosphorylation during apoptosis of activated B cells. Nat Commun. 2016;7:12526.
Xu Z, Isaji T, Fukuda T, Wang Y, Gu J. O-GlcNAcylation regulates integrin-mediated cell adhesion and migration via formation of focal adhesion complexes. J Biol Chem. 2019;294:3117–24.
Chubanov V, Gudermann T. Mapping TRPM7 function by NS8593. Int J Mol Sci. 2020;21:7017.
Elbatrawy AA, Kim EJ, Nam G. O-GlcNAcase: emerging mechanism, substrate recognition and small-molecule inhibitors. ChemMedChem. 2020;15:1244–57.
Chen YF, Lin PC, Yeh YM, Chen LH, Shen MR. Store-operated Ca2+ entry in tumor progression: from molecular mechanisms to clinical implications. Cancers (Basel). 2019;11:899.
Voringer S, Schreyer L, Nadolni W, Meier MA, Woerther K, Mittermeier C, et al. Inhibition of TRPM7 blocks MRTF/SRF-dependent transcriptional and tumorigenic activity. Oncogene. 2020;39:2328–44.
Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai JY, Grenett MH, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–19.
Andrés-Bergós J, Tardio L, Larranaga-Vera A, Gómez R, Herrero-Beaumont G, Largo R. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J Biol Chem. 2012;287(40):33615–28.
Acknowledgements
We would like to acknowledge Professors Davor Solter and Barbara Knowles for their comments on project and Sirinart Buasamrit for her assistance with laboratory management and administration.
Funding
This work was supported by grants from the Royal Golden Jubilee Ph.D. Programme (PHD/0085/2558, to PS), Thailand Research Fund (RSA6280103, to SL), Commission on Higher Education (CHE-RES-RG-49, to SI) and NIH (R01-ES022968, to YR; P20-RR016440 and P30-RR032138/GM103488, to WVU Animal Models & Imaging Facility).
Author information
Authors and Affiliations
Contributions
PS performed bioinformatics analysis, carried out in vitro and animal experiments, and helped to draft the manuscript. SL conceived the study, designed research, participated in the data analysis, coordinated the project and drafted the manuscript. YR participated in the design of the study. SI supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All cell lines used in this study were purchased from American Type Culture Collection (ATCC, Manassas, VA). All animal experiments were performed in accordance with Guidelines for Animal Experiments at West Virginia University with the approval of the Institutional Animal Care and Use Committee (#1702005551).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table S1.
The Oligos sequences of single guide RNA (sgRNA).
Additional file 2: Fig. S1.
Schematic representation of small molecule inhibitors (SMIs) targeting Ca2+ influx channels used in this study. Fig. S2. Small molecule inhibition of Ca2+ influx channels do not affect cell viability of multiple myeloma (MM) cells. Fig. S3. Small molecule inhibition of Ca2+ influx channels do not affect cell cycle and apoptosis of MM cells. Fig. S4. Small molecule inhibition of Ca2+ influx channels dose-dependently inhibits MM cell motility. Fig. S5. Ca2+ influx channels sgRNA decreases the protein levels of TRPM7, ORAI1, and STIM1, respectively, in RPMI8226 cells. Fig. S6. Knockdown of TRPM7, ORAI1, and STIM1 do not affect cell viability, cell cycle, and apoptosis of human MM-derived RPMI8226 cells. Fig. S7. Expression of Ca2+ influx channels are not affected by increasing levels of O-GlcNAc modification. Fig. S8. Hyper-O-GlcNAcylation by PugNAc treatment inhibits MM cell motility. Fig. S9. Hyper-O-GlcNAcylation by thiamet G treatment inhibits MM cell motility. Fig. S10. Hyper-O-GlcNAcylation by sgRNA against MGEA5 in the CRISPR/Cas9 system inhibit MM cell motility. Fig. S11. Hyper-O-GlcNAcylation by OGA inhibition does not affect cell viability, cell cycle, and apoptosis of RPMI8226 cells. Fig. S12. Knockdown of OGA does not affect cell viability, cell cycle, and apoptosis of human MM-derived RPMI8226 cells. Fig. S13. Elevated O-GlcNAcylation similarly reduces the migration and invasion of human MM-derived NCI-H929 cells. Fig. S14. O-GlcNAcylation acts as a down-stream of Ca2+ influx channels. Fig. S15. Inhibition of Ca2+ influx channels and OGA knockdown impair MM cell dissemination in vivo. Fig. S16. Inhibition of Ca2+ influx channels and OGA knockdown inhibit MM spreading to BM. Fig. S17. TRPM7 depletion inhibits MM cell dissemination in vivo. Fig. S18. OGA depletion inhibits MM cell dissemination in vivo. Fig. S19. Elevated O-GlcNAcylation does not significantly associate with integrin β1 (ITGB1), β3 (ITGB3), α5 (ITGA5) and αV (ITGAV), E-cadherin (E-cad), N-cadherin (Ncad), and β-catenin (B-cat). Fig. S20. Knockdown of ITGA4 and ITGB7 does not affect cell viability, proliferation and apoptosis of human MM-derived RPMI8226 cells. Fig. S21. Matrix metalloproteinase (MMP) activity upon an inhibition of Ca2+ influx channels or induction of O-GlcNAcylation. Fig. S22. Upregulation of mRNA expression of Ca2+ influx channels is associated with advanced MM clinical stages. Fig. S23. mRNA expression of Ca2+ influx channels, O-GlcNAc cycling enzymes, and integrins were analyzed using microarray data available on Gene Expression Omnibus (GEO) under accession number (GSE5900 and GSE2658).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Samart, P., Luanpitpong, S., Rojanasakul, Y. et al. O-GlcNAcylation homeostasis controlled by calcium influx channels regulates multiple myeloma dissemination. J Exp Clin Cancer Res 40, 100 (2021). https://doi.org/10.1186/s13046-021-01876-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-01876-z