
Abstract
Extrachromosomal DNA (ecDNA), gene-encoding extrachromosomal particles of DNA, is often present in tumor cells. Recent studies have revealed that oncogene amplification via ecDNA is widespread across a diverse range of cancers. ecDNA is involved in increasing tumor heterogeneity, reverting tumor phenotypes, and enhancing gene expression and tumor resistance to chemotherapy, indicating that it plays a significant role in tumorigenesis. In this review, we summarize the characteristics and genesis of ecDNA, connect these characteristics with their concomitant influences on tumorigenesis, enumerate the oncogenes encoded by ecDNA in multiple cancers, elaborate the roles of ecDNA in tumor pathogenesis and progression, and propose the considerable research and therapeutic prospects of ecDNA in cancer.
Similar content being viewed by others


Background
Genetic material guarantees the transmission of genetic information from parents to their offspring. With the exception of some viruses, DNA is the carrier of genetic material, which was first identified by Frederick Griffith in a Streptococcus pneumoniae transformation experiment [1,2,3]. DNA is present in a compacted and dynamic complex called chromatin in the cell nucleus. During the metaphase of cell division, chromatin organizes into highly condensed chromosomes [4]. DNA also exists in organelles including mitochondria and chloroplasts, which probably evolved from microorganisms such as α-proteobacteria and cyanobacterium invading host cells and starting a symbiotic life form [5,6,7,8]. In addition, it has been reported that extrachromosomal particles of DNA exist, the size of which varies from dozens to millions of base pairs [9, 10]. These particles have been proved to be circular by biophysical methods and DNA sequencing. Verhaak et al. divided these particles into two types based on the size and resultant functions [11]. Small particles, which are called extrachromosomal circular DNA (eccDNA), are usually less than 1 kb; consequently, they are undetectable by light microscopy and lack full-length genes [12,13,14]. Investigations of eccDNA have revealed its association with aging, as eccDNA containing ribosomal DNA genes accumulates in old cells and contributes to the aging of yeast cells. The probable mechanism is described by the titration hypothesis, which indicates that the accumulated eccDNA might precipitate the components of replication and/or transcription elements, and eventually trigger the senescence and the eventual death of old cells [15, 16]. Cohen et al. revealed that small polydispersed circular DNA (spcDNA), one type of eccDNA, is related to chromosomal instability (CIN), which is a characteristic of malignant cells since the spcDNA molecules were frequently found in either intrinsically unstable cells including tumor cells or in cells exposed to an external carcinogen treatment, indicating that spcDNA may be a possible marker of CIN [17, 18]. Extrachromosomal DNA (ecDNA), the focus of this review, includes relatively large particles generally ranging from 1 to 3 Mb, with a median size of 1.26 Mb, which are therefore visible by light microscopy and encompass whole genes as well as regulatory regions [11, 19,20,21]. ecDNA was first discovered as paired small chromatin bodies in neuroblastoma (NB) cells called double minutes (DMs) [10]. Jack et al. performed a scanning electron microscopy (SEM) study of DMs and demonstrated that the identical sister minutes appeared as two spherical chromatin bodies interconnected by chromatin fibers [22]. Later, Turner et al. performed whole-genome sequencing (WGS), structural modeling and cytogenetic analyses of 17 different cancer types and found that only 30% of ecDNA in tumor cells is paired; thus, ecDNA is used as a general term to refer to large, gene-containing extrachromosomal particles of DNA, including both DMs and single body forms, with single minutes showing no apparent partners nearby. The existence of ecDNA has been confirmed in various types of cancers and cancer cell lines [23]. Unlike the smaller eccDNA particles, ecDNA is sufficiently large enough to contain one or multiple full genes, including oncogenes, and oncogenes located on ecDNA were first reported to map MYCN to DMs in NB [24]. Subsequent studies confirmed the existence of oncogene amplification on ecDNA in multiple types of cancer [25,26,27,28]. Although oncogene amplification via ecDNA has been recognized, ecDNA was thought to be rare in cancer for a long time; therefore, the role of ecDNA in tumorigenesis has received little attention [29, 30]. This perception was maintained until an unbiased approach was developed to detect ecDNA by integrating WGS and cytogenetic image analysis. The results showed that oncogene amplification via ecDNA is widespread in different cancers, thus arousing people’s interest in extrachromosomal oncogene amplification when exploring tumorigenesis [23]. In this review, we focus on the characteristics and genesis of ecDNA and emphasize its role in tumorigenesis.
Characteristics of ecDNA
The rapid development of new techniques enables determination of the unique characteristics of ecDNA. Compared to chrDNA, ecDNA is acentric with uneven segregation at cell division, leading to increased intratumoral heterogeneity and enhanced fitness to changing environments. In addition, ecDNA can be eliminated via micronucleus formation to reverse the tumor phenotype. Furthermore, ecDNA is circular with high chromatin accessibility and ultra-long-range chromatin contact. We will summarize the unique characteristics of ecDNA and resultant outcomes in cancer (Table 1).
ecDNA is acentric with uneven segregation at cell division
The centromere, which consists of repetitive DNA, is the region where sister chromatids remain connected until mitosis and has a unique histone—CenpA. The centromere is the ‘landing platform’ for the kinetochore, a large protein structure that binds to microtubules in mitosis and ensures accurate sister chromatid separation and chromosome segregation [40, 41]. Because centromeres lack ecDNA, ecDNA segregates unevenly at cell division, allowing daughter cells to possess up to twice as many ecDNA particles as their corresponding mother cells [31]. Uneven segregation of ecDNA leads to genetic material distribution uncertainty, which may result in a rapid increase in oncogene copy number in a short period and increase intratumoral heterogeneity, enhancing tumor fitness to changing environments [32].
The resultant heterogeneity in cancer with ecDNA
deCarvalho et al. revealed that ecDNA presented divergent inheritance patterns and clonal selection dynamics through an extensive genomic and transcriptomic analysis of 13 glioblastoma (GBM) tumor samples and neurosphere-forming cultures and orthotopic xenograft models established from these samples, inferring the uneven inheritance of ecDNA between offspring cells, which may explain the rapidly enhanced genomic heterogeneity during GBM evolution [42]. Xu et al. characterized DMs in paired diagnosis and relapse tumors from 4 GBM patients and found that identical oncogenes could amplify on different DMs at diagnosis and relapse, reflecting the primed evolution and rapidly increasing heterogeneity of DMs [43].
ecDNA can be eliminated by micronucleus formation
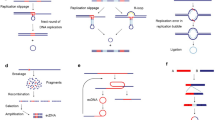
Oncogenes on DMs are unstable, and DMs can be eliminated in tumor cells by incorporating DMs into the cytoplasmic micronucleus when they are exposed to DNA replication inhibitors such as a low dose of hydroxyurea (HU) and radiation [34, 35]. In addition, inhibition of ERK1/2 activation, one of the main mitogen-activated protein kinase (MAPK) signaling pathways, also breaks DNA to eliminate DMs [36]. Acentric DMs were found to segregate by adhering to the chromosome arm during cell division, a process is called ‘hitchhiking’ (Fig. 1.a). When cells are exposed to a low dose of HU, DMs aggregate and detach from the chromosome; subsequently, the aggregated DMs generate micronuclei (Fig. 1.b). The micronuclei may be eliminated, religated, or integrated into the main nucleus or generate giant DMs or homogeneously staining regions (HSRs) [37]. Since a decrease in oncogene amplification in tumor cells to revert the tumor phenotype has been reported in various cancers [44,45,46,47,48,49], elimination and morphological transformation of DMs may result in a reduction in tumorigenicity and a loss of malignant properties. However, HU treatment does not decrease the copy number of oncogenes amplified on HSRs, another form of oncogene on chromosomes, indicating that oncogene amplification is unstable on ecDNA but stable on HSRs [38]. In addition to being eliminated by DNA replication inhibitors, DMs can be eliminated by suppressing the expression of genes residing on DMs. Suppression decreases the number of DMs and reduces oncogene amplification on DMs through micronucleus formation, resulting in a reduction in tumor proliferation and invasion [39].

Generation of DM-type micronuclei. a. DMs are located on the nuclear periphery during the G1 phase and move to the interior during their replication in the early S phase. Acentric DMs segregate by adhering to the chromosome arm during the M phase, which is called ‘hitchhiking.’ b. In cells exposed to a low dose of HU during the early S phase, DMs tend to aggregate and detach from the chromosome during the subsequent M phase. Then, the aggregated DMs generate micronuclei during the subsequent interphase
The resultant tumor reversion in cancer with ecDNA
Ambros et al. used fluorescence in situ hybridization (FISH) with a MYCN-specific probe in NB cell lines and revealed spontaneous elimination of extrachromosomally amplified MYCN in F-cells with significant biological features and loss of the malignant phenotype and properties. Therefore, elimination of extrachromosomally amplified MYCN in NB is postulated to be associated with tumor cell reversion [46].
ecDNA is circular with highly accessible chromatin and ultra-long-range chromatin contacts
Based on biophysical methods and DNA sequencing, ecDNA is thought to be circular [50]. Recently, the distinct structure of ecDNA was demonstrated to be circular through the use of ultrastructural imaging, computational WGS analyses, long-range optical mapping and superresolution three-dimensional structured illumination microscopy (3D-SIM), scanning electron microscopy (SEM) and transmission electron microscopy (TEM). ecDNA is packaged into chromatin with nucleosome units, which mainly contain active histone marks, and its chromatin landscape is more accessible than that of chromosomal DNA (chrDNA) due to its nucleosomal organization being less compact than that of chrDNA (Fig. 2). Since the number of transcripts is markedly increased when oncogenes are present in circular ecDNA, even after normalization for the DNA copy number, the transcription levels are still high, indicating that the active chromatin state and highly accessible chromatin may be associated with the high transcription level of ecDNA. Moreover, ultra-long-range chromatin contact can occur on ecDNA, revealing the potential influence of ecDNA on distal gene expression [19].

Structures of ecDNA and chrDNA. a. The nucleosomal organization of chrDNA is compact, and during cell division, DNA is packaged into chromosomes with centromeres. b. The nucleosomal organization of ecDNA is less compact than that of chrDNA, and ecDNA is circular without centromeres
The resultant distal chromatin contact in cancer with ecDNA
Morton et al. resolved EGFR ecDNA amplicon patterns from primary GBM sequencing and discovered that amplified noncoding regions are inclined to incorporate enhancers and that ecDNA structures always include adjacent enhancers, indicating that enhancers are co-selected with the coding regions of oncogenes during the formation of ecDNA. Furthermore, they revealed that the EGFR locus amplified upon ecDNA contact with the distal enhancer by chromosome conformation experiments and confirmed that these co-selected enhancers are partly outside the topologically associated domain (TAD) of the original chromosome [51].
Genesis of ecDNA
Although the precise ecDNA genesis mechanism is still unknown, some models have been established to suggest the possible mechanism, including the episome model, the translocation-excision-deletion-amplification model, chromothripsis and a multistep evolutionary process (Fig. 3 and Table 2).

Models of the possible ecDNA genesis mechanism. a. ‘Episome’ model. Bidirectional replication of DNA leads to the existence of two replication forks, and the region between them is named the replication bubble. When an error in replication occurs, the replication forks arrest, and the replication bubble drops and transforms into an episome, which further replicates and recombines to form DMs. b. ‘Translocation-excision-deletion-amplification’ model. Two chromosomes translocate before excision and deletion occur in close proximity to the translocation breakpoints. Then, amplification and circularization of the separate fragments lead to the formation of DMs. c. ‘Chromothripsis’ model. The mis-segregation of a chromosome results in the chromosome shattering and subsequent random repair to form DMs, allowing non-contiguous DNA fragments to fuse in the DMs. d. A multistep evolutionary process in the GLC1 cell line. Recombination and duplication of two single-chromosomal ancestral DMs can occur first, and then the new DMs undergo various types of deletion and recombination with another DM of a single-chromosomal source, resulting in multiple DM subpopulations
Episome in ecDNA
One of the classic models is the episome model, where replication fork stalling elicits replication fork collapse, and the replication bubble subsequently falls off the chromosome, interconnects and transforms into an extrachromosomal circular molecule named the episome, with autonomous replication and recombination of episomes leading to the formation of DMs (Fig. 3.a) [52, 53]. The definitive evidence underpinning the episome model is the genesis of MYC-containing DMs in acute myeloid leukemia (AML). Storlazzi performed FISH analysis on metaphase chromosomes of bone marrow samples derived from the 30 AML patients with MYC-containing DMs and discovered that 23 samples showed a deletion of the MYC gene region on chromosome 8. DNA sequencing revealed that the linker sequences of DMs were as the same as the linker sequences of chromosome 8 with a deletion of the MYC gene region, indicating that the gene carried by DMs originated from chromosome 8, which is consistent with the episome model [54]. Later, the episome model was suggested to also be applicable to solid tumors, including NB and small cell lung cancer (SCLC) [55].
Translocation-excision-deletion-amplification in ecDNA
Van Roy et al. discovered the co-localization of MYC and ATBF1 on the same DMs in the NB cell line SJNB-12 and proposed another model called translocation-excision-deletion-amplification. The model was prompted by the reciprocal translocation between chromosomes 8 and 16 on which MYC and ATBF1 were located, with subsequent excisions and deletions occurring in close proximity to the translocation breakpoints, and then the isolated sequence amplified and circularized to form DMs (Fig. 3.b).
Chromothripsis in ecDNA
Cancer genome sequencing identified a new model called chromothripsis in which a chromosome mis-segregates and ends up in a micronucleus where it has less access to replication proteins, leading to replication stress, chromosome breaks and subsequent faulty repair (Fig. 3.c) [57, 58]. The model was supported by a xenografted human oligodendroglioma where co-amplification of EGFR and MYC existed in the form of DMs. Vogt et al. discovered that the small fragments localized hundreds of kilobases apart on the chromosome were associated with contigs in the DMs, and that the various junctions were associated with fusions between non-contiguous sequences in the normal reference genome through FISH and WGS, indicating that the presence of rearrangements during the formation of DMs is related to the chromothripsis model [59].
A multistep evolutionary process in ecDNA
L’Abbate et al. investigated the SCLC cell line GLC1 through an integrated approach combining next-generation sequencing technologies (NGS) and single-nucleotide polymorphism (SNP) array, polymerase chain reaction (PCR) and FISH techniques and proposed the presence of multiple DM subpopulations with the variety of shared structural variations (SVs), with some consisting of a single chromosome and others carrying combinations of different chromosomes, suggesting that a multistep evolutionary process starting from single-chromosome ancestral DMs can better explain processes involving more than two chromosomes. For example, in the GLC1 cell line, single-chromosome ancestral DMs derived from chromosome 1, 8 or 21 undergo a series of mutation events, including recombinations, deletions and duplications, to transform into multiple DM subpopulations. The ancestral DMs derived from chromosomes 1 and 8 recombined and duplicated first to form a new DM, and then various types of deletion and recombination events occurred between the new DM and ancestral DM derived from chromosome 21, leading to multiple DM subpopulations (Fig. 3.d) [62, 63].
ecDNA in cancer
ecDNA is abundant in cancer cells but rare in normal cells, and the copy number of ecDNA varies in different types of cancer, with the highest prevalence in GBM [23]. Notably, ecDNA is a vehicle for oncogene amplification, and the role of oncogenes in tumorigenesis is well studied in vitro and in vivo, indicating that ecDNA plays a considerable role in tumorigenesis. We will review recent reports describing our understanding of the biological functions of ecDNA in various types of cancer and explore new mechanisms for tumor pathogenesis and evolution (Fig. 4 and Table 3).

ecDNA-mediated oncogene amplification promotes tumor progression. In NB, MYCN encoded by ecDNA is closely associated with tumor progression. In glioblastoma, the amplification of oncogenes, such as MYC, EGFR, PDGFRα, ERBB2, CDK4, and MDM2, on ecDNA promotes tumorigenesis. In breast cancer, HER2 encoded on ecDNA contributes to tumorigenesis. In ovarian cancer, ecDNA containing MYCN and EIF5AR stimulates tumorigenesis. In colon cancer, DHFR amplification via ecDNA is also involved in tumorigenesis
ecDNA in neuroblastoma
ecDNA was first discovered in metaphase spreads of NB cells [10]. In addition, Alt et al. used cloning methods to demonstrate that a new oncogene MYCN was mapped to DMs in the human NB cell line IMR-32 [24]; this is the first report confirming the presence of oncogenes in ecDNA. Ambros et al. investigated the NB cell lines with DMs in detail and discovered that when exposed to low-dose HU, the cells manifested an enlarged and flattened morphology and increased granularity and expressed senescence-associated-β-galactosidase (SA-β-GAL), all of which are features of cell senescence, indicating that low-dose HU is an effective senescence activator for tumor cells with DMs [71]. A recent study revealed that circular DNAs, including ecDNA, eccDNA and neochromosomes, unexpectedly remodel the chromosomal genome. The remodeling arises from the generation of circular DNA with the subsequent reintegration into chromosomal genomic loci, affecting chromosomal gene expression, including aberrant expression of oncogenes and tumor suppressors. Specifically, the adjacent circle integration of the oncogene TERT is associated with its augmented expression, and the integration of circle fragments into the tumor suppressor DCLK1 is associated with its repressed expression. Moreover, Kaplan–Meier analysis comparing the survival of 22 NB patients with circle-derived remodeling to that of 59 NB patients lacking such remodeling revealed that circle-derived remodeling was associated with adverse outcomes, which may explain the clinical heterogeneity observed in NBs [66].
ecDNA in glioblastoma
Zhou et al. used two syngeneic primary cultures of GBM that differed in the presence or absence of EGFR-encoding DMs and found that the former had higher levels of invasiveness, heterogeneity and radioresistance. However, whether the presence of EGFR-coding DMs or upregulation of EGFR leads to these observations requires further research [62]. Nathanson et al. revealed the reversible suppression of ecDNA containing a functional mutation in epidermal growth factor receptor vIII (EGFRvIII) to evade targeted therapy—epidermal growth factor receptor (EGFR) inhibitors. Specifically, they discovered an unexpected phenomenon in which EGFR inhibitors decreased the number of tumor cells with high levels of EGFRvIII in vitro and in vivo, and after drug removal, the tumor cells containing a high copy number of EGFRvIII returned, which exceeded the classical genetics. Then, through observation of cells in metaphase, they found the possible mechanism through which EGFRvIII almost completely amplified on ecDNA; moreover, the number of ecDNAs containing EGFRvIII is reduced by EGFR inhibitors and then reemerged 1–2 weeks after drug removal [63]. Later, Nikolaev et al. developed a model called amplification-linked extrachromosomal mutations (ALEMs) to explain the above observation. ALEMs are a novel type of cancer variant originating from the extrachromosomal region and can be eliminated from cancer cells. The model is based on the fact that proliferation-promoting oncogenes reside on ecDNA, and ecDNA increases the mutational possibility. ALEMs occur not only in EGFR but also in PDGFRα and ERBB2 according to exome sequencing of 7 GBM patients. Moreover, an analysis of 4198 tumors indicated the presence of ALEMs in various tumor types, indicating that ALEMs may be the foundation of resistance to therapies in multiple types of tumors [65]. The enhancers in the noncoding regions, which are co-selected with the coding regions of oncogene EGFR during the formation of ecDNA, are outside the TAD of the original chromosome, which may reflect the ability of ultra-long-range chromatin contacts in ecDNA. Moreover, by ablating individual enhancer activity by CRISPR interference, these enhancers are rewired in ecDNA to enhance oncogene EGFR expression and tumor fitness, suggesting a novel mechanism of enhancers in regulating oncogene amplification [51].
ecDNA in colon cancer
Gene amplification is a frequent manifestation of genomic instability in human tumors and plays a critical role in tumor progression and drug resistance [72]. Methotrexate (MTX) resistance through dihydrofolate reductase (DHFR) gene amplification is the most common mechanism of drug resistance [71]. Morales et al. demonstrated that in colon cancer HT29 cells with a high MTX dose, the DHFR gene copy number is markedly increased via extrachromosomal DMs. Furthermore, they discovered that the loss of the DHFR amplicon occurs contemporaneously with the withdrawal of MTX in MTX-sensitive cells, and these cells present a decreased drug resistance capacity when they are re-exposed to MTX, providing a potential second-round treatment option for patients with drug resistance occurring via gene amplification [66]. Moreover, Meng et al. discovered that nonhomologous end joining (NHEJ) plays an important role in the formation of DMs; therefore, depletion of DNA-PKc, a key NHEJ protein, decreases the amplification of DHFR, resulting in increased MTX sensitivity. This discovery suggests that NHEJ may be targeted for the treatment of MTX-resistant colon cancer [67]. Subsequently, the same authors investigated the function of homologous recombination, another DSB repair pathway involved in gene amplification, and found that in comparison to MTX-sensitive cells, MTX-resistant cells present increased homologous recombination activity. Silencing BRCA1, a key player in homologous recombination, decreased the number of DMs and the copy number of oncogenes amplified on DMs, and the sensitivity to MTX increased in DM-containing MTX-resistant cells. However, BRCA1 silencing had no influence on the copy number of oncogenes amplifying on HSRs and had no effect on the sensitivity to MTX in HSR-containing MTX-resistant cells; these findings indicate that the homologous recombination pathway may also be a target for the improvement of chemotherapeutic effects by decreasing extrachromosomal oncogene amplification [68].
ecDNA in ovarian cancer
Jin et al. proposed the considerable effect of noncoding regions on DMs. They discovered 5 matrix attachment regions (MARs) in a 682 kb DM from human ovarian cancer cell line UACC-1598 by sequencing and bioinformatics analyses and determined that these MARs can interact with the nuclear matrix both in vitro and in vivo through electrophoretic mobility shift assay (EMSA) and PCR. Moreover, the transfection of MARs constructs into human embryonic kidney 293 T cells revealed the upregulation of oncogenes MYCN and EIF5A2, which localized near MARs, indicating that noncoding regions on DMs may regulate gene expression and play a crucial role in oncogene activation [69].
ecDNA in breast cancer
Approximately 20% of breast cancers demonstrate amplification of the proto-oncogene HER2, a receptor tyrosine kinase (RTK) belonging to the epidermal growth factor receptor (EGFR) family, and approximately 30% of HER2-positive tumors show DM amplification [70, 73]. Although these tumors can respond to therapies against HER2 directly, they often develop resistance and resume their progression [74]. In various models with resistance to anti-HER2 therapy, the number of DMs containing HER2 has been shown to be preserved, even when the resistance is acquired by the loss of HER2 protein expression, suggesting that the loss of HER2 protein expression due to the loss of DMs containing HER2 is not a likely mechanism underlying resistance to anti-HER2 therapy [70].
Conclusion and perspectives
Recent years have witnessed tremendous advancements in the field of ecDNA. A wide array of oncogene amplification via ecDNA has been uncovered, reverting the concentration of research on the association between ecDNA and tumorigenesis [23]. However, the understanding of ecDNA and its influence on tumors remains limited. Fortunately, the rapid development of biotechnology has enabled various methods to be used to detect and construct ecDNA, offering an opportunity to confirm the structure of ecDNA and speculate its role in tumorigenesis [19].
Notably, many studies have revealed the features of ecDNA and proposed that ecDNA plays a considerable role in tumorigenesis, including NB, GBM, colon cancer, ovarian cancer and breast cancer [64, 67, 69, 75,76,77]. The elimination of ecDNA is confirmed and it can decrease oncogene amplification on ecDNA to revert the tumor phenotype [34, 37, 78]. Although the current methods for ecDNA elimination lack specificity, ecDNA is expected to be a good therapeutic target in the future. Circular and acentric ecDNA contributes to increased oncogene copy number and tumor heterogeneity, thus providing tumor cells with the ability to respond rapidly to changing environments, including treatment [19, 79]. A contradictory finding is that tumor cells can develop drug resistance through the increased copy number of oncogenes on ecDNA, such as DHFR, while resistance can also be acquired through the downregulation of the expression of oncogenes on ecDNA, such as EGFR, indicating that different methods exist to evade treatment [63, 66]. However, the mechanisms underlying these different methods are unknown. Tumor cells are hypothesized to tend to have a survival advantage according to the specific environment, called “passive choice”. Another possible mechanism is that tumor cells perceive the environment and then change the quantity of ecDNA that they carry to fit the environment, called “initiative regulation”. Understanding the underlying molecular mechanism of ecDNA in evading therapy is essential for exploring novel therapies.
The perception that ecDNA possesses a unique structure and function, the discovery that ecDNA drives oncogene amplification and the profound significance of ecDNA in cancer increase the understanding of current cancer genome maps, rendering ecDNA a hotspot for the investigation of tumor pathogenesis and evolution. To develop novel tumor therapies, numerous endeavors are required for a thorough understanding of ecDNA. Knowledge of the precise mechanism of the formation, maintenance, replication and influence of ecDNA in tumorigenesis merely represent the tip of the iceberg, offering a direction for future exploration, and continuous advancement of technology and unremitting exploration are necessary to eventually unveil the mysteries of ecDNA.
Availability of data and materials
Not applicable.
Abbreviations
- 3D-SIM:
-
Three-dimensional structured illumination microscopy
- ALEMs:
-
Amplification-linked extrachromosomal mutations
- AML:
-
Acute myeloid leukemia
- chrDNA:
-
Chromosomal DNA
- CIN:
-
Chromosomal instability
- Circle-seq:
-
Circle-sequencing
- DHFR :
-
Dihydrofolate reductase
- DMs:
-
Double minutes
- DSBs:
-
Double-strand breaks
- eccDNA:
-
Extrachromosomal circular DNA
- ecDNA:
-
Extrachromosomal DNA
- EGFR :
-
Epidermal growth factor receptor
- EGFRvIII :
-
Epidermal growth factor receptor vIII
- EMSA:
-
Electrophoretic mobility shift assay
- FISH:
-
Fluorescence in situ hybridization
- GBM:
-
Glioblastoma
- HSRs:
-
Homogeneously staining regions
- HU:
-
Hydroxyurea
- MAPK:
-
Mitogen-activated protein kinase
- MARs:
-
Matrix attachment regions
- MTX:
-
Methotrexate
- NB:
-
Neuroblastoma
- NGS:
-
Next generation sequencing technologies
- NHEJ:
-
Nonhomologous end joining
- PCR:
-
Polymerase chain reaction
- RTK:
-
Receptor tyrosine kinase
- SA-β-GAL:
-
Senescence-associated-β-galactosidase
- SCLC:
-
Small cell lung carcinoma
- SEM:
-
Scanning electron microscopy
- SNP:
-
Single nucleotide polymorphism
- spcDNA:
-
Small polydispersed circular DNA
- SVs:
-
Structural variations
- TAD:
-
Topologically associated domain
- TEM:
-
Transmission electron microscopy
- TKIs:
-
Tyrosine kinase inhibitors
- WGS:
-
Whole-genome sequencing
References
Griffith F. The Significance of Pneumococcal Types. J Hyg (Lond). 1928;27:113–59..
Hehlmann R, Schetters H, Kreeb G, Erfle V, Schmidt J, Luz A. RNA-tumorviruses, oncogenes, and their possible role in human carcinogenesis. Klin Wochenschr. 1983;61:1217–31.
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44.
Novais-Cruz M, Alba Abad M, van IJcken WF, Galjart N, Jeyaprakash AA, Maiato H, Ferrás C. Mitotic progression, arrest, exit or death relies on centromere structural integrity, rather than de novo transcription. Elife. 2018;7:e36898.
Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–42.
Smith DR. Evolution: in chloroplast genomes, Anything Goes. Curr Biol. 2017;27:R1305–r07.
Gray MW. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 2012;4:a011403.
Kato Y, Sakamoto W. FtsH protease in the thylakoid membrane: physiological functions and the regulation of protease activity. Front Plant Sci. 2018;9:855.
Hotta Y, Bassel A. Molecular size and circularity of DNA in cells of mammals and higher plants. Proc Natl Acad Sci U S A. 1965;53:356–62.
Cox D, Yuncken C, Spriggs AI. Minute chromatin bodies in malignant tumours of childhood. Lancet. 1965;1:55–8.
Verhaak RGW, Bafna V, Mischel PS. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer. 2019;19:283–8.
Shoura MJ, Gabdank I, Hansen L, Merker J, Gotlib J, Levene SD, Fire AZ. Intricate and Cell Type-Specific Populations of Endogenous Circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3 (Bethesda). 2017;7:3295–303.
Moller HD, Mohiyuddin M, Prada-Luengo I, Sailani MR, Halling JF, Plomgaard P, Maretty L, Hansen AJ, Snyder MP, Pilegaard H, et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun. 2018;9:1069.
Kumar P, Dillon LW, Shibata Y, Jazaeri AA, Jones DR, Dutta A. Normal and cancerous tissues release Extrachromosomal circular DNA (eccDNA) into the circulation. Mol Cancer Res. 2017;15:1197–205.
Moller HD, Parsons L, Jorgensen TS, Botstein D, Regenberg B. Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci U S A. 2015;112:E3114–22.
Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–42.
Cohen S, Regev A, Lavi S. Small polydispersed circular DNA (spcDNA) in human cells: association with genomic instability. Oncogene. 1997;14:977–85.
Schmidt H, Taubert H, Lange H, Kriese K, Schmitt WD, Hoffmann S, Bartel F, Hauptmann S. Small polydispersed circular DNA contains strains of mobile genetic elements and occurs more frequently in permanent cell lines of malignant tumors than in normal lymphocytes. Oncol Rep. 2009;22:393–400.
Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019;575:699–703.
Schoenlein PV, Gottesman MM. Extrachromosomal DNA in human cancers. J Natl Cancer Inst. 1990;82:1798–800.
Zirbel RM, Mathieu UR, Kurz A, Cremer T, Lichter P. Evidence for a nuclear compartment of transcription and splicing located at chromosome domain boundaries. Chromosom Res. 1993;1:93–106.
Jack EM, Waters JJ, Harrison CJ. A scanning electron microscopy study of double minutes from a human tumour cell line. Cytogenet Cell Genet. 1987;44:49–52.
Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017;543:122–5.
Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, Alt FW. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell. 1983;35:359–67.
Fan Y, Mao R, Lv H, Xu J, Yan L, Liu Y, Shi M, Ji G, Yu Y, Bai J, et al. Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J Appl Genet. 2011;52:53–9.
Stark GR, Debatisse M, Giulotto E, Wahl GM. Recent progress in understanding mechanisms of mammalian DNA amplification. Cell. 1989;57:901–8.
Schimke RT. Gene amplification in cultured animal cells. Cell. 1984;37:705–13.
Solovei I, Kienle D, Little G, Eils R, Savelyeva L, Schwab M, Jager W, Cremer C, Cremer T. Topology of double minutes (dmins) and homogeneously staining regions (HSRs) in nuclei of human neuroblastoma cell lines. Genes Chromosomes Cancer. 2000;29:297–308.
Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–45.
Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37.
Kanda T, Otter M, Wahl GM. Mitotic segregation of viral and cellular acentric extrachromosomal molecules by chromosome tethering. J Cell Sci. 2001;114:49–58.
Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, Ji HP, Maley CC. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22:105–13.
Lundberg G, Rosengren AH, Hakanson U, Stewenius H, Jin Y, Stewenius Y, Pahlman S, Gisselsson D. Binomial mitotic segregation of MYCN-carrying double minutes in neuroblastoma illustrates the role of randomness in oncogene amplification. PLoS One. 2008;3:e3099.
Von Hoff DD, Waddelow T, Forseth B, Davidson K, Scott J, Wahl G. Hydroxyurea accelerates loss of extrachromosomally amplified genes from tumor cells. Cancer Res. 1991;51:6273–9.
Sanchez AM, Barrett JT, Schoenlein PV. Fractionated ionizing radiation accelerates loss of amplified MDR1 genes harbored by extrachromosomal DNA in tumor cells. Cancer Res. 1998;58:3845–54.
Sun W, Quan C, Huang Y, Ji W, Yu L, Li X, Zhang Y, Zheng Z, Zou H, Li Q, et al. Constitutive ERK1/2 activation contributes to production of double minute chromosomes in tumour cells. J Pathol. 2015;235:14–24.
Oobatake Y, Shimizu N. Double-strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes Chromosomes Cancer. 2020;59:133–43.
Shimizu N, Itoh N, Utiyama H, Wahl GM. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol. 1998;140:1307–20.
Ji W, Bian Z, Yu Y, Yuan C, Liu Y, Yu L, Li C, Zhu J, Jia X, Guan R, et al. Expulsion of micronuclei containing amplified genes contributes to a decrease in double minute chromosomes from malignant tumor cells. Int J Cancer. 2014;134:1279–88.
Harasymiw LA, Tank D, McClellan M, Panigrahy N, Gardner MK. Centromere mechanical maturation during mammalian cell mitosis. Nat Commun. 2019;10:1761.
McKinley KL, Cheeseman IM. The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol. 2016;17:16–29.
deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, Koeman J, Seth S, Protopopov A, Felicella M, et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nature Genetics. 2018;50:708–17.
Xu K, Ding L, Chang TC, Shao Y, Chiang J, Mulder H, Wang S, Shaw TI, Wen J, Hover L, et al. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol. 2019;137:123–37.
Helias-Rodzewicz Z, Pedeutour F, Coindre JM, Terrier P, Aurias A. Selective elimination of amplified CDK4 sequences correlates with spontaneous adipocytic differentiation in liposarcoma. Genes Chromosomes Cancer. 2009;48:943–52.
Eckhardt SG, Dai A, Davidson KK, Forseth BJ, Wahl GM, Von Hoff DD. Induction of differentiation in HL60 cells by the reduction of extrachromosomally amplified c-myc. Proc Natl Acad Sci U S A. 1994;91:6674–8.
Ambros IM, Rumpler S, Luegmayr A, Hattinger CM, Strehl S, Kovar H, Gadner H, Ambros PF. Neuroblastoma cells can actively eliminate supernumerary MYCN gene copies by micronucleus formation--sign of tumour cell revertance? Eur J Cancer. 1997;33:2043–9.
Von Hoff DD, McGill JR, Forseth BJ, Davidson KK, Bradley TP, Van Devanter DR, Wahl GM. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci U S A. 1992;89:8165–9.
Shimizu N, Nakamura H, Kadota T, Kitajima K, Oda T, Hirano T, Utiyama H. Loss of amplified c-myc genes in the spontaneously differentiated HL-60 cells. Cancer Res. 1994;54:3561–7.
Valent A, Benard J, Clausse B, Barrois M, Valteau-Couanet D, Terrier-Lacombe MJ, Spengler B, Bernheim A. In vivo elimination of acentric double minutes containing amplified MYCN from neuroblastoma tumor cells through the formation of micronuclei. Am J Pathol. 2001;158:1579–84.
Kuttler F, Mai S. Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. Semin Cancer Biol. 2007;17:56–64.
Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, Piazza MS, Allan KC, Mack SC, Wang X, Gimple RC, et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell. 2019;179:1330–41 e13.
Carroll SM, DeRose ML, Gaudray P, Moore CM, Needham-Vandevanter DR, Von Hoff DD, Wahl GM. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol. 1988;8:1525–33.
Wahl GM, Robert de Saint Vincent B, DeRose ML. Effect of chromosomal position on amplification of transfected genes in animal cells. Nature. 1984;307:516–20.
Storlazzi CT, Fioretos T, Surace C, Lonoce A, Mastrorilli A, Strombeck B, D'Addabbo P, Iacovelli F, Minervini C, Aventin A, et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet. 2006;15:933–42.
Storlazzi CT, Lonoce A, Guastadisegni MC, Trombetta D, D'Addabbo P, Daniele G, L'Abbate A, Macchia G, Surace C, Kok K, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res. 2010;20:1198–206.
Van Roy N, Vandesompele J, Menten B, Nilsson H, De Smet E, Rocchi M, De Paepe A, Påhlman S, Speleman F. Translocation-excision-deletion-amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes Chromosomes Cancer. 2006;45:107–17.
Ly P, Cleveland DW. Rebuilding chromosomes after catastrophe: emerging mechanisms of Chromothripsis. Trends Cell Biol. 2017;27:917–30.
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40.
Vogt N, Gibaud A, Lemoine F, de la Grange P, Debatisse M, Malfoy B. Amplicon rearrangements during the extrachromosomal and intrachromosomal amplification process in a glioma. Nucleic Acids Res. 2014;42:13194–205.
L'Abbate A, Macchia G, D'Addabbo P, Lonoce A, Tolomeo D, Trombetta D, Kok K, Bartenhagen C, Whelan CW, Palumbo O, et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res. 2014;42:9131–45.
Zheng S, Fu J, Vegesna R, Mao Y, Heathcock LE, Torres-Garcia W, Ezhilarasan R, Wang S, McKenna A, Chin L, et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev. 2013;27:1462–72.
Zhou YH, Chen Y, Hu Y, Yu L, Tran K, Giedzinski E, Ru N, Gau A, Pan F, Qiao J, et al. The role of EGFR double minutes in modulating the response of malignant gliomas to radiotherapy. Oncotarget. 2017;8:80853–68.
Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343:72–6.
Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, Chamorro R, Munoz-Perez N, Puiggros M, Dorado Garcia H, et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet. 2020;52:29–34.
Nikolaev S, Santoni F, Garieri M, Makrythanasis P, Falconnet E, Guipponi M, Vannier A, Radovanovic I, Bena F, Forestier F, et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun. 2014;5:5690.
Morales C, Garcia MJ, Ribas M, Miro R, Munoz M, Caldas C, Peinado MA. Dihydrofolate reductase amplification and sensitization to methotrexate of methotrexate-resistant colon cancer cells. Mol Cancer Ther. 2009;8:424–32.
Meng X, Qi X, Guo H, Cai M, Li C, Zhu J, Chen F, Guo H, Li J, Zhao Y, et al. Novel role for non-homologous end joining in the formation of double minutes in methotrexate-resistant colon cancer cells. J Med Genet. 2015;52:135–44.
Cai M, Zhang H, Hou L, Gao W, Song Y, Cui X, Li C, Guan R, Ma J, Wang X, et al. Inhibiting homologous recombination decreases extrachromosomal amplification but has no effect on intrachromosomal amplification in methotrexate-resistant colon cancer cells. Int J Cancer. 2019;144:1037–48.
Jin Y, Liu Z, Cao W, Ma X, Fan Y, Yu Y, Bai J, Chen F, Rosales J, Lee KY, et al. Novel functional MAR elements of double minute chromosomes in human ovarian cells capable of enhancing gene expression. PLoS One. 2012;7:e30419.
Vicario R, Peg V, Morancho B, Zacarias-Fluck M, Zhang J, Martinez-Barriocanal A, Navarro Jimenez A, Aura C, Burgues O, Lluch A, et al. Patterns of HER2 gene amplification and response to anti-HER2 therapies. PLoS One. 2015;10:e0129876.
Singer MJ, Mesner LD, Friedman CL, Trask BJ, Hamlin JL. Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proc Natl Acad Sci U S A. 2000;97:7921–6.
Kim JA, Anurag M, Veeraraghavan J, Schiff R, Li K, Wang XS. Amplification of TLK2 induces genomic instability via impairing the G2-M checkpoint. Mol Cancer Res. 2016;14:920–7.
Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25:282–303.
Gradishar WJ. HER2 therapy--an abundance of riches. N Engl J Med. 2012;366:176–8.
Sanborn JZ, Salama SR, Grifford M, Brennan CW, Mikkelsen T, Jhanwar S, Katzman S, Chin L, Haussler D. Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res. 2013;73:6036–45.
DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin. 2017;67:439–48.
Huh YO, Tang G, Talwalkar SS, Khoury JD, Ohanian M, Bueso-Ramos CE, Abruzzo LV. Double minute chromosomes in acute myeloid leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia are associated with micronuclei, MYC or MLL amplification, and complex karyotype. Cancer Genet. 2016;209:313–20.
Raymond E, Faivre S, Weiss G, McGill J, Davidson K, Izbicka E, Kuhn JG, Allred C, Clark GM, Von Hoff DD. Effects of hydroxyurea on extrachromosomal DNA in patients with advanced ovarian carcinomas. Clin Cancer Res. 2001;7:1171–80.
Shimizu N. Extrachromosomal double minutes and chromosomal homogeneously staining regions as probes for chromosome research. Cytogenet Genome Res. 2009;124:312–26.
Acknowledgements
Not applicable.
Funding
This work was supported by The Science and Technology Commission of Shanghai (17DZ2260100, 19JC1410200).
Author information
Authors and Affiliations
Contributions
SG and XF provided direction and guidance throughout the preparation of this manuscript. XG collected and interpreted studies and was a major contributor to the writing and editing of the manuscript. JY reviewed and made significant revisions to the manuscript. PC assisted in the revision of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, X., Yu, J., Chai, P. et al. Novel insights into extrachromosomal DNA: redefining the onco-drivers of tumor progression. J Exp Clin Cancer Res 39, 215 (2020). https://doi.org/10.1186/s13046-020-01726-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-020-01726-4