
Abstract
Pancreatic cancer is one of the most malignant tumors worldwide, and pancreatic ductal adenocarcinoma is the most common type. In pancreatic cancer, glycolysis is the primary way energy is produced to maintain the proliferation, invasion, migration, and metastasis of cancer cells, even under normoxia. However, the potential molecular mechanism is still unknown. From this perspective, this review mainly aimed to summarize the current reasonable interpretation of aerobic glycolysis in pancreatic cancer and some of the newest methods for the detection and treatment of pancreatic cancer. More specifically, we reported some biochemical parameters, such as newly developed enzymes and transporters, and further explored their potential as diagnostic biomarkers and therapeutic targets.
Similar content being viewed by others


Background
Pancreatic cancer is an extremely aggressive malignant tumor associated with age. It is the fourth leading cause of mortality among all cancers in the US. It is predicted that pancreatic ductal adenocarcinoma (PDAC) will become the second leading cause of cancer-related death among all cancers [1, 2], and morbidity peaks by the age of 80 years old. PDAC is the primary exocrine pancreatic cancer and the most common type of pancreatic cancer, accounting for almost 90% of cases. Mucinous tumors, which are the second most common type of pancreatic cancer, account for less than 10% of cases [1, 2]. Because the majority of pancreatic cancer cases are PDAC, this article mainly discusses glycometabolic rearrangements in PDAC. PDAC classification based on genetic alterations is commonly discussed right now, and researchers count on these subdivided groups, which could provide prognostic measurements for the detection and treatments of PDAC. There are several methods for molecular classification, such as relying on single-genetic marker classification, genomic aberrations, and transcriptomic subtypes. Single-genetic marker classification aims to identify a single gene mutation, and these mutations have either a sufficient rate of occurrence or relate to beneficial treatment decisions [3]. PDAC can be divided into four subgroups according to genomic aberrations: stable genomes (< 50 structural variants per genome), scattered genomes (50–200 structural variants per genome), locally rearranged genomes (> 200 structural variants clustered on < 3 chromosomes), and unstable genomes (> 200 structural variants across the genome); these classifications can be divided into two groups: locally rearranged ones and unstable ones [4]. Transcriptomic subtypes enable unbiased classification and can demonstrate the preferential expression of genes. Based on a multiplatform method including transcriptomic profiles (mRNA and microRNA), whole-genome sequencing, and proteomics studies, a novel clinical subgroup was proposed in 2016. A total of 456 resected PDAC specimens were divided into 4 gene expression-based subgroups: squamous, pancreatic progenitor, immunogenic and aberrantly differentiated endocrine exocrine (ADEX) [5]. Nevertheless, in the following year, a minor mistake was found in this novel method; another similar analysis of 150 resected PDAC samples suggested that the ADEX and immunogenic subgroups of this classification system were derived from nonneoplastic cell contamination [6, 7]. Recently, after analyzing the genomic profiles, transcriptomic profiles, and clinical data of 325 PDAC patients, researchers identified four clinical subgroups: quiescent, glycolytic, cholesterogenic, and mixed [8]. This specific classification system should be explored and researched further because it indicates different clinical outcomes correlated with specific molecular mechanisms.
PDAC is associated with inherited mutations in many genes, including KRAS, TP53, HIF-1A, PIK3CA, EGFR and other mutated genes [9,10,11]. However, the most common driver mutations are in KRAS and TP53, contributing to more than 90 and 50% of cases, respectively [12]. Mutant genes modulate the expression and behaviors of PDAC, especially those related to metabolism, which is called metabolic reprograming (or rearrangements). KRAS stimulates glucose uptake by increasing glucose transporter levels on the cancer cell membrane, which promotes glucose uptake and further increases glycolysis in PDAC [13]; p53, the gene product of TP53, was demonstrated to increase glycolysis mediated by TP53-induced glycolysis and apoptosis regulator (TIGAR) [14]. Under hypoxic conditions, KRAS mutations occur in PDAC cells, which stabilize HIF1A and HIF2A to regulate pH and glycolysis [15]. The glycometabolic rearrangements occurring in PDAC are the main way energy is produced in PDAC and have been shown to be closely connected with the tumorigenesis of PDAC. Because of the harsh microenvironment and inherited changes of PDAC, PDAC strongly tends to generate energy through aerobic glycolysis, which is also known as the Warburg effect [16, 17].
.The microenvironment is a cellular environment where tumors can exist, grow and invade, and tumors can interact with the surrounding microenvironment [18]. Environmental factors also affect the emergence of pancreatic cancer, and 10% of PDAC patients have a relevant inherited predisposition [1, 19,20,21,22]. The dense stroma of pancreatic cancer consists of acellular (fibrin, collagen, fibronectin, and hyaluronan) and cellular (endothelial cells, cancer-associated fibroblasts, nerve cells, pancreatic satellite cells, and immune cells) components that occupy over half the size of the tumor mass. PDAC has an impenetrable desmoplastic stroma and hypovascular microenvironment. Due to the rigid stroma, the vessels are compressed, leading to low perfusion and hypervascularity [23, 24]. This harsh microenvironment provides PDAC with a unique hypoxic and nutrient-deprived environment, which requires PDAC to utilize glucose in an oxygen-deficient manner.
Patients with PDAC are generally diagnosed in an advanced stage because they usually do not present any noticeable clinical symptoms in early phases. Thus, approximately 80% of patients with a confirmed diagnosis of PDAC die within a year [22] (Fig. 1). In regard to diagnostic methods, tumor biomarkers exhibit unsatisfactory specificity and sensitivity [26], and computed tomography (CT) examination displays limitations, such as a low resolution for small lesions [20, 22]. Although pathological examination is the gold standard for PDAC diagnosis, its invasiveness restricts its application. Thus, the development of a precise strategy to detect PDAC in an early stage, which will benefit patient prognosis, is urgently needed. Moreover, conventional treatments such as chemotherapy and radiotherapy produce disappointing results, and the indication for surgery, which is the only method to achieve a clinical cure, is quite restricted [27]. Thus, we propose seeking other effective means for PDAC therapy. In conclusion, this article mainly discusses interactions between glycometabolic rearrangements, mainly glycolysis, in PDAC and characterizes factors for early detection of pancreatic cancer as well as therapeutic methods for achieving better clinical outcomes.

Pancreatic cancer is the most dangerous cancer in the world. The most common form of pancreatic cancer is PDAC, the five-year survival rate of which is 8%. It is difficult to detect PDAC because of the anatomically inaccessible location of the pancreas and difficult to cure PDAC because it is chemo- and radioresistant. Surgery is the only method to achieve a complete cure. Promisingly, olaparib, an inhibitor of PARP, is the newest standard for BRCA1/2-mutant pancreatic cancer patients [25].
Glycometabolic rearrangements in pancreatic cancer
Glycolysis
Carbohydrates are the main ingredient in the daily diet, and cancer applies aerobic glycolysis as a carbon supplement [28, 29]. Glucose is transferred through the cell membrane into the cytoplasm by glucose transporters (GLUTs), including GLUT1, GLUT2, GLUT3, GLUT4, and GLUT5 [30]. GLUT1 and GLUT3 are extensively distributed in all cells as primary transporters to transfer glucose into the cytoplasm [31]. However, most GLUT2 exists in hepatocytes and pancreatic β-cells, and GLUT4 mainly exists in adipocytes and cardiomyocytes, while GLUT5 is distributed in the epithelial cells of the small intestine. Interestingly, GLUT1 is commonly overexpressed in PDAC cells, accompanied by cellular invasiveness induced by the upregulation of matrix metalloproteinase-2 (MMP-2) expression via p53 mutation [32, 33]. Several studies have indicated that MMP-2 overexpression is likely modulated by GLUT1 via the PI3K/AKT and JNK/MEKKI pathways. GLUT1 is transactivated by KRAS, cMyc, and HIF-1α but inhibited by p53. Furthermore, GLUT1 is negatively associated with the expression of PTEN in thyroid cancer cells [31, 32]. Similarly, GLUT3 is activated through the IKK/NF-KB axis and inhibited by p53. Due to the correlation between the overexpression of GLUTs and rising uptake of glucose, glucose can be utilized for positron emission tomography (PET) [32, 33]. Glucose transferred into the cytoplasm by a GLUT is catalyzed by hexokinase (HK) and converted into glucose-6-phosphate. Then, phosphohexose isomerase (PGI) converts glucose-6-phosphate into fructose-6-phosphate (F-6-P). F-6-P is catalyzed by phosphofructokinase (PFK) to generate fructose-1,6-biphosphate (F-1,6-BP) [34]. F-1,6-BP is split into 3-phosphoglyceraldehyde (GA3P) and dihydroxy-acetone phosphate (DHAP). Then, the latter phosphotriose is transformed into the former by triose phosphate isomerase (TPI), and 3-phosphoglyceraldehyde is oxidized to 1,3-diphosphoglyceric acid (1,3-DPG) by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [35] and then catalyzed into 3-phosphoglycerate (3-PG) by phosphoglycerate kinase. 3-PG is converted into 2-PG by phosphoglycerate mutase (PGAM) [36], after which it is dehydrated to form phosphoenolpyruvate (PEP), which is catalyzed by enolase [37]. Following this step, pyruvate kinase catalyzes the transformation of PEP into pyruvate and adenosine triphosphate (ATP) [38]. Then, lactate dehydrogenase (LDH) catalyzes pyruvate conversion into L-lactate, which is the last step of glycolysis [30] (Fig. 2). Glycolysis is the primary metabolic rearrangement in PDAC cells under aerobic conditions, according to Otto Warburg [39].

Metabolic reaction in PDAC cells, especially aerobic glycolysis in pancreatic cancer cells. Glycolysis is the main form of energy production in pancreatic cancer, even under normoxia. However, there are also some cancer cells that produce energy through oxidative phosphorylation. GLUT: glucose transporter; G-6-P: glucose-6-phosphate; F-6-P: fructose-6-phosphate; F-1,6-BP: fructose-1,6-biphosphate. 1: Hexokinase; 2: phosphohexose isomerase; 3: phosphofructokinase; 4: aldolase; 5: triose phosphate isomerase; 6: glyceraldehyde 3-phosphate dehydrogenase; 7: phosphoglycerate kinase; 8: phosphoglycerate mutase; 9: enolase; 10: pyruvate kinase 11: lactate dehydrogenase
Glycolysis and the microenvironment
Hypoxic microenvironment
Aerobic glycolysis means that PDAC cells produce ATP even under aerobic conditions, which is called the Warburg effect, because this process avoids generating reactive oxygen species (ROS). It is a faster way to supply the needs for the biosynthesis of PDAC cells during proliferation, invasion, migration, and metastasis [28]. PDAC as a malignant tumor occurs with universal somatic evolution, which resembles Darwinian processes. Cells that are more suitable for the microenvironment survive in competition, whereas inadaptable cells gradually become extinct. Therefore, what precisely is meant by ‘suitable’? [40] Premalignant PDAC lesions are characterized as the hypovascular type caused by the desmoplastic stroma, and the capillaries in the extracellular matrix (ECM) are basically nonfunctional [41, 42]. In addition, the microcapillary in premalignant lesions is separate from the lesion, such as in carcinoma in situ, and the blood supply, such as a branch of capillary, is encircled by the vascular stroma and separated from the lesion by the basement membrane [40]. The microcapillaries are compressed, making it difficult for them to retract and transport substances. Therefore nutrient substances, such as glucose and oxygen, cannot be transported into tumor tissue directly but instead diffuse from the vessels slowly through the membrane and stroma into tumor cells [43]. This process results in solid tumors that are poorly oxygenated and hypoxic, which might also occur in tumors as a result of fluctuating blood flow [40, 42, 44]. Therefore, the special hypoxic microenvironment facilitates metabolic rearrangements in cancers to make the best use of the available oxygen when the oxygen level is quite low, such as in PDAC [45] (Fig. 3).

The microenvironment of pancreatic cancer. The stroma consists of cellular components (including immune cells, PSCs, CAFs and cancer cells) and noncellular components (hyaluronic acid, collagen, capillaries and fibronectin). This compact tissue lacking feeding vessels to provide nutrition blocks the diffusion of oxygen and transportation of glucose. In this situation, cells away from the vessels start to convert into glycolytic types. The dense stroma also contributes to chemo/radiotherapy resistance to drugs and resistance to T cells. There is a heated debate on the concrete function of the dense stroma: does it provide more benefits than disadvantages in PDAC? It is worth expending more efforts to unravel this mystery
Organisms need oxygen to sustain energy production. When oxygen is lacking, it causes immense stress to cells. Hypoxia-inducible factor (HIF) has been widely found to be expressed at elevated levels under hypoxia to match the oxygen supply [46,47,48]. HIF-1α is unstable under normoxic conditions due to immediate proteasomal degradation [49]. However, under hypoxia, HIF-1α and HIF-2α are both stable [50, 51]. HIF-1α is extensively expressed in all cells; however, HIF-2α is restricted to only several cell types, including the vascular endothelium, hepatocytes, and kidney epithelial cells [52]. HIF-1α is transported into the nucleus and binds to HIF-1β (heterodimeric counterpart), integrating with the coactivators CBP and p300 to generate activated HIF-1 combined with hypoxia-responsive elements as a complex to regulate hypoxia-related gene expression [53, 54]. Additionally, the overexpression of HIF-1α was proven to be connected with a poor prognosis in several cancers in a study [55]. The authors discovered that HIF-1α regulates glucose metabolism-related rearrangements in PDAC and that the expression of HIF-1α in tumor tissue is higher than that in surrounding normal tissues even under normoxic conditions [55]. Upregulation of HIF-1α expression has been associated with relatively early relapse and shortened overall survival (OS) [56, 57]. The poor clinical outcomes associated with HIF-1α may be related to epithelial-to-mesenchymal transition (EMT), producing essential effects on the invasion and metastasis of PDAC. EMT is a transition from an epithelial phenotype to a mesenchymal phenotype. This process is accompanied by the loss of epithelial biomarkers, such as E-cadherin, desmoplankins, claudins, and cytokeratins, and the gain of mesenchymal biomarkers, such as N-cadherin, vimentin, and fibronectin. E-cadherin, as a glycoprotein, can boost adhesion among cells and maintain cytoskeletal organization, and loss of E-cadherin leads to decreased cell adhesiveness and promotes EMT and metastasis [58]. Together with increasing biomarker levels in EMT, transcriptional repressors of E-cadherin are also highly expressed, such as zinc-finger E-box binding homeobox (ZEB), SNAIL1 (SNAIL), SNAIL2 (SLUG), TWIST, and FOXC2, which are regulated by the hypoxic status to suppress E-cadherin expression for EMT initiation [59]. The process of EMT involves several pathways actually modulated by HIFs under hypoxia. First, HIF-1α mediates the TGF-β1-induced pathway, synergistically activating the transcription factors SNAIL, ZEB, and TWIST in a Smad-dependent manner, which leads to suppression of E-cadherin and regulation of TGF-β target genes [60]. Another pathway involved in the hypoxia-EMT process is the Notch signaling pathway. The Notch pathway enhances SNAIL expression in cooperation with HIF-1α under hypoxia via two synergistic manners: directly upregulating SNAIL expression through intracellular Notch binding to the SNAIL promoter and indirectly upregulating SNAIL expression through intracellular Notch recruitment to the LOX promoter to enhance LOX and SNAIL production [61]. Studies have suggested that the PI3K/AKT pathway is also involved in the EMT process related to HIF-1α under hypoxia. In hepatocellular carcinoma, activated HIF-1α is indicated to be a downstream regulator of the PI3K/AKT pathway and is accompanied by differences in EMT biomarkers [62]. The other signaling pathway related to hypoxia-EMT is the Wnt/β-catenin signaling cascade. HIF-2α induces the Wnt/β-catenin pathway in PDAC, which then mediates the activation of SNAIL, inhibiting E-cadherin production to induce the EMT process [63]. Nuclear factor-κB (NF-κB) is an omnipresent transcription factor in cancer cells involved in many cellular actions, including EMT. NF-κB activates the transcriptional repressors TWIST and SNAIL in a HIF-dependent manner, which inhibits the expression of E-cadherin and increases the expression of N-cadherin and vimentin, indicating the activation of EMT [51, 64]. Recently, some studies have established the importance of the hypoxic microenvironment and EMT; immune and inflammatory cells and their secreted factors in the microenvironment have tight relationships with the formation of EMT. For instance, tumor necrosis factor-α (TNF-α) secreted by tumor-associated macrophages induces activation of the SNAIL promoter and promotes the EMT process [65, 66].
Overexpressed HIF-1α fuels the expression and activity of GLUTs, HK, PFK-L, ALD-A, GAPDH, PGAM-B, ENO-α, and PKM-2, which increase glycolysis and glucose uptake in PDAC [67, 68]. In a hypoxic microenvironment, mutant KRAS stabilizes HIF1A and HIF2A to augment the expression of carbonic anhydrase 9 to regulate pH and glycolysis in PDAC [15]. Moreover, transforming growth factor beta-induced (TGFBI) boosts the focal adhesion kinase (FAK) signaling pathway by binding to the integrin αVβ5, a cell membrane protein that stabilizes HIF-1α and then facilitates glycolysis in PDAC [69]. Recently, chromatin immunoprecipitation assays were applied to detect the occupancy of HIF-1α on the CTPS1 and TKT gene promoters. In human PDAC, CA IX (a HIF-1α activity symbol) was colocalized with TKT and CTPS. HIF-1α stabilized by MUC1, a highly expressed transmembrane protein, could modulate the expression of TKT, CTPS1, and other genes related to glycolysis, leading to gemcitabine and FOLFIRINOX resistance and increasing glycolysis in PDAC [70]. After knocking down MUC1 expression, reductions in HIF-1α protein expression, glucose uptake, and lactate release were observed [71]. Another study demonstrated that MUC1 facilitated HIF-1α translocation into the nucleus to act as a transcription factor for genes responsible for PDAC proliferation and invasion [72]. Moreover, the colocalization of the HIF-1α protein and VEGF mRNA identified via immunohistochemical analysis suggested that HIF-1α mediates the transcription of VEGF to induce angiogenesis in PDAC [73]. On the one hand, receptor for advanced glycation end products (RAGE) binding to KRAS fuels the activation of HIF-1α and promotes tumor development and glycolysis in PDAC under hypoxia. On the other hand, hypoxia induces HIF-1α-independent RAGE expression, which in turn activates KRAS signaling pathways (RAF-MEK-ERK and PI3K-AKT), stabilizing and activating HIF-1α [74]. Furthermore, the expression of miR-125a was shown to be significantly increased when HIF-1α expression was knocked down in the PANC-1 cell line, and HIF-1α was found to negatively regulate miR-125a, which plays vital roles in apoptosis, invasion, and metabolic rearrangements [75]. The mutant gene profiles in addition to hypoxic and hypovascular conditions, especially with the regulation of HIF-1α, contribute to the distinctive metabolic rearrangements related to aerobic glycolysis through multiple intersecting molecular pathways. Interestingly, upregulated glycolysis in PDAC can promote EMT phenotypes in PDAC cells by maintaining low ROS levels [76], which suggests a direction for inhibiting PDAC metastasis.
Given the increasing experimental information available, the stroma and tumor microenvironment (TME) are now considered to exert significant effects on PDAC metabolic rearrangements, carcinogenesis, metastasis, and resistance to radio/chemotherapy, suggesting a poor prognosis [24, 41]. Hypoxia and the hypovascular microenvironment cause tumor cell glycolysis and hinder immune cell infiltration and drug penetration, leading to immunosuppression and chemoresistance [77]. In this situation, the stiff stroma is also quite difficult for oxygen and glucose to pass through [78]. Recently, it was demonstrated that SerpinB2, which regulates CAF interactions and engagement with collagen in the matrix and is required for normal collagen remodeling associated with local invasion, is lost in pancreatic cancer [41]. However, there is a heated dispute over the contradictory functions of the dense stroma in PDAC carcinogenesis. Depleting the stroma in PDAC mice by degrading HA with enzymes or an inhibitor of Sonic Hedgehog (IPI926) restrains the development of PDAC [79, 80]. However, some clinical data have presented different conclusions, finding that the stiff stroma restrains the development of pancreatic cancer rather than facilitates progression due to the limited space [81]. The concrete function and molecular mechanisms of the stiff stroma in PDAC are still unclear, but the stroma is tightly correlated with aerobic glycolysis.
Pancreatic stellate cells and the stroma
Glycolysis emerges under microenvironmental selection [44], and it can produce energy for proliferation, invasion, migration, and even metastasis in the hypoxic state regulated by HIF-1α [70]. However, this process is quite deficient, accounting for only 31% of aerobic respiration [82]. Interestingly, with further research, a new theory called the ‘reverse Warburg effect’ was recently developed by studying metastatic breast cancer, which claims that metabolic reprogramming occurs in cancer-associated fibroblasts (CAFs), which also undergo aerobic glycolysis [83]. Nonetheless, anabolic products, such as pyruvate and L-lactate, can be transferred into cancer cells through mono-carboxylate transporters 4 (MCT4) and mono-carboxylate transporters 1 (MCT1) to the mitochondria for oxidative phosphorylation [84, 85]. L-lactate can be converted into pyruvate by LDHB in cancer cells [84]. ROS generated by the mitochondria in cancer cells diffuse into CAFs, inducing an increase in glycolysis regulated by upregulated HIF-1α expression [23]. This process is a more efficient way for tumors to produce energy than aerobic glycolysis [82]. In summary, the reverse Warburg and Warburg effects demonstrate the great plasticity and flexibility of cancer glycometabolic rearrangements. This inspired us to develop diverse therapeutic strategies, such as targeting MCT and suppressing glycolysis, addressing both pathways to inhibit the growth and invasion of PDAC [84, 85]. The reverse Warburg effect is considered to be similar to the Warburg effect because increased glycolysis occurs under normoxia in tumors [85].
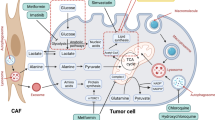
.Pancreatic stellate cells (PSCs), first isolated and cultured in 1998 [86], are the fundamental constituent of the TME and the most significant type of cancer-associated fibroblast (CAF) that induces the underlying molecular mechanisms of pancreatic cancer [87]. Under normal conditions, PSCs are localized in the basolateral aspect of pancreatic acinar cells as quiescent pancreatic stellate cells (qPSCs), generating ECM proteins and relevant enzymes [88]. They are reprogrammed into activated pancreatic satellite cells (aPSCs) in PDAC by environmental stress, molecular pathways, risk factors, and cellular factors, leading to the accumulation of abundant ECM proteins and a desmoplastic stroma [89]. In PDAC, to sustain biosynthesis for tumor growth and proliferation, PSCs and PDAC cells highly utilize glucose via the Warburg effect and reverse Warburg effect [23]. The complete interactions between PDAC cells and PSCs remain to be explored. However, some data indicate that Zeb1 in PSCs is responsible for KRAS activation in PSCs [90]. Coincidently, the oncogene KRAS mutates, causing enhanced glucose uptake and activating pivotal enzymes in glycolysis in both PDAC cells and PSCs. Furthermore, mutant KRAS signaling also promotes Sonic Hedgehog (SHH) secretion by PDAC cells, which then facilitates the expression of GAS6, IGF1, GM-CSF and other cytokines in PSCs [91]. This progress is responsible for feedback signaling via the IGF1R/AXL axis, which induces the phosphorylation of PI3K/AKT, increasing the mitochondrial respiration capability of PDAC cells. Additionally, inhibiting the ERK1/2 pathway in PSCs can suppress PDAC cell-stroma interactions and metabolic rearrangements [78]. Interestingly, exosomes secreted by PSCs containing miRNAs, mRNAs and metabolites, such as lactate and amino acids, also lead to the growth of PDAC cells through the exosomal transport of contents, such as miR-21, miR-1246 and miR-1290, which are responsible for the progression of PDAC [92,93,94]. Furthermore, the metabolites in exosomes can enter PDAC cells and provide supplements for oxidative phosphorylation to increase energy production in the mitochondria. MiR-210 can also modulate the interplay between PDAC cells and PSCs [95]. PSCs have effects on PDAC cells, such as metabolic rewiring, microenvironmental homeostasis maintenance, immunosuppression and immune evasion to regulate PDAC survival and metastasis (Fig. 4). These interactions between PDAC cells and PSCs offer researchers novel strategies to disrupt the pancreatic stroma to suppress the growth of PDAC and provide an accurate target for PDAC therapy.

Interaction between a PSC and PDAC cell. Reverse Warburg effect: PSCs utilize glucose to conduct glycolysis and produce pyruvate and lactate, which can enter PDAC cells and undergo mitochondrial respiration to produce energy. The metabolites of glycolysis in PSCs can be transported into pancreatic cancer cells for oxidative phosphorylation via MCT-4 and MCT-1. ROS generated by pancreatic cancer cells then spread into PSCs, inducing an increase in HIF-1α expression to further augment glycolysis in the PSCs. Exosomes secreted by PSCs containing gene products and lipids are internalized by pancreatic cancer cells. This interaction plays a vital role in the progression of pancreatic cancer and contributes to immune escape
Metabolites and oxidative phosphorylation
As continuous upregulation of aerobic glycolysis occurs in PDAC cells under hypoxia/anoxia [45], lactic acid generated from the process of glycolysis causes enormous extracellular and intracellular pH changes caused by hydrogen ions [40]. Normal and stromal tissues will be affected by the acidic microenvironment. Apoptosis and autophagy are induced by acidosis in healthy cells, and the structure of the stromal tissue is modified [96, 97]. However, PDAC cells can survive in this microenvironment due to some adaptive mutations, such as mutations in TP53 and KRAS. The acidic anaerobic microenvironment is a selective pressure that allows most malignant tumor cells to survive in competition for finite substrates and living areas with normal cells, as well as to migrate, invade and metastasize more easily [19, 28, 40, 98].
Interestingly, accumulating literature has demonstrated that oxidative phosphorylation (OXPHOS) is upregulated in some glycolytic cancers, including PDAC, which might be driven by the oncogene KRAS and the loss of LKB1 [99]. According to the reverse Warburg effect, PSCs and CAFs undergo glycolysis and transport metabolites into PDAC cells that are used for mitochondrial respiration to generate energy [84]. Meanwhile, pancreatic cancer stem cells rely on mitochondrial OXPHOS, which may be correlated with the suppression of MYC and the MYC/PGC-1α ratio, so mitochondrial agents and genetic therapy can easily target this phenotype [31]. Nonetheless, some studies have revealed that OXPHOS is not consistently suppressed. Instead, it can be reactivated under some conditions, such as activation of the PI3K-AKT-mTOR and LKB1-AMPK-p53 pathways [99]. As accumulating lactate is released into the microenvironment, glycolysis may be affected by the acidic microenvironment. When glycolysis is inhibited, glycolytic PDAC cells transport pyruvate into the mitochondria for OXPHOS, as the acidified microenvironment makes the reprogrammed microenvironment transport glucose and oxygen more efficiently and easily, which may also be associated with PDAC progression [100]. Intriguingly, mitochondrial OXPHOS accounts for more than 70% of the overall ATP production in cervical and breast cancer cell lines under normoxia, but it is reduced to less than 40% under hypoxic conditions [99]. However, the concrete relationship between OXPHOS and PDAC progression is still unclear, and the specific molecular mechanisms are under investigation. Although the mechanisms are unclear, some data verify that OXPHOS inhibitors can serve as promising therapeutic agents in PDAC [101]. Overall, proliferative PDAC acts as a glycolysis-dominant metabolic cancer with a possible alternative OXPHOS pathway being activated when glycolysis is inhibited. These two metabolic pathways render PDAC aggressive, allowing it to adapt to different microenvironmental conditions. Targeting or inhibiting OXPHOS should be thoroughly considered and verified because the plastic reprogramming of PDAC metabolism may switch OXPHOS cells into more aggressive glycolytic cells.
Immune cells in the cancer microenvironment, including T cells, B cells, natural killer cells, dendritic cells, neutrophils, and macrophages, should convert into glycolytic types to adapt to increasing biosynthetic needs for their anabolic functions and rapid growth in activated states [102,103,104,105,106]. The secretion of lactate and depletion of glucose by cancer cells can inhibit the functions of immune cells, and PDAC cells are able to escape the immune response [107, 108]. Mechanistically, MCTs exist in stromal cells, such as PSCs and immune cells, as mentioned before; to sustain ceaseless glycolysis in PDAC, the lactate and H+ released into the extracellular stroma are transported into immune cells, which induces intracellular acidification and inhibits glycolysis, ultimately resulting in functional damage to the immune cells [109]. Moreover, the lactate levels in PDAC are not as high as we hypothesized, possibly because the abundant CAFs and PSCs as well as immune cells utilize lactate via conversion into pyruvate for OXPHOS [110]. However, regulatory T cells with distinctly lower glycolytic characteristics increase resistance to the low-glucose, high-lactate tumor milieu and retain immunosuppressive functions that may be related to peripheral tolerance in the low-glucose, high-lactate tissue environment [111]. Based on the negative association between glycolysis and immune cells, a novel metabolism-tumor-stroma (MeTS) score was proposed to guide therapy selection for different metabolic classifications. There are four different type: MeTS1, OXPHOS tumors with a high T cell proportion, also called “hot”; MeTS2, reverse Warburg tumor cells with OXPHOS tumor cells and glycolytic stromal cells; MeTS3, a mixed classification having both OXPHOS and glycolytic cells; and MeTS4, glycolytic cancers with a low T cell proportion, also called “cold” [112]. However, not all solid tumors are identified as a specific glycolytic type because of tumor heterogeneity. Furthermore, even in a given tumor, not all cancer cells have the same metabolic identity. PDAC cells have significant glycolytic metabolism features with the reverse Warburg effect observed in PSCs and OXPHOS occurring in PDAC cells, so PDAC should be sorted as MeTS2 or MeTS3 depending on individual characteristics, according to the sorting method.
Clinical subgroups and glycolysis
New publications classify PDAC into 4 subtypes: quiescent, glycolytic, cholesterogenic, and mixed according to metabolic reprogramming features. Among the subtypes, the glycolytic subtype with high expression of glycolysis-related genes exhibits glycolysis-predominant metabolism with the worst clinical outcomes, and the cholesterogenic subtype with high expression of sterol biosynthesis- or cholesterogenesis-related genes promotes pyruvate entry into the mitochondria through mitochondrial pyruvate carriers (MPCs) to produce acetyl CoA for cholesterol biosynthesis and has the best clinical outcomes [113]. The quiescent and mixed subtypes with low and high expression of both metabolic pathways, respectively, show intermediate clinical outcomes. Mechanistically, the glycolytic subtype has a tight correlation with the oncogene KRAS and a weak correlation with MYC. Moreover, HIF1A and related genes, such as LDHA and SLC16A3, exhibit upregulated expression in the glycolytic subtype, which may indicate aggressive features, and the expression of the transcriptional regulator sterol regulatory element binding transcription factor 2 (SREBF2) is upregulated in the cholesterogenic subtype [8]. SREBF2 was proven to attenuate the activation of the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) promoter, which is upregulated in pancreatic intraepithelial neoplasia driven by KRASG12D and may be associated with relatively good outcomes [114].
Pivotal enzymes in glycolysis
Intriguingly, almost every enzyme in glycolysis plays a dual-function role during the progression of pancreatic cancer. The enzymes usually perform their catalytic activity in the cytoplasm and regulate transcription factors in the nucleus. The two processes are both crucial in the proliferation, invasion, migration, and metastasis of PDAC, and some new tactics for therapy and detection of PDAC may be developed from this information.
Glucose is converted into glucose-6-phosphate (G-6-P) by HK, which is the first step of glycolysis and rate limiting. HK has four isoenzymes, and glucokinase is the fourth type of HK that mainly exists in the liver and pancreatic β-cells [115]. The product, G-6-P, and long-chain fatty-acyl-coA inhibit the activation of HK. HK2 binds to the outer mitochondrial membrane through voltage-dependent anion channels (VDACs), and HK2 binding to the mitochondria increases the glycolytic capacity and promotes the immortalization of PDAC [27]. Interestingly, the upregulation of HK2 expression is mediated by HIF-1α, as mentioned before. Recently, PTEN/p53-deficient prostate cancer cells were found to exhibit increased expression of HK2, in which PTEN loss activated the AKT-mTORC1-4EBP1 axis to increase HK2 mRNA translation and loss of p53 inhibited miR-143 biogenesis to enhance HK2 mRNA stability. Moreover, miR-34a was shown to directly target HK1 and HK2, which suppressed glycolysis and promoted mitochondrial respiration [35]. Currently, KRAS4A, the unique palmitoylation-depalmitoylation cycle of the RAS isoform, is known to colocalize with HK1 on the outer mitochondrial membrane, and HK1, as a downstream effector of KRAS4A, can enhance glycolytic flux and cancer progression. Intriguingly, KRAS4A displays twice the effect of KRAS4B at the same gene expression level. When HK1 is silenced, however, the difference disappears. This indicates that KRAS4A directly binds to HK1 and acts downstream of KRAS4A to mediate transcription, but palmitoylated KRAS4A inhibits binding with HK1 [116]. Based on the mechanisms linking KRAS4A and HK, targeting KRAS4A to reduce HK levels should be thoroughly studied in clinical research. The expression of HK is linked to increasing glycolysis and an unfavorable clinical outcome in PDAC. 2-Deoxy-D-glucose (2-DG) is an analog of glucose that has recently been used as an HK inhibitor because it can be phosphorylated by HKs but not catalyzed further [117]. Another small molecule, 3-bromopyruvate (3-BP), and the novel agent methyl jasmonate (MJ) can suppress glycolysis and cancer growth in cancer patients [118, 119]. It was concluded that the lncRNA hox transcript antisense RNA (HOTAIR) is tightly linked with HK2 in PDAC patients. Furthermore, HOTAIR promotes the expression of HK2, and HOTAIR and HK2 are overexpressed in both the serum and tumor tissues of PDAC patients [120]. Therefore, their detection may indicate PDAC progression, and targeting HOTAIR to reduce HK2 expression may lead to the discovery of novel therapeutic strategies for PDAC.
Phosphofructokinase (PFK), which catalyzes the second committed step of glycolysis that involves the conversion of F-6-P into F-1,6-BP, is activated by fructose-2,6-biphosphate (F-2,6-BP), which is the strongest allosteric activator of all (including but not limited to ADP, AMP, F-1,6-BP, and F-2,6-BP), and is inhibited by high concentrations of ATP and citrate [28, 34, 121]. Interestingly, F-2,6-BP is catalyzed and hydrolyzed by a dual-function kinase that has two separate catalytic centers, phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFBs) [35]. More precisely, the level of PFKFB3 but not that of other PFKFBs, as a target of HIF-1α that displays the highest phosphofructo-2-kinase activity, is significantly elevated in aggressive cancers, such as PDAC, colon cancer, and breast carcinoma [27, 122]. Similarly, PFKFB4 was also found to have a homologous role in PDAC. PFKFB3 and PFKFB4 are both induced by HIF-1α, hypoxia and augmented expression of the GLUT and VEGF genes [122]. The glucagon-mediated activation of cAMP-dependent protein kinase (AMPK) is responsible for phosphorylation and inducing attenuation of the phosphofructo-2-kinase activity and augmentation of fructose-2,6-biphosphatase activity in PFKFB3 [123]. Additionally, PFKFB3 is located in both the cytoplasm and nucleus, suggesting that it correlates with the proliferation and invasion of PDAC via transcriptional regulatory effects in addition to glycolytic catalysis, similar to other enzymes (pyruvate kinase) in glycolysis. Nuclear localization of PFKFB3 is correlated with enhanced expression of vital cycle proteins, such as cyclin-dependent kinase-1, cyclin-dependent-25c, and cyclin D3, and reduced expression of the cell cycle inhibitor p27 [124]. These specific characteristics of PFKFB3 can be inhibited by 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (also known as 3-PO) to downregulate the synthesis of F-2,6-BP and reduce glycolytic flux [125]. It has been suggested that inhibiting the HIF-1α/PFKFB3/PFK-1 axis with metformin could suppress glycolysis and impair cancer growth in hepatocellular carcinoma, but whether similar effects can be achieved in other cancers is not yet clear [126]. Recently, miR-135 expression was shown to be elevated in PDAC as a result of glutamine deprivation. This upregulation is dependent on glutamine deficiency and mutant p53 activated by ROS, which directly promotes miR-135 expression. MiR-135 directly targets PFK expression and leads to decreased mRNA and protein levels, thereby suppressing aerobic glycolysis and increasing OXPHOS flux for attenuation of glutamine dependence to promote PDAC survival in a low-glutamine environment [127]. This information indicates that suppressing miR-135 in the context of enhanced PFK expression and increased glycolysis in PDAC still represses tumor growth. However, we hypothesize that the novel strategy of targeting miR-135 in combination with inhibitors of glutaminase (constructing a glutamine-deficient condition) may create a promising treatment for aggressive PDAC. Similarly, PFK-1 is O-GlcNAcylated at serine 529, which inhibits F-2,6-BP binding with PFK-1 to suppress PFK-1 activity under hypoxia. With reduced PFK-1 activity and glycolytic flux, cancer cells redirect glucose from glycolysis to the pentose phosphate pathway, which shuttles glucose into pathways for protein and DNA synthesis. Conversely, blocking PFK-1 glycosylation at serine 529 impairs cancer formation and proliferation in vivo and in vitro [128].
Notably, pyruvate kinase (PK) catalyzes the conversion of phosphoenolpyruvate into pyruvate and is the last rate-limiting step in glycolysis [35]. F-1,6-BP acts as an allosteric activator of pyruvate kinase. The allosteric inhibitors of pyruvate kinase include acetyl-CoA, ATP, and long-chain fatty acids. Posttranslational modification (PTM) of pyruvate kinase can also regulate its activity. Pyruvate kinase consists of four isoforms: PKLR encodes PKL and PKR; PKM encodes PKM-1 and PKM-2 via a relatively opposite swapped splicing mechanism. PKL is present in the kidneys and healthy liver, PKR exists in erythrocytes, PKM-1 is abundant in differentiated somatic cells, such as brain and muscle cells, and PKM-2 is expressed in fetal tissue and proliferating cells. A previous study showed that PKM-2 is the central kinase in cancer cells in a dimeric form and has no catalytic affinity for phosphoenolpyruvate [30]. In this situation, there are increasing levels of glycolytic intermediates that could be utilized as precursors for metabolic biosynthesis (such as synthesis pathways for lipids, amino acids, and nucleotides), which could perfectly meet the changing growth requirements in PDAC. Mechanistically, PKM-2 catalyzes a reaction in glycolysis and regulates gene transcription related to proliferation and invasion in PDAC, and it has been demonstrated that high polypyrimidine tract binding protein expression promotes PKM splicing to confer drug (gemcitabine) resistance to PDAC cells [38]. With the transformation of PKM-2 from a dimer to a tetramer, PKM-2 has a high affinity for phosphoenolpyruvate, producing a low concentration of metabolic precursors. This transition can be activated by thieno [3,2-b] pyrrole [3,2-d] pyridazinone (TEPP-46), which activates PKM-2 and impairs tumor growth and proliferation, revealing obvious antitumor activity [129]. In addition, PKM-2 induces gemcitabine resistance by downregulating p38 mitogen–activated protein kinase activity, and silencing PKM-2 strongly enhances gemcitabine-resistant cell apoptosis [130].
The lactate dehydrogenase (LDH)-mediated catalysis of pyruvate into lactate is significantly increased in PDAC cells. Previously, the expression of LDH-A (also called LDH-M and encoded by LDHA), which catalyzes the conversion of pyruvate into lactate during the last step in glycolysis, was found to be increased in PDAC and other aggressive tumors. However, the levels of LDH-B (also known as LDH-H and encoded by LDHB), which preferentially performs the reversible conversion of lactate and pyruvate, were also shown to be increased in PDAC [121, 131]. Nevertheless, there is still an argument to be made for targeting LDH-B, which suppresses the glycolytic subtype of PDAC cells and inhibits the proliferation, invasion, and migration of PDAC [132]. Moreover, a clinical study noted that LDH-A in the serum was associated with a poor prognosis after surgery, which might be correlated with a relatively low pH facilitating tumor relapse, as mentioned above (acidosis selects cells that can survive in that microenvironment and leads to mutations, and low pH stimulates tumor invasion and migration) [19, 23, 24]. Another study reported that Forkhead box protein M1 (FOXM1) promotes glycolysis, glucose consumption, and lactate production in PDAC by upregulating LDHA gene expression and augmenting LDHA activity. Unsurprisingly, the FOXM1/LDH-A pathway is also responsible for the progression and growth of PDAC [39]. Targeting LDH-A with siRNA and small molecule inhibitors leads to decreased tumor growth. The selective inhibitor 3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid (FX-11) impairs the progression of lymphoma and PDAC xenografts in combination with FK866, a synthetic NAD+ inhibitor [133]. Recently, FX-11 in combination with TEPP-46 was employed to treat PDAC cells and demonstrated augmented antitumor activity without obvious toxicity. A phase III trial evaluating the combined treatment has been completed, and the combination is a promising tactic for PDAC therapy [129]. It has been shown that acetylation of LDH-A at lysine 5 (K5) occurs. Acetylation inhibits LDH-A activity, reduces protein levels and promotes the degradation of LDH-A through chaperone-regulated autophagy, which is related to low-efficiency glycolysis by blocking the conversion of pyruvate into lactate, impairing the growth and progression of tumors. Intriguingly, LDH-A strongly accumulates in PDAC, which is accompanied by strongly decreased LDH-A K5 acetylation [134]. Resuming or accelerating the acetylation of K5 in LDH-A in PDAC may produce promising therapeutic results. Novel N-hydroxyindole-based (NHI) inhibitors targeting LDH-A impair proliferation, growth, and migration in PDAC. Moreover, when an NHI inhibitor is synergistically cultured with gemcitabine, it shows enhanced anticancer activity against PDAC [135]. Krüppel-like factor 4 (KLF4), a zinc-finger transcription factor, negatively regulates the transcriptional activity of LDH-A, thus impacting glycolysis and tumorigenesis in PDAC. The KLF4/LDH-A axis has close correlations with glycolysis and the progression of PDAC, as evidenced by both clinical data and experimental data [136].
Aldolase is an enzyme in aerobic glycolysis, and aldolase gene expression is significantly elevated in pancreatic cancer. Aldolase includes three isozymes, aldolase A, aldolase B, and aldolase C, encoded by three different genes, ALDOA, ALDOB, and ALDOC, respectively. Aldolase A is present in muscle tissues; aldolase B is extensively expressed in the kidneys, liver, stomach, and intestine; and aldolase C is expressed in the brain. Remarkably, aldolase A is highly relevant to various malignant cancers, including PDAC, and its expression is significantly increased in metastatic PDAC, in which it is used as a crucial indicator for detection. After treatment with transforming growth factor-β, PANC-1 cells exhibit increased ALDOA expression, leading to enhanced glycolysis. Furthermore, silencing aldolase A decreases aerobic glycolysis and ROS generation and inhibits the proliferation and metastasis (as indicated by EMT markers such as E-cadherin, N-cadherin, and vimentin) of PDAC in vivo and in vitro [137]. Similar to other enzymes involved in glycolysis, aldolase A is speculated to play dual-function roles in PDAC: catalysis in the cytoplasm and transcriptional regulation in the nucleus, which has been clarified in the pathogen Francisella [138]. A study showed that aldolase A could be inhibited by the hypoxic cytotoxin 3-[2-hydroxyethyl (methyl)amino]-2-quinoxalinecarbonitrile 1,4-dioxide (TX-2098). TX-2098 treatment suppressed the expression of HIF-1α, vascular endothelial cell growth factor, GLUT1, and aldolase A, leading to distinct antitumor efficacy in a xenograft PDAC model [139]. Moreover, naphthalene-2,6-diyl bisphosphate (ND1) is an active site substrate mimic that acts as an effective inhibitor of aldolase A, but it can be hydrolyzed easily. To address this shortcoming, a series of analogs of ND1 were extensively researched to identify covalent and noncovalent inhibitors. The most stable noncovalent inhibitor identified is NDB, which has two difluoromethylene insertions that do not impair the binding affinity. However, biochemical assays showed that methylene insertion weakened the effect of ND1 [140]. It is clear that the lncRNA DIO3OS promotes the growth of PDAC tumors with low expression of miR-122 and high expression of aldolase A [141]. Therefore, the DIO3OS/miR-122/aldolase A axis could be exploited as a therapeutic target in PDAC progression.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), another vital enzyme in glycolysis, exhibits increased expression in PDAC at both the mRNA and protein levels. Acetylation of lysine 254 augments the activity of GAPDH in glycolysis and promotes the proliferation of cancer cells [35]. Increased GAPDH mRNA and protein expression is associated with increased glycolytic flux in PDAC. It was demonstrated that GAPDH exerts its effects through DNA repair, autophagy, apoptosis, iron metabolism and transcriptional regulation in addition to catalysis in glycolysis, so this enzyme can be detected in the cytosol, membrane, nucleus, Golgi, and endoplasmic reticulum. Moreover, mutant p53 enhances glycolytic GAPDH activity and induces the formation of the SIRT1-GAPDH complex, which can stabilize cytosolic GAPDH for glycolysis and induce tumor growth and survival in PDAC [10]. The natural product koningic acid (KA) is an established selective inhibitor of GAPDH that exerts bioactivity mainly on tumors, with little effect on normal tissue. It inhibits the activity of GAPDH, leading to low glycolytic flux and a low cytotoxic response in highly glycolytic tumor cells [142]. Another inhibitor of GAPDH is iodoacetate; when PANC-1 cells are cultured with iodoacetate, low glycolytic flux is observed, accompanied by reduced cell survival. However, there is little alteration in the signaling machinery [143].
Phosphoglycerate kinase (PGK), as the first enzyme in glycolysis that produces ATP, includes two forms of PGK, the extensively expressed form PGK-1 and the testis-expressed form PGK-2. It has been demonstrated by immunohistochemistry that PGK-1 is highly expressed in PDAC [144]. Interestingly, PGK-1 has been proven to affect DNA duplication and repair in the mammalian nucleus. The mRNA and protein expression of PGK-1 is relevant to poor outcomes and a poor prognosis [145]. Accordingly, PTMs play vital roles in regulating the function of PGK-1 during tumorigenesis. More specifically, promoter methylation has been negatively associated with the mRNA expression of PGK-1; phosphorylation of the PGK-1 protein was associated with the clinical outcomes of PDAC patients [144]. Interestingly, another study reported that pyruvate dehydrogenase kinase 1 (PDHK-1) is phosphorylated by mitochondria-translocated PGK-1 (acting as a protein kinase), which activates PDHK-1 and augments glycolysis but inhibits OXPHOS [146]. Under glutamine deprivation and hypoxia in cancers, PGK-1 has an enhanced interaction with the acetyl-transferase ARD1, leading to acetylation of PGK-1 at lysine388. Subsequently, PGK-1 (acting as a protein kinase) phosphorylates Beclin1 without affecting the formation of Beclin1/VPS34/ATG14L, which causes an altered conformation and increased activity of VPS34 as well as augmented autophagy for tumor homeostasis [147]. However, HIF-1α upregulates the expression of PGK-1 but not its subcellular distribution under hypoxia. Increased expression of PGK-1 can result in the catalytic phosphorylation of troxacitabine, converting troxacitabine into the triphosphate form and causing increasing cytotoxicity to PANC-1 cells [148]. When PDAC is treated with troxacitabine, creating PGK-1 overexpression conditions to increase the cytotoxicity of troxacitabine may be a potent method for PDAC therapy. In a study, when FOXM1 expression was knocked down, PGK-1 levels were also decreased significantly, similar to the changes in LDHA levels mentioned before; however, the author did not specify the concrete mechanisms related to FOXM1 and PGK-1 [39]. Nuclear Factor of Activated T Cells 5 is highly expressed in PDAC patients and is associated with tumor progression by positively modulating PGK-1 [149]. SMAD4 and PTEN are distinctly silenced in PDAC. Loss of SMAD4 in PDAC is responsible for the high glycolytic capacity and tumor progression by upregulating PGK-1 expression, which has been shown to have dual roles in glycolytic catalysis and transcription factor activity in metastasis. Nuclear PGK-1 induces EMT by repressing E-cadherin expression, thus contributing to the migratory and metastatic potential. Cytoplasmic PGK-1 affects the metabolic type of PDAC by regulating the ratio of glycolysis and mitochondrial oxidative phosphorylation. Moreover, SMAD4-silenced PDAC patients can be classified by the subcellular localization of PGK-1 into nuclear PGK-1 positive or negative and high or low cytoplasmic PGK-1 to differentiate patient prognoses [150]. The new classification and interaction between SMAD4 and PGK-1 suggests some predictions and an instructive strategy for PDAC treatment, but these conclusions need to be thoroughly confirmed before a clinical application is proposed. In addition, PGK-1 is activated through autophosphorylation at tyrosine 324 (Y324), which enhances glycolysis and proliferation. PTEN exerts its protein phosphatase effect and dephosphorylates PGK-1, but the loss of PTEN found in glioblastoma is strongly associated with a poor prognosis [151]. Thus, regulating the PTMs of PGK-1 to block glycolysis and proliferation in tumors may provide novel methods for cancer therapy.
Phosphoglycerate mutase (PGAM) displays relatively high expression and activities in many malignant cancers, including PDAC. Via proteomic techniques, PGAM-1 is statistically indicated to be associated with a relatively poor prognosis in PDAC [152]. After knockdown of PGAM-1 expression, decreased glycolytic flux and reduced cell invasion were observed by detecting 3-PG and 2-PG levels and performing invasion experiments, respectively, but the association with proliferation was independent [36]. A study reported that PGAM-1, which is downstream of the PI3K/Akt/mTOR pathway, promoted EMT by stimulating the Wnt/β-catenin pathway, offering a potential application in PDAC through targeting a related signaling pathway to regulate the activity or expression of PGAM [153]. Moreover, acetylated PGAM-1 displays augmented enzymatic activity, and SIRT-1, an NAD+-dependent deacetylase, inhibits enzymatic activity by deacetylating PGAM [154]. Another NAD+-dependent deacetylase, SIRT-2, induces deacetylation at lysines 100/106/113/138 of PGAM-2 to inhibit enzymatic activity and repress tumor proliferation [155]. Interestingly, the phosphorylation of PGAM-1 at tyrosine 26 (Y26) was proven to activate enzymatic activity to alter glycolytic flux and induce migration [156]. Therefore, targeting PTMs, such as dephosphorylation and deacetylation, at some sites in PGAM-1 could attenuate PGAM-1 enzymatic activity and tumorigenesis for PDAC treatment. Recently, a newly developed and promising compound, KH3, was found to act as an allosteric suppressor of PGAM-1, showing satisfactory drug effects to downregulate glycolysis and cell proliferation with limited cytotoxicity to PDAC cells [152].
Enolase is the enzyme that generates PEP in glycolysis, and there are five forms of enolase in mammalian tissue composed of three immunological subunits, alpha, beta, and gamma. The alpha subunit exists in many tissues; the beta subunit is present only in skeletal and heart muscles, and the gamma subunit is localized in neurons [157]. Alpha enolase (ENO-1) levels are elevated in PDAC cells [37, 157, 158], which correlates with the migration and invasion of PDAC cells by inducing plasminogen activation into plasmin to degrade the compact ECM in a plasminogen-dependent process, and this process can be strongly blocked by using an adeno-associated virus/anti-ENO-1 antibody construct [159]. In addition, aberrant expression of ENO-1 is positively correlated with Ki67 and negatively correlated with p53 in PDAC, which indicates that ENO-1 exerts its effects on proliferation and metastasis by regulating the Ki67 and p53 pathways. In addition, increased ENO-1 expression is relevant to HIF-1α under hypoxia [37]. ENO-1 acts as the receptor of plasminogen in addition to a glycolytic enzyme, and silencing ENO-1 attenuates adhesion, invasion, and metastasis in PDAC [160]. Similarly, PTMs can also regulate ENO-1 activity, and phosphorylating ENO-1 peptide-MHC complexes at serine 419 can induce T cell signaling and autoantibody production in PDAC [158]. Accordingly, SF2312 produced by Micromonospora actinomycetes was first used as an antibiotic and is considered a specific inhibitor of enolase [161]. ENO-1 DNA vaccination has been established for PDAC, and additional treatments, such as ENO-1 inhibitor application, immune cell activation, or chemotherapy, in combination with ENO-1 vaccination could amplify the therapeutic response in PDAC [162].
Metabolic reprogramming interactions
The oxidative phenotype exists in PDAC, and cells with the glycolytic phenotype can also choose OXPHOS to sustain metabolic needs when glycolysis is inhibited in PDAC, as we mentioned before. Increased OXPHOS can be characterized by activated enzymes and increasing levels of products of the TCA cycle; moreover, mitochondrial dynamics and the mitochondrial membrane potential can be evaluated to confirm this phenotype [100]. The ROS level is the major factor that affects the tumorigenesis of PDAC, and low levels of ROS, which are produced by respiratory complex IV, activate vital redox signaling pathways; additionally, respiratory complexes I, II, and III produce superoxide, which may cause oxidative stress and mitochondrial dysfunction [163]. Mitochondrial respiration unifies several important bioenergetic pathways; it produces precursors for lipid, amino acid, and nucleotide biosynthesis and contributes to glutamine metabolism (Fig. 2). In addition to glucose, glutamine, a dispensable amino acid, is another nutrient fuel for cancer energy production and biosynthesis. Of note, glutamine deprivation is greatly associated with tumor suppression [164]. Glutamine is transferred into cells through alanine/serine/cysteine-preferring transporter 2 (ASCT2) in PDAC cells [165], and then it is converted into glutamate by glutaminase (correlated with EMT in hepatocellular carcinoma and encoded by GLS1 [166]) and metabolized to α-ketoglutarate as a carbon donor for the TCA cycle via amino acid transaminase or glutamate dehydrogenase, which is also accepted as anaplerosis. In addition, glutamine provides nitrogen for nucleotide synthesis and synthesis of other dispensable amino acids (such as arginine, asparagine, serine, alanine, aspartate, glycine, and cysteine) [167]. De novo lipogenesis is a prominent feature of PDAC cells and is essential for fatty acid biosynthesis. Lipid metabolism provides aggressive PDAC cells with enough lipids for the cell membrane, energy production, and second messenger and signaling molecule generation. Another intermediate in the TCA cycle, citrate, is transformed into acetyl-coenzyme A (CoA) via ATP-citrate lyase (ACLY) and then converted into malonyl-CoA, which is mediated by acetyl-CoA carboxylase (ACC) (encoded by ACACA or ACACB), finally producing fatty acids through fatty acid synthesis (FASN). The expression of ACLY, ACC, and FASN is augmented in some PDAC patients, who show a shortened survival time, chemoresistance, and a poor prognosis. Downregulating the activity and expression of these proteins reduces lipid generation while suppressing tumor proliferation and inhibiting tumorigenesis [168,169,170]. In hepatocellular carcinoma, it has been proven that inhibiting the AMPK-induced phosphorylation of ACC causes larger lesions and progression in hepatocellular carcinoma. The opposite results are observed when treatment with the ACC inhibitor ND-654, which imitates the effects of phosphorylating ACC, is applied [171]. Expression of FASN is mediated by transcriptional factor sterol regulatory element-binding protein 1c (SREBP1c), and downregulating PI3K and MAPK pathways can inhibit SREBP1c to decrease FASN transcription and suppress lipogenesis and PDAC progression. Furthermore, after FASN and SREBP1c expression was analyzed, SREBP1c was demonstrated to directly or coordinately regulate enzymes and FASN in lipogenesis [172]. The hexosamine biosynthetic pathway (HBP) is greatly interlinked with other metabolic pathways, such as glucose, lipid, nucleotide, and amino acid metabolism, in PDAC. The end product of the HBP, uridine diphosphate N-acetyl glucosamine (UDP-GlcNAc), is generated from 3 materials, glucose, glutamine, and glucosamine, and acts as a substrate for O-GlcNAcylation. The HBP and glycolysis share the first two steps from glucose to F-6-P generation, and then F-6-P and glutamine are converted into glucosamine-6-phosphate via glutamine fructose-6-phosphate amidotransferase (GFAT); meanwhile, glucosamine is converted into glucosamine-6-phosphate by GlcNAc kinase. Then, glucosamine-6-phosphate is catalyzed to UDP-GlcNAc with the assistance of intermediates including acetyl-CoA and UTP from de novo lipogenesis and nucleotide metabolism, respectively [173]. O-GlcNAcylation is a PTM on some proteins that leads to metastatic potential and aggressive reactions, and transcriptional regulation is widely disrupted (both O-GlcNAcase and O-GlcNAc transferase levels are increased) in PDAC to promote tumor growth [174]. The activated HBP is not only highly interconnected with other metabolic pathways but is also closely related to growth, PTMs, invasion, EMT, and aggressiveness in PDAC [173].
Clinical applications of glycolysis in pancreatic cancer
Detection
Recent detection approaches for PDAC include biomarker evaluations, imaging assays, and oncogene mutation evaluations, but none of these can act as an independent confirmatory diagnostic measurement. The anatomically inaccessible location of the pancreas leads to an incomplete diagnosis [29]. Some serum biomarkers, such as CA-125/CEA/CA19–9, are correlated with poor surgical outcomes and a poor prognosis [175,176,177]. However, conventional tumor markers, such as CA19–9 and CEA, lack sensitivity and specificity. However, traditional imaging examinations such as CT and magnetic resonance imaging (MRI) can only reflect the visual tumor size measured by a computer [178]. These measurements lack the foresight needed to predict the occurrence of PDAC and provide additional information related to malignancy. Thus, analysis of metabolic rearrangements in glycolysis could be considered a relatively precise diagnostic means for the detection of the metabolic phenotypes of pancreatic cancer to indicate malignancy. Specifically, this section summarizes alterations in enzymes and serum metabolites in glycolysis differing between PDAC patients and healthy people to explore if these parameters could be prospective measurements for PDAC (Table 1).
Augmented enzyme expression and activity indicate an increase in glycolysis, which also acts as a vital sign of PDAC [202]. These changes in proteins can be detected by mass spectrometry (MS), which identifies molecules by measuring the mass-to-charge ratio, and the instrument includes an ion source, a mass analyzer, a detector, and a data system [203]. Moreover, GLUTs accumulate in PDAC, so more glucose is conveyed into the cytoplasm through these transporters. Upregulation of GLUT expression is generally associated with a poor prognosis and can be detected by the MS technique [26, 204]. Similarly, this tool has been developed to analyze proteomics, including large-scale protein expression and PTMs in glucose metabolism. Dysfunction of the enzymes in glycolysis is usually induced by PTMs, which are involved in the formation, proliferation, and invasion of PDAC, such as acetylation, phosphorylation, O-methylation, and glycosylation, and these modifications can also be detected by LC-MS/MS. [205] However, not all molecules can be detected by MS, and biomarkers in glycolysis with a low abundance in PDAC, such as HIF-1α, cannot be detected by MS. [26].
Presently, many researchers have deemed metabolism to be a vital characteristic for detecting and treating pancreatic cancer, so the metabolic tumor burden (MTB), especially total lesion glycolysis (TLG), is a new parameter to discriminate pancreatic carcinoma from benign pancreatic diseases. In addition to discriminating PDAC, TLG can also predict clinical outcomes and prognosis because it is related to poor OS and recurrence-free survival (RFS) in PDAC [206]. Interestingly, these indexes, such as the MTB and metabolic tumor volume (MTV), are detectable by 18-fluoro-deoxyglucose positron emission tomography computed tomography (18FDG-PET/CT) [20], which uses 18FDG as a tracer because of the elevated uptake of glucose. Notably, 13C-labeled metabolites can translate into cancer cells for reprogramming metabolism, such as glycolysis, and the metabolic process can be detected by hyperpolarized magnetic resonance spectroscopy (MRS), which can be used to discover multiple metabolic processes via labeled metabolites of glucose, amino acids, lipids, and nucleotides in PDAC [29]. For example, 1-13C-labeled pyruvate was converted into lactate in a preclinical PDAC model and detected by hyperpolarized MRS to discriminate cancer cells from normal cells [207]. The safety of hyperpolarized MRS has been proven in clinical trials for prostate cancer [208]. In addition, a new study reported that the specific preoperative neutrophil-lymphocyte ratio in the serum was associated with OS after resection in PDAC patients [209].
.In summary, conventional imaging examinations such as CT and MRI can be used for surgical resectability and range; biomarkers in body fluids, such as the serum and urine, can be evaluated to monitor for PDAC; the expression of glycolytic enzymes and regulators and the concentrations of glycolytic metabolites in the serum can be used to predict prognosis and malignancy; PET and MRS can be used to evaluate the metabolic properties and metastasis of pancreatic cancer; and TLG and MTV can be used to predict prognosis and clinical outcomes. We believe that an integrated method together with genomic profiling of PDAC may provide a prognostic and personalized diagnosis for PDAC patients.
Therapy
Traditional treatment strategies include chemotherapy, radiation therapy, and molecular targeted therapy. However, surgery is the only possibly curative treatment [210]. After surgery, neoadjuvant chemoradiation is increasingly used to treat PDAC patients [211]. Furthermore, metabolic rearrangements can not only reflect characteristics and hallmarks for diagnosis and prognosis but also lead to the development of new therapeutic strategies as biomarkers for PDAC. This review summarizes inhibitors of glycolytic enzymes and regulators to explore whether they could be prospective treatments for PDAC (Table 2).
Recently, the fructo-1,6-bisphosphatase (FBP)-mediated rate-limiting step in gluconeogenesis has been reported to inhibit the Warburg effect and KRAS signaling. Furthermore, FBP-2 and FBP-1 were found to attenuate soft tissue sarcomas (STS) and breast cancer, respectively. In STS, the authors re-expressed FBP-2 through overexpression vectors and found that FBP-2 inhibited glycolysis in the cytoplasm and suppressed mitochondrial respiration, biogenesis and the tricarboxylic acid (TCA) cycle in the nucleus by inhibiting the function of the transcription factor c-Myc in the nucleus. In breast cancer, restoration of FBP-1 expression intensely inhibited glycolysis by catalyzing the function and silencing activity of HIF [34]. However, the exact role of FBP-2 in PDAC is still obscure; the precise mechanism remains unknown. In addition, targeted therapy aimed at common gene mutations consistently exhibits a lack of efficacy because the most commonly mutated genes in pancreatic cancer are known to be KRAS and TP53, which can hardly be made into drug targets [3]. Promisingly, AMG-510, the first inhibitor of KRAS in clinical development, caused a decline in KRASG12C tumor growth and augmented the efficacy of chemotherapy and targeted agents [213]. Moreover, in clinical data, approximately 8% of PDAC samples had germline and somatic mutations in the DNA damage repair genes BRCA2, PALB2, and ATM [6]. Inspiringly, a recent study noted that olaparib, a poly ADP-ribose polymerase (PARP) inhibitor, prolonged PFS among germline BRCA-mutant PDAC patients [214,215,216]. Olaparib has been approved by the FDA for metastatic pancreatic cancer, breast cancer and recurrent ovarian cancer with BRCA1/2 mutation [214]. Although olaparib exhibits a therapeutic effect, the application of olaparib is limited to patients with mutations in the DNA damage repair gene BRCA2, and further studies will be required to identify a detailed approach for treating PDAC.
Conclusion
In conclusion, we first discussed the cause and characteristics of glycolysis in PDAC, especially the microenvironment and metabolic subgroup. Next, we summarized the vital kinases in the glycolytic process. Notably, glycolysis in PDAC can be easily suppressed by inhibiting the activation of critical enzymes such as HK, PFK, and PK. These kinases catalyze the rate-limiting steps in glycolysis, so we analyzed diagnostic and therapeutic means focused on these enzymes to target PDAC. Moreover, we also presented the latest findings related to clinical therapy and the diagnosis of PDAC. However, the precise molecular mechanisms of glycolysis and PDAC are still unknown. Therefore, the processes of clinical applications and molecular mechanisms should also be explored to maximize the proportion of PDAC patients who will derive benefit.
Availability of data and materials
Data available upon request.
Abbreviations
- 1,3-DPG:
-
1,3-diphosphoglyceric acid
- 2-DG:
-
2-deoxy-D-glucose
- 3-BP:
-
3-bromopyruvate
- 3-PG:
-
3-phosphoglycerate
- 3-PO:
-
3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
- ACLY:
-
ATP-citrate lyase
- AMPK:
-
cAMP-dependent protein kinase
- aPSCs:
-
Activated pancreatic satellite cells
- ASCT2:
-
Alanine/serine/cysteine-preferring transporter 2
- ATP:
-
Adenosine triphosphate
- CAF:
-
Cancer-associated fibroblast
- CoA:
-
Acetyl-coenzyme A
- CT:
-
Computed tomography
- DHAP:
-
Dihydroxy-acetone phosphate
- ECM:
-
Extracellular matrix
- F-1,6-BP:
-
Fructose-1,6-biphosphate
- F-2,6-BP:
-
Fructose-2,6-biphosphate
- F-6-P:
-
Fructose-6-phosphate
- FASN:
-
Fatty acid synthesis
- FBP:
-
Fructo-1,6-bisphosphatase
- FOXM1:
-
Forkhead box protein M1
- FX-11:
-
3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid
- G-6-P:
-
Glucose-6-phosphate
- GA3P:
-
3-phosphoglyceraldehyde
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GFAT:
-
Glutamine fructose-6-phosphate amidotransferase
- GK:
-
Glucokinase
- GLUT:
-
Glucose transporter
- HBP:
-
Hexosamine biosynthetic pathway
- HK:
-
Hexokinase
- HMGCR:
-
3-hydroxy-3-methylglutaryl coenzyme A reductase
- HOTAIR:
-
Hox transcript antisense RNA
- KLF4:
-
Krüppel-like factor 4
- LDH:
-
Lactate dehydrogenase
- MCT:
-
Mono-carboxylate transporter
- MMP-2:
-
Metalloproteinase-2
- MRI:
-
Magnetic resonance imaging
- MRS:
-
Magnetic resonance spectroscopy
- MS:
-
Mass spectrometry
- MTB:
-
Metabolic tumor burden
- MTV:
-
Metabolic tumor volume
- OS:
-
Overall survival
- OXPHOS:
-
Oxidative phosphorylation
- PARP:
-
Poly ADP-ribose polymerase
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDHK:
-
Pyruvate dehydrogenase kinase
- PEP:
-
Phosphoenolpyruvate
- PET:
-
Positron emission tomography
- PFK:
-
Phosphofructokinase
- PFKFB:
-
Phosphofructo-2-kinase/fructose-2,6-biphosphatase
- PGAM:
-
Phosphoglycerate mutase
- PGI:
-
Phosphohexose isomerase
- PK:
-
Pyruvate kinase
- PSC:
-
Pancreatic stellate cell
- PTM:
-
Posttranslational modification
- qPSCs:
-
Quiescent pancreatic stellate cells
- RAGE:
-
Receptor for advanced glycation end products
- RFS:
-
Recurrence-free survival
- ROS:
-
Reactive oxygen species
- SREBF2:
-
Sterol regulatory element binding transcription factor 2
- SREBP:
-
Sterol regulatory element-binding protein
- STS:
-
Soft tissue sarcomas
- TCA:
-
Tricarboxylic acid
- TEPP-46:
-
Thieno [3,2-b] pyrrole [3,2-d]pyridazinone
- TIGAR:
-
TP53-induced glycolysis and apoptosis regulator
- TLG:
-
Total lesion glycolysis
- TME:
-
Tumor microenvironment
- TPI:
-
Triose phosphate isomerase
- UDP-GlcNAc:
-
Uridine diphosphate N-acetyl glucosamine
- VDAC:
-
Voltage-dependent anion channel
References
Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2(12):897–909.
Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer Epidemiol Biomark Prev. 2005;14(7):1766–73.
Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16(4):207–20.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185–203.e13.
Pihlak R, Weaver JMJ, Valle JW, McNamara MG. Advances in molecular profiling and categorisation of pancreatic adenocarcinoma and the implications for therapy. Cancers. 2018;10(1):17.
Karasinska JM, Topham JT, Kalloger SE, Jang GH, Denroche RE. Altered gene expression along the glycolysis-cholesterol synthesis Axis is associated with outcome in pancreatic cancer. Clin Cancer Res. 2020;26(1):135–46.
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science (New York, NY). 2009;325(5947):1555–9.
Butera G, Pacchiana R, Mullappilly N, Margiotta M, Bruno S, Conti P, et al. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 2018;1865(12):1914–23.
Sikdar N, Saha G, Dutta A, Ghosh S, Shrikhande SV, Banerjee S. Genetic alterations of Periampullary and pancreatic ductal adenocarcinoma: an overview. Curr Genomics. 2018;19(6):444–63.
Yang J, Ren B, Yang G, Wang H, Chen G, You L, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77(2):305–21.
Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39(2):91–100.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–20.
McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology. 2019;157(3):823–37.
Hu LP, Zhang XX, Jiang SH, Tao LY, Li Q, Zhu LL. Targeting Purinergic receptor P2Y2 prevents the growth of pancreatic ductal adenocarcinoma by inhibiting cancer cell glycolysis. Clin Cancer Res. 2019;25(4):1318–30.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–30.
Wang JJ, Lei KF, Han F. Tumor microenvironment: recent advances in various cancer treatments. Eur Rev Med Pharmacol Sci. 2018;22(12):3855–64.
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85.
Fox RG, Lytle NK, Jaquish DV, Park FD, Ito T, Bajaj J, et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534(7607):407–11.
Soreide K, Sund M. Epidemiological-molecular evidence of metabolic reprogramming on proliferation, autophagy and cell signaling in pancreas cancer. Cancer Lett. 2015;356(2 Pt A):281–8.
Zhang Q, Chen S, Zeng L, Chen Y, Lian G, Qian C, et al. New developments in the early diagnosis of pancreatic cancer. Expert Rev Gastroenterol Hepatol. 2017;11(2):149–56.
Fu Y, Liu S, Zeng S, Shen H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol Cancer. 2018;17(1):62.
Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: a complex relationship. Cancers. 2018;10(9):316.
Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–48.
Zhou W, Liotta LA, Petricoin EF. Cancer metabolism and mass spectrometry-based proteomics. Cancer Lett. 2015;356(2 Pt A):176–83.
Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. 2015;35(Suppl):S129–S50.
Bose S, Le A. Glucose Metab Cancer. 2018;1063:3–12.
Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31(1):5–19.
Satriano L, Lewinska M. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16(12):748–66.
Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22(4):590–605.
Ito H, Duxbury M, Zinner MJ, Ashley SW, Whang EE. Glucose transporter-1 gene expression is associated with pancreatic cancer invasiveness and MMP-2 activity. Surgery. 2004;136(3):548–56.
Reske SN, Grillenberger KG, Glatting G, Port M, Hildebrandt M, Gansauge F, et al. Overexpression of glucose transporter 1 and increased FDG uptake in pancreatic carcinoma. J Nuclear Med. 1997;38(9):1344–8.
Huangyang P, Li F, Lee P, Nissim I, Weljie AM, Mancuso A, et al. Fructose-1,6-Bisphosphatase 2 inhibits sarcoma progression by restraining mitochondrial biogenesis. Cell Metab. 2019;31:174–188.e7.
Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2015;73(2):377–92.
Liu X, Weng Y, Liu P, Sui Z, Zhou L, Huang Y, et al. Identification of PGAM1 as a putative therapeutic target for pancreatic ductal adenocarcinoma metastasis using quantitative proteomics. Onco Targets Ther. 2018;11:3345–57.
Sun L, Guo C, Cao J, Burnett J, Yang Z, Ran Y, et al. Over-expression of alpha-Enolase as a prognostic biomarker in patients with pancreatic cancer. Int J Med Sci. 2017;14(7):655–61.
Calabretta S, Bielli P, Passacantilli I, Pilozzi E, Fendrich V, Capurso G, et al. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene. 2016;35(16):2031–9.
Cui J, Shi M, Xie D, Wei D, Jia Z, Zheng S, et al. FOXM1 promotes the Warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin Cancer Res. 2014;20(10):2595–606.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9.
Harris NLE, Vennin C, Conway JRW, Vine KL, Pinese M, Cowley MJ, et al. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene. 2017;36(30):4288–98.
Ntellas P, Dadouli K, Perivoliotis K, Sogka E, Pentheroudakis G, Ioannou M, et al. Microvessel density and impact of angiogenesis on survival of resected pancreatic cancer patients: a systematic review and meta-analysis. Pancreas. 2019;48(2):233–41.
Hirabayashi K, Kurokawa S, Maruno A, Yamada M, Kawaguchi Y, Nakagohri T, et al. Sex differences in immunohistochemical expression and capillary density in pancreatic solid pseudopapillary neoplasm. Ann Diagn Pathol. 2015;19(2):45–9.
Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:295–321.
Shukla SK, Mulder SE, Singh PK. Hypoxia-mediated in vivo tumor glucose uptake measurement and analysis. Methods Mol Biol. 2018;1742:107–13.
Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304–8.
Påhlman S, Mohlin S. Hypoxia and hypoxia-inducible factors in neuroblastoma. Cell Tissue Res. 2018;372(2):269–75.
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309.
Dabral S, Muecke C, Valasarajan C, Schmoranzer M, Wietelmann A, Semenza GL, et al. A RASSF1A-HIF1α loop drives Warburg effect in cancer and pulmonary hypertension. Nat Commun. 2019;10(1):2130.
Franovic A, Holterman CE, Payette J, Lee S. Human cancers converge at the HIF-2alpha oncogenic axis. Proc Natl Acad Sci U S A. 2009;106(50):21306–11.
Tirpe AA, Gulei D, Ciortea SM. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci. 2019;20(24):6140.
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–75.
Chen J, Zhao S, Nakada K, Kuge Y, Tamaki N, Okada F, et al. Dominant-negative hypoxia-inducible factor-1α reduces Tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am J Pathol. 2003;162(4):1283–91.
Yin S, Kaluz S, Devi NS, Jabbar AA, de Noronha RG, Mun J, et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1α interaction with cofactors p300/CBP. Clin Cancer Res. 2012;18(24):6623–33.
Shibaji T, Nagao M, Ikeda N, Kanehiro H, Hisanaga M, Ko S, et al. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003;23(6c):4721–7.
Shi CY, Fan Y, Liu B, Lou WH. HIF1 contributes to hypoxia-induced pancreatic cancer cells invasion via promoting QSOX1 expression. Cell Physiol Biochem. 2013;32(3):561–8.
Zhao X, Gao S, Ren H, Sun W, Zhang H, Sun J, et al. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014;74(9):2455–64.
Gulei D, Magdo L, Jurj A, Raduly L, Cojocneanu-Petric R. The silent healer: miR-205-5p up-regulation inhibits epithelial to mesenchymal transition in colon cancer cells by indirectly up-regulating E-cadherin expression. Cell Death Dis. 2018;9(2):66.
Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172(7):973–81.
Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11(8):943–50.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105(17):6392–7.
Jiao M, Nan KJ. Activation of PI3 kinase/Akt/HIF-1α pathway contributes to hypoxia-induced epithelial-mesenchymal transition and chemoresistance in hepatocellular carcinoma. Int J Oncol. 2012;40(2):461–8.
Zhang Q, Lou Y, Zhang J, Fu Q, Wei T, Sun X, et al. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol Cancer. 2017;16(1):119.
Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of hypoxia-induced NF-kappaB. Mol Cell Biol. 2010;30(20):4901–21.
Dong R, Wang Q, He XL, Chu YK, Lu JG, Ma QJ. Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial-mesenchymal transition of MCF-7 cells. Brazilian J Med Biol Res= Revista brasileira de pesquisas medicas e biologicas. 2007;40(8):1071–8.
Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: a review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018;80:23–32.
Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–71.
Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009;9(9):1084–101.
Costanza B, Rademaker G, Tiamiou A, De Tullio P, Leenders J, Blomme A, et al. Transforming growth factor beta-induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int J Cancer. 2019;145(6):1570–84.
Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32(1):71–87 e7.
Sahraei M, Roy LD, Curry JM, Teresa TL, Nath S, Besmer D, et al. MUC1 regulates PDGFA expression during pancreatic cancer progression. Oncogene. 2012;31(47):4935–45.
Goudarzi H, Iizasa H, Furuhashi M, Nakazawa S, Nakane R, Liang S, et al. Enhancement of in vitro cell motility and invasiveness of human malignant pleural mesothelioma cells through the HIF-1alpha-MUC1 pathway. Cancer Lett. 2013;339(1):82–92.
Buchler P, Reber HA, Buchler M, Shrinkante S, Buchler MW, Friess H, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26(1):56–64.
Kang R, Hou W, Zhang Q, Chen R, Lee YJ, Bartlett DL, et al. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 2014;5:e1480.
Pan L, Zhou L, Yin W, Bai J, Liu R. miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int J Oncol. 2018;53(1):124–36.
Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen Q, et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J Cell Mol Med. 2017;21(9):2055–67.
DuFort CC, DelGiorno KE, Hingorani SR. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology. 2016;150(7):1545–57.e2.
Yan Z, Ohuchida K, Fei S, Zheng B, Guan W, Feng H, et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J Exp Clin Cancer Res. 2019;38(1):221.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (New York, NY). 2009;324(5933):1457–61.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47.
Tourmente M, Villar-Moya P, Rial E, Roldan ER. Differences in ATP generation via glycolysis and oxidative phosphorylation and relationships with sperm motility in mouse species. J Biol Chem. 2015;290(33):20613–26.
Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the "reverse Warburg effect" in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer's disease, and "neuron-glia metabolic coupling". Aging. 2010;2(4):185–99.
Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813–25.
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001.
Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–32.
Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–32.
Roy I, Boyle KA, Vonderhaar EP, Zimmerman NP, Gorse E, Mackinnon AC, et al. Cancer cell chemokines direct chemotaxis of activated stellate cells in pancreatic ductal adenocarcinoma. Lab Invest. 2017;97(3):302–17.
Koikawa K, Ohuchida K, Takesue S, Ando Y, Kibe S, Nakayama H, et al. Pancreatic stellate cells reorganize matrix components and lead pancreatic cancer invasion via the function of Endo180. Cancer Lett. 2018;412:143–54.
Sangrador I, Molero X, Campbell F, Franch-Exposito S, Rovira-Rigau M, Samper E, et al. Zeb1 in stromal Myofibroblasts promotes Kras-driven development of pancreatic cancer. Cancer Res. 2018;78(10):2624–37.
Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell. 2016;165(4):910–20.
Takikawa T, Masamune A, Yoshida N, Hamada S, Kogure T, Shimosegawa T. Exosomes derived from pancreatic stellate cells: MicroRNA signature and effects on pancreatic cancer cells. Pancreas. 2017;46(1):19–27.
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250.
Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36(13):1770–8.
Takikawa T, Masamune A, Hamada S, Nakano E, Yoshida N, Shimosegawa T. miR-210 regulates the interaction between pancreatic cancer cells and stellate cells. Biochem Biophys Res Commun. 2013;437(3):433–9.
Alikhani A, Gharooni M, Abiri H, Farokhmanesh F, Abdolahad M. Tracing the pH dependent activation of autophagy in cancer cells by silicon nanowire-based impedance biosensor. J Pharm Biomed Anal. 2018;154:158–65.
Dong B, Zhang X, Fan Y, Cao S, Zhang X. Acidosis promotes cell apoptosis through the G protein-coupled receptor 4/CCAAT/enhancer-binding protein homologous protein pathway. Oncol Lett. 2018;16(5):6735–41.
Sun S, Li H, Chen J, Qian Q. Lactic acid: no longer an inert and end-product of glycolysis. Physiology (Bethesda). 2017;32(6):453–63.
Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24(11):2482–90.
Shiratori R, Furuichi K, Yamaguchi M, Miyazaki N, Aoki H, Chibana H, et al. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci Rep. 2019;9(1):18699.
Zhou C, Sun H, Zheng C, Gao J, Fu Q, Hu N, et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018;9(2):161.
Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125(1):194–207.
Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15(8):749–57.
Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192(8):3626–36.
Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5(8):e1191731.
Kesarwani P, Kant S, Prabhu A, Chinnaiyan P. The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro-oncology. 2017;19(10):1308–15.
Choi SY, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol. 2013;230(4):350–5.
Stefan W, Kristina G, Nadine V, Wolfgang M-K. Immunological and translational aspects of glycolytic metabolism in various human tumor entities. Clin Immunol Endocrine Metab Drugs (Discontinued). 2017;4(1):37–46.
Marin E, Bouchet-Delbos L, Renoult O, Louvet C, Nerriere-Daguin V, Managh AJ, et al. Human Tolerogenic dendritic cells regulate immune responses through lactate synthesis. Cell Metab. 2019;30(6):1075–90.e8.
Sullivan MR, Danai LV, Lewis CA. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife. 2019;8:e44235.
Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25(6):1282–93.e7.
Siska PJ, Singer K, Evert K, Renner K. The immunological Warburg effect: can a metabolic-tumor-stroma score (MeTS) guide cancer immunotherapy? Immunol Rev. 2020;295(1):187–202.
Mehla K, Singh PK. Metabolic subtyping for novel personalized therapies against pancreatic cancer. Clin Cancer Res. 2020;26(1):6–8.
Deng YZ, Cai Z, Shi S, Jiang H, Shang YR, Ma N, et al. Cilia loss sensitizes cells to transformation by activating the mevalonate pathway. J Exp Med. 2018;215(1):177–95.
Bedoya FJ, Matschinsky FM, Shimizu T, O'Neil JJ, Appel MC. Differential regulation of glucokinase activity in pancreatic islets and liver of the rat. J Biol Chem. 1986;261(23):10760–4.
Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen WC, Zhou M, et al. KRAS4A directly regulates hexokinase 1. Nature. 2019;576(7787):482–6.
Tan VP, Miyamoto S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. 2015;11(6):963–4.
Abbaszadeh H, Valizadeh A, Mahdavinia M, Teimoori A, Pipelzadeh MH, Zeidooni L, et al. 3-Bromopyruvate potentiates TRAIL-induced apoptosis in human colon cancer cells through a reactive oxygen species- and caspase-dependent mitochondrial pathway. Can J Physiol Pharmacol. 2019;97(12):1176–84.
Sucu BO, Ipek OS, Kurtulus SO, Yazici BE, Karakas N, Guzel M. Synthesis of novel methyl jasmonate derivatives and evaluation of their biological activity in various cancer cell lines. Bioorg Chem. 2019;91:103146.
Ma Y, Hu M, Zhou L, Ling S, Li Y, Kong B, et al. Long non-coding RNA HOTAIR promotes cancer cell energy metabolism in pancreatic adenocarcinoma by upregulating hexokinase-2. Oncol Lett. 2019;18(3):2212–9.
Han HS, Kang G, Kim JS, Choi BH, Koo SH. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med. 2016;48:e218.
Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol. 2014;20(38):13705–17.
Mendoza EE, Pocceschi MG, Kong X, Leeper DB, Caro J, Limesand KH, et al. Control of glycolytic flux by AMP-activated protein kinase in tumor cells adapted to low pH. Transl Oncol. 2012;5(3):208–16.
Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem. 2009;284(36):24223–32.
Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, et al. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J Transl Med. 2012;10:95.
Hu ZZ, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci. 2019;239:116966.
Yang Y, Ishak Gabra MB, Hanse EA, Lowman XH, Tran TQ, Li H, et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat Commun. 2019;10(1):809.
Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (New York, NY). 2012;337(6097):975–80.
Mohammad GH, Vassileva V, Acedo P, Olde Damink SWM, Malago M, Dhar DK, et al. Targeting pyruvate kinase M2 and lactate dehydrogenase a is an effective combination strategy for the treatment of pancreatic cancer. Cancers. 2019;11(9):1372.
Kim DJ, Park YS, Kang MG, You YM, Jung Y, Koo H, et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res. 2015;336(1):119–29.
Poggiogalle E, Jamshed H, Peterson CM. Circadian regulation of glucose, lipid, and energy metabolism in humans. Metab Clin Exp. 2018;84:11–27.
Cui J, Quan M, Jiang W, Hu H, Jiao F, Li N, et al. Suppressed expression of LDHB promotes pancreatic cancer progression via inducing glycolytic phenotype. Med Oncol. 2015;32(5):143.
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase a induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–42.
Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase a and is decreased in pancreatic cancer. Cancer Cell. 2013;23(4):464–76.
Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R, et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer. 2014;110(1):172–82.
Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, et al. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. 2014;20(16):4370–80.
Ji S, Zhang B, Liu J, Qin Y, Liang C, Shi S, et al. ALDOA functions as an oncogene in the highly metastatic pancreatic cancer. Cancer Lett. 2016;374(1):127–35.
Ziveri J, Tros F, Guerrera IC, Chhuon C, Audry M, Dupuis M, et al. The metabolic enzyme fructose-1,6-bisphosphate aldolase acts as a transcriptional regulator in pathogenic Francisella. Nat Commun. 2017;8(1):853.
Miyake K, Nishioka M, Imura S, Batmunkh E, Uto Y, Nagasawa H, et al. The novel hypoxic cytotoxin, TX-2098 has antitumor effect in pancreatic cancer; possible mechanism through inhibiting VEGF and hypoxia inducible factor-1alpha targeted gene expression. Exp Cell Res. 2012;318(13):1554–63.
Qi R, Walker B, Jing Z. Computational and experimental studies of inhibitor Design for Aldolase A. J Phys Chem B. 2019;123(28):6034–41.
Cui K, Jin S, Du Y, Yu J, Feng H, Fan Q, et al. Long noncoding RNA DIO3OS interacts with miR-122 to promote proliferation and invasion of pancreatic cancer cells through upregulating ALDOA. Cancer Cell Int. 2019;19:202.
Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, et al. A predictive model for selective targeting of the Warburg effect through GAPDH inhibition with a natural product. Cell Metab. 2017;26(4):648–59.e8.
Bhardwaj V, Rizvi N, Lai MB, Lai JC, Bhushan A. Glycolytic enzyme inhibitors affect pancreatic cancer survival by modulating its signaling and energetics. Anticancer Res. 2010;30(3):743–9.
Shao F, Yang X, Wang W, Wang J, Guo W, Feng X, et al. Associations of PGK1 promoter hypomethylation and PGK1-mediated PDHK1 phosphorylation with cancer stage and prognosis: a TCGA pan-cancer analysis. Cancer Commun (Lond). 2019;39(1):54.
Hwang TL, Liang Y, Chien KY, Yu JS. Overexpression and elevated serum levels of phosphoglycerate kinase 1 in pancreatic ductal adenocarcinoma. Proteomics. 2006;6(7):2259–72.
Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, et al. Mitochondria-Translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell. 2016;61(5):705–19.
Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang Y, et al. Phosphoglycerate kinase 1 phosphorylates Beclin1 to induce autophagy. Mol Cell. 2017;65(5):917–31.e6.
Lam W, Leung CH, Bussom S, Cheng YC. The impact of hypoxic treatment on the expression of phosphoglycerate kinase and the cytotoxicity of troxacitabine and gemcitabine. Mol Pharmacol. 2007;72(3):536–44.
Jiang Y, He R, Jiang Y, Liu D, Tao L, Yang M, et al. Transcription factor NFAT5 contributes to the glycolytic phenotype rewiring and pancreatic cancer progression via transcription of PGK1. Cell Death Dis. 2019;10(12):948.
Liang C, Shi S, Qin Y, Meng Q, Hua J, Hu Q, et al. Localisation of PGK1 determines metabolic phenotype to balance metastasis and proliferation in patients with SMAD4-negative pancreatic cancer; 2019.
Qian X, Li X, Shi Z, Xia Y, Cai Q, Xu D, et al. PTEN suppresses glycolysis by dephosphorylating and inhibiting autophosphorylated PGK1. Mol Cell. 2019;76(3):516–27.e7.
Wen CL, Huang K, Jiang LL, Lu XX, Dai YT. An allosteric PGAM1 inhibitor effectively suppresses pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2019;116(46):23264–73.
Liu X, Tan X, Liu P, Wu Y, Qian S, Zhang X. Phosphoglycerate Mutase 1 (PGAM1) promotes pancreatic ductal adenocarcinoma (PDAC) metastasis by acting as a novel downstream target of the PI3K/Akt/mTOR pathway. Oncol Res. 2018;26(7):1123–31.
Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem. 2012;287(6):3850–8.
Tsusaka T, Guo T, Yagura T, Inoue T, Yokode M, Inagaki N, et al. Deacetylation of phosphoglycerate mutase in its distinct central region by SIRT2 down-regulates its enzymatic activity. Genes Cells. 2014;19(10):766–77.
Hitosugi T, Zhou L, Fan J, Elf S, Zhang L, Xie J, et al. Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat Commun. 2013;4:1790.
Iwase K, Kato K, Nagasaka A, Miura K, Kawase K, Miyakawa S, et al. Immunohistochemical study of neuron-specific enolase and CA 19-9 in pancreatic disorders. The value of neuron-specific enolase as a marker for islet cell and nerve tissue. Gastroenterology. 1986;91(3):576–80.
Capello M, Caorsi C, Bogantes Hernandez PJ, Dametto E, Bertinetto FE, Magistroni P, et al. Phosphorylated alpha-enolase induces autoantibodies in HLA-DR8 pancreatic cancer patients and triggers HLA-DR8 restricted T-cell activation. Immunol Lett. 2015;167(1):11–6.
Principe M, Ceruti P, Shih NY, Chattaragada MS, Rolla S, Conti L, et al. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget. 2015;6(13):11098–113.
Principe M, Borgoni S, Cascione M, Chattaragada MS, Ferri-Borgogno S, Capello M, et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J Hematol Oncol. 2017;10(1):16.
Leonard PG, Satani N, Maxwell D, Lin YH, Hammoudi N, Peng Z, et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat Chem Biol. 2016;12(12):1053–8.
Cappello P, Curcio C, Mandili G, Roux C, Bulfamante S, Novelli F. Next generation immunotherapy for pancreatic cancer: DNA vaccination is seeking new combo partners. Cancers. 2018;10(2):51.
Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1(1):304–12.
Fuchs BC, Bode BP. Stressing out over survival: glutamine as an apoptotic modulator. J Surg Res. 2006;131(1):26–40.
Sun HW, Yu XJ, Wu WC, Chen J, Shi M, Zheng L, et al. GLUT1 and ASCT2 as predictors for prognosis of hepatocellular carcinoma. PLoS One. 2016;11(12):e0168907.
Soukupova J, Malfettone A, Hyroššová P, Hernández-Alvarez MI, Peñuelas-Haro I, Bertran E, et al. Role of the transforming growth factor-β in regulating hepatocellular carcinoma oxidative metabolism. Sci Rep. 2017;7(1):12486.
Reyes-Castellanos G, Masoud R, Carrier A. Mitochondrial metabolism in PDAC: from better knowledge to new targeting strategies. Biomedicines. 2020;8(8):270.
Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24(41):6314–22.
Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017;77(20):5503–17.
Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers. 2017;10(1):3.
Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses Lipogenesis and hepatocellular carcinoma. Cell Metab. 2019;29(1):174–82.e5.
Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–77.
Akella NM, Ciraku L, Reginato MJ. Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019;17(1):52.
Qian K, Wang S, Fu M, Zhou J, Singh JP, Li MD, et al. Transcriptional regulation of O-GlcNAc homeostasis is disrupted in pancreatic cancer. J Biol Chem. 2018;293(36):13989–4000.
Wu L, Huang P, Wang F, Li D, Xie E, Zhang Y, et al. Relationship between serum CA19-9 and CEA levels and prognosis of pancreatic cancer. Ann Transl Med. 2015;3(21):328.
Gu YL, Lan C, Pei H, Yang SN, Liu YF, Xiao LL. Applicative value of serum CA19-9, CEA, CA125 and CA242 in diagnosis and prognosis for patients with pancreatic cancer treated by concurrent Chemoradiotherapy. Asian Pac J Cancer Prev. 2015;16(15):6569–73.
Ni XG, Bai XF, Mao YL, Shao YF, Wu JX, Shan Y, et al. The clinical value of serum CEA, CA19-9, and CA242 in the diagnosis and prognosis of pancreatic cancer. European J Surg Oncol. 2005;31(2):164–9.
Singhi AD, Koay EJ, Chari ST, Maitra A. Early detection of pancreatic cancer: opportunities and challenges. Gastroenterology. 2019;156(7):2024–40.
Anderson M, Marayati R, Moffitt R, Yeh JJ. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget. 2017;8(34):56081–94.
Cai X, Ding H, Liu Y, Pan G, Li Q, Yang Z, et al. Expression of HMGB2 indicates worse survival of patients and is required for the maintenance of Warburg effect in pancreatic cancer. Acta Biochim Biophys Sin. 2017;49(2):119–27.
Lyshchik A, Higashi T, Hara T, Nakamoto Y, Fujimoto K, Doi R, et al. Expression of glucose transporter-1, hexokinase-II, proliferating cell nuclear antigen and survival of patients with pancreatic cancer. Cancer Invest. 2007;25(3):154–62.
Paggi MG, Fanciulli M, Del Carlo C, Citro G, Bruno T, Floridi A. Glucose utilization by tumor cells: a post-translational modification of mitochondrial hexokinase may play a regulatory role. Biochem Biophys Res Commun. 1991;178(2):648–55.
Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-gastroenterology. 2008;55(88):2016–27.
Grimm M, Cetindis M, Lehmann M, Biegner T, Munz A, Teriete P, et al. Association of cancer metabolism-related proteins with oral carcinogenesis - indications for chemoprevention and metabolic sensitizing of oral squamous cell carcinoma? J Transl Med. 2014;12:208.
Malina A, Bryant SK, Chang SH, Waldrop GL, Gilman SD. Capillary electrophoresis-based assay of phosphofructokinase-1. Anal Biochem. 2014;447:1–5.
Li C, Zhao Z, Zhou Z, Liu R. PKM2 promotes cell survival and invasion under metabolic stress by enhancing Warburg effect in pancreatic ductal adenocarcinoma. Dig Dis Sci. 2016;61(3):767–73.
Yu L, Teoh ST, Ensink E, Ogrodzinski MP, Yang C, Vazquez AI, et al. Cysteine catabolism and the serine biosynthesis pathway support pyruvate production during pyruvate kinase knockdown in pancreatic cancer cells. Cancer Metab. 2019;7:13.
Mohammad GH, Olde Damink SW, Malago M, Dhar DK, Pereira SP. Pyruvate kinase M2 and lactate dehydrogenase a are overexpressed in pancreatic cancer and correlate with poor outcome. PLoS One. 2016;11(3):e0151635.
Mohammad GH, Vassileva V. Targeting pyruvate kinase M2 and lactate dehydrogenase a is an effective combination strategy for the treatment of pancreatic cancer. Cancers (Basel). 2019;11(9):1372.
Tas F, Karabulut S, Ciftci R, Sen F, Sakar B, Disci R, et al. Serum levels of LDH, CEA, and CA19-9 have prognostic roles on survival in patients with metastatic pancreatic cancer receiving gemcitabine-based chemotherapy. Cancer Chemother Pharmacol. 2014;73(6):1163–71.
Cui Y, Tian M, Zong M, Teng M, Chen Y, Lu J, et al. Proteomic analysis of pancreatic ductal adenocarcinoma compared with normal adjacent pancreatic tissue and pancreatic benign cystadenoma. Pancreatology. 2009;9(1–2):89–98.
Mikuriya K, Kuramitsu Y, Ryozawa S, Fujimoto M, Mori S, Oka M, et al. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int J Oncol. 2007;30(4):849–55.
Schek N, Hall BL, Finn OJ. Increased glyceraldehyde-3-phosphate dehydrogenase gene expression in human pancreatic adenocarcinoma. Cancer Res. 1988;48(22):6354–9.
Guo C, Liu S, Sun MZ. Novel insight into the role of GAPDH playing in tumor. Clin Transl Oncol. 2013;15(3):167–72.
Patwa TH, Li C, Poisson LM, Kim HY, Pal M, Ghosh D, et al. The identification of phosphoglycerate kinase-1 and histone H4 autoantibodies in pancreatic cancer patient serum using a natural protein microarray. Electrophoresis. 2009;30(12):2215–26.
Rezaei M, Nikeghbalian S, Mojtahedi Z, Ghaderi A. Identification of antibody reactive proteins in pancreatic cancer using 2D immunoblotting and mass spectrometry. Oncol Rep. 2018;39(5):2413–21.
Tomaino B, Cappello P, Capello M, Fredolini C, Sperduti I, Migliorini P, et al. Circulating autoantibodies to phosphorylated alpha-enolase are a hallmark of pancreatic cancer. J Proteome Res. 2011;10(1):105–12.
Yabushita S, Fukamachi K, Tanaka H, Fukuda T, Sumida K, Deguchi Y, et al. Metabolomic and transcriptomic profiling of human K-ras oncogene transgenic rats with pancreatic ductal adenocarcinomas. Carcinogenesis. 2013;34(6):1251–9.
Xu Y, Li F, Lv L, Li T, Zhou X, Deng CX, et al. Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res. 2014;74(13):3630–42.
Sharma A, Smyrk TC, Levy MJ, Topazian MA, Chari ST. Fasting blood glucose levels provide estimate of duration and progression of pancreatic cancer before diagnosis. Gastroenterology. 2018;155(2):490–500.e2.
Cortese N, Capretti G, Barbagallo M, Rigamonti A, Takis PG. Metabolome of pancreatic juice delineates distinct clinical profiles of pancreatic cancer and reveals a link between glucose metabolism and PD-1(+) cells. Cancer Immunol Res. 2020;8(4):493–505.
Hosein AN, Beg MS. Pancreatic cancer metabolism: molecular mechanisms and clinical applications. Curr Oncol Rep. 2018;20(7):56.
Tyanova S, Temu T, Carlson A, Sinitcyn P, Mann M, Cox J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics. 2015;15(8):1453–6.
MacDougall G, Anderton RS, Mastaglia FL, Knuckey NW, Meloni BP. Proteomic analysis of cortical neuronal cultures treated with poly-arginine peptide-18 (R18) and exposed to glutamic acid excitotoxicity. Mol Brain. 2019;12(1):66.
Miyoshi E, Kamada Y. Application of glycoscience to the early detection of pancreatic cancer. Cancer Sci. 2016;107(10):1357–62.
Xiang J, Liu L, Wang W, Xu H, Wu C, Xu J, et al. Metabolic tumor burden: a new promising way to reach precise personalized therapy in PDAC. Cancer Lett. 2015;359(2):165–8.
Serrao EM, Kettunen MI, Rodrigues TB, Dzien P, Wright AJ, Gopinathan A, et al. MRI with hyperpolarised [1-13C] pyruvate detects advanced pancreatic preneoplasia prior to invasive disease in a mouse model. Gut. 2016;65(3):465–75.
Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci Transl Med. 2013;5(198):198ra08.
Wei K, Wang M, Zhang W, Mu H, Song TQ. Neutrophil-lymphocyte ratio as a predictor of outcomes for patients with hepatocellular carcinoma undergoing TAE combined with Sorafenib. Med Oncol (Northwood, London, England). 2014;31(6):969.
McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2018;24(43):4846–61.
Heinrich S, Lang H. Neoadjuvant therapy of pancreatic cancer: definitions and benefits. Int J Mol Sci. 2017;18(8):1622.
Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs. 2008;17(10):1533–45.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–23.
Park PH, Yamamoto TM, Li H, Alcivar AL. Amplification of the mutation-carrying BRCA2 allele promotes RAD51 loading and PARP inhibitor resistance in the absence of reversion mutations; 2019.
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–27.
Jain PG, Patel BD. Medicinal chemistry approaches of poly ADP-ribose polymerase 1 (PARP1) inhibitors as anticancer agents - a recent update. Eur J Med Chem. 2019;165:198–215.
Acknowledgements
The authors would like to express our gratitude to American Journal Experts (http://bit.ly/AJE-HS) for editing the English text of a draft of manuscript that greatly improved it.
Funding
This work was supported by the NSFC (grants from the Natural Science Foundation of China 81902484), the China Postdoctoral Science Foundation (2020 M670864), and the Medical and Health Talents Project of Jilin Province (2019SCZT0032019SCZT013; 2019SCZT007).
Author information
Authors and Affiliations
Contributions
LC and XZ contributed to the conception and design of the study; LC wrote the first draft of the manuscript; LC wrote sections of the manuscript; and XZ provided critical comments. LC, WJ, XQ, JS, MC, SL, XH, YX, and BL contributed to the revised manuscript. Specifically, XQ and JS looked up references for the reviewers suggestions, and replenished the manuscript with other metabolic pathways. MC and SL answered all the questions of reviewers and made manuscript more coherent to read. XH re-wrote the BACKGROUND part. YX modified the four figures and prepared high quality Figs. LC, WJ, and BL made the two new tables and corrected al the errors of manuscript. The authors critically reviewed the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cao, L., Wu, J., Qu, X. et al. Glycometabolic rearrangements--aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications. J Exp Clin Cancer Res 39, 267 (2020). https://doi.org/10.1186/s13046-020-01765-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-020-01765-x