
Abstract
Tumor heterogeneity results in differential response to therapy due to the existence of plastic tumor cells, called cancer stem cells (CSCs), which exhibit the property of resistance to therapy, invasion and metastasis. These cells have a distinct, signaling network active at every stage of progression. It is difficult to envisage that the CSCs will have a unique set of signaling pathways regulating every stage of disease progression. Rather, it would be easier to believe that a single pivotal pathway having significant contribution at every stage, which can further turn on a battery of signaling mechanisms specific to that stage, would be instrumental in regulating the signaling network, enabling easy transition from one state to another. In this context, we discuss the role of RhoC which has contributed to several phenotypes during tumor progression.
RhoC (Ras homolog gene family member C) has been widely reported to regulate actin organization. It has been shown to impact the motility of cancer cells, resultantly affecting invasion and metastasis, and has contributed to carcinoma progression of the breast, pancreas, lung, ovaries and cervix, among several others. The most interesting finding has been its indispensable role in metastasis. Also, it has the ability to modulate various other phenotypes like angiogenesis, motility, invasion, metastasis, and anoikis resistance. These observations suggest that RhoC imparts the plasticity required by tumor cells to exhibit such diverse functions based on microenvironmental cues. This was further confirmed by recent reports which show that it regulates cancer stem cells in breast, ovary and head and neck cancers. Studies also suggest that the inhibition of RhoC results in abolition of advanced tumor phenotypes.
Our review throws light on how RhoC, which is capable of modulating various phenotypes may be the apt core signaling candidate regulating disease progression. Additionally, mice studies show that RhoC is not essential for embryogenesis, giving scope for its development as a possible therapeutic target. This review thus stresses on the need to understand the protein and its functioning in greater detail to enable its development as a stem cell marker and a possible therapeutic target.
Similar content being viewed by others


Background
Despite major advances in molecular and diagnostic sciences, and the emergence of personalized treatment, challenges remain due to the non-availability of personalized medicine across all cancers and the ever-evolving nature of this form of therapy. Therefore, the study and exploration of signaling pathways has intensified in the quest for novel therapeutic targets. The role of Notch, Wnt, Tumor Growth Factor-beta (TGF-beta) and Nuclear Factor kappa-light-chain-enhancer of activated B cells (NFκB) amongst several other signaling pathways, has been well studied over the years and across several tumors. Consequently, several candidates (like Epidermal Growth Factor Receptor (EGFR) for lung cancer) have been developed as molecular targets for personalized medicine. Another signaling pathway that has been shown to contribute extensively to tumor progression in several tumor types is the Ras homolog gene family member C (RhoC) signaling pathway. RhoC belongs to the Rho family of small Guanosine Triphosphatases (GTPases) [1]. Rho GTPases are small signaling G-proteins that regulate cytoskeletal organization and thus affect multiple cellular functions, including cell motility, polarity and division by switching between the Guanosine Triphosphate (GTP) and Guanosine Diphosphate (GDP) bound states, as shown in Fig. 1 [2,3,4,5]. This switch in states is tightly regulated by RhoGAPs (Rho GTPase Activating Proteins), RhoGEFs (Rho Guanine Exchange Factors) and RhoGDIs (Rho Guanine Dissociation Inhibitors) [6]. RhoGAPs support the intrinsic GTPase activity of RhoGTPases, converting them from the GTP-bound state to GDP-bound, thereby leading to their deactivation [7]. RhoGEFs on the other hand help maintaining RhoGTPases in the active state by facilitating their switch from the GDP-bound form to the GTP-bound form [8]. The third regulator protein, the RhoGDIs, stabilize the RhoGTPases in the GDP form, consequently playing an important role in determining localization of the protein [9]. Active forms of the protein, GTP-bound, regulate the actin cytoskeleton, cell cycle, membrane trafficking and transcription [10]. Significantly, the activity of each RhoGTPase is governed by regulators specific to each of them, with reports suggesting that the activity of RhoC in particular is regulated by GEFs like p190RhoGEF, ARHGEF10, ARHGEF12 and GAPs like p190RhoGAP, DLC1 to name a few [11,12,13,14]. Although the Rho isoforms have more than 90% sequence homology with each other and are known to regulate actin organization, several studies have proven that they have vastly distinct functions [15]. For example, RhoA and RhoC localize in the cytoplasm while RhoB localizes to the endosomal membrane [16]. In mouse embryonic fibroblasts, RhoA is dispensable for actomyosin regulation; however, it is important for mitosis [17, 18]. RhoC, too, has been shown to be responsible for cytoskeletal reorganization and cellular motility. Nevertheless, RhoA and RhoC have distinct roles in invasion, as they act through different targets [19]. In the context of viral infections, the process of cell contraction through the viral protein F11, is seen to be dependent on ROCK signaling via activation by RhoC and not RhoA. Additionally, this effect is seen to be abrogated by recruitment of Pak6 to the cellular membrane by another RhoGTPase, RhoD [20]. An interesting study by Hakem et al. showed that RhoC is dispensable for embryogenesis but is essential for metastasis [21]. While RhoC has an important contribution to metastasis, RhoB has been reported, using mice models, to be a tumor suppressor [22]. A study in colorectal cancer indicated a strong correlation of both RhoA and RhoC in metastasis and invasion [23], whereas other studies in breast and colon cancer have suggested that RhoA often inhibits cell invasion, while RhoC, on the other hand, enhances cell invasion [24, 25]. In 1989, Chardin and colleagues showed that RhoC affects actin microfilaments in Vero cells [26]. Following this report, there have been incessant efforts to understand the role of this molecule in both physiological and patho-physiological conditions. Here, we provide a comprehensive account of the work done on this molecule in the context of cancer progression and resistance to therapy, followed by an outline of the work that remains to be performed to enable better understanding of the working of this protein within the cell.

Cycling of Ras homolog gene family member C (RhoC) between active and inactive forms: The switching of RhoC between the inactive GDP-bound form to the active GTP-bound form is regulated by Guanine Nucleotide Exchange Factors (GEFs), GTPase Activating Proteins (GAPs) and Guanine Dissociation Inhibitors (GDIs). Binding to GTP changes the conformation of the molecule, thus allowing the binding of various downstream effectors of RhoC like Diaphanous Related Formin (mDia) and Rho Associated Coiled-Coil Containing Protein Kinase (ROCK), thereby facilitating various downstream signaling pathways
RhoC in tumor phenotypes and molecular pathways
The role of RhoC in carcinoma progression has been extensively clarified by several research groups over the years. The first report, which suggested that RhoC contributed to progression of cancer, was by Suwa et al. in the year 1998. This group investigated changes in the expression levels of the Rho family of genes—RhoA, B and C in pancreatic ductal carcinoma. It was discovered that the expression of the RhoC gene was significantly higher in metastatic tumors than in primary tumors, whereas RhoA and RhoB did not show significant changes in expression under these conditions. Also, increased RhoC expression significantly correlated with poor prognosis of patients, unlike RhoA and RhoB, which showed no such correlation [27]. Following this study, several other groups reported the role of RhoC in numerous other cancers, including those of the breasts, skin, ovaries, liver and head and neck, among several others [28,29,30,31,32,33]. The increased expression of RhoC is therefore positively correlated to poor prognosis. However, activation of the molecule is necessary to enable its downstream effects. MyoGEF, a molecule responsible for activation of RhoA and RhoC was found to regulate both polarity and invasive phenotypes of MDA-MB-231 (an invasive breast cancer cell line) [34]. On the contrary, p190RhoGAP which converts GTP-bound Rho to the inactive GDP-bound form, is associated with reduced proliferation, migration and invasion in breast and pancreatic cancer models, thus acting as the antithesis of MyoGEF in this context [35, 36]. The role of RhoC as a transforming oncogene was postulated by van Golen et al. This group demonstrated that the stable transfectants of human mammary epithelial cells over-expressing RhoC not only gained tumorigenic properties but were also highly invasive [32]. In the year 2013, Xie et al. showed that stable transfection of the RhoC expression vector into a normal hepatocyte cell line, imparted tumor phenotypes like proliferation, anchorage-independent growth, migration, invasion, increased expression of matrix metalloproteases like MMP2 and MMP9, and elevated levels of Vascular Endothelial Growth Factor (VEGF), further cementing RhoC’s role as an oncogene [37]. Additionally, RhoC was found to have a positive association with dedifferentiation and the phosphorylated form of p70s6k, a protein well known for its role in promoting survival and proliferation, thus making it a probable marker for carcinogenesis and the progression of ovarian epithelial carcinoma [38].
MicroRNAs (miRNAs), which have diverse cellular functions, have been shown to regulate RhoC expression. Chen X et al., showed in 2015 that increased miR-93-5P (specific to RhoC) resulted in decreased tumorigenesis and the progression of epithelial ovarian carcinoma [39]. Another microRNA, miR-10b, inhibits the translation of homeobox D10. This process leads to increased RhoC expression, resulting in increased invasion and metastasis of breast cancer [40]. The tumorigenesis and progression of ovarian epithelial carcinoma was also seen to be inhibited by miR 106b, which binds to the 3′ UTR of RhoC [41]. Long non-coding RNA (lncRNA) TDRG1 increases RhoC expression, consequently leading to tumorigenesis in the ovarian epithelial carcinoma model via miR-93 [42]. Similarly, over-expression of lncRNA ABHD11-AS1 correlates with the progression of epithelial ovarian carcinoma by regulating RhoC [43]. Signaling pathways regulated by RhoC are also involved in regulating the expression of certain lncRNAs. The expression of HOTAIR, an lncRNA known to be a negative prognostic marker, is under the influence of RhoC-ROCK signaling in breast cancer cells [44]. On the other hand, the proliferation, invasion and metastasis of gastric cancer was blocked by miR-493, which was proven to directly target RhoC [45]. Likewise, miR-372 over-expression led to G1 arrest and apoptosis, along with a suppression of tumor growth and the metastasis of endometrial carcinoma via inhibition of RhoC [46].
The switch from a locally confined tumor to an invasive, metastatic form is the most damaging alteration in a tumor; allowing it to disseminate, eventually leading to a poor prognosis. Epithelial to mesenchymal transition (EMT) is a prerequisite to metastasis [47,48,49,50,51]. Interestingly, DNA array analysis of metastatic melanoma cells revealed that RhoC was important for metastasis [52]. RhoGTPases are also known to regulate the activity of myocardin-related transcription factors MRTFA/B, which are upstream of genes necessary for metastasis [53]. Inhibition of MRTF using a pharmacological inhibitor CCG-203971, led to decreased lung metastases in mice injected with the highly invasive, RhoC overexpressing melanoma cell line SK-Mel-147 [54].Bellovin et al. showed that Ets-1 increases RhoC expression in LIM1863 colon cancer cells, resulting in increased EMT and cell migration [24]. Interestingly, Zhou X et al., demonstrated that HIF (Hypoxia Inducible Factor), a protein known to be associated with abnormal growth and invasion, acts via trasncriptionally altering the RhoC-ROCK1 pathway in the pancreatic cancer model [55]. RhoC also regulates EMT in cervical cancer, wherein the inhibition of Notch1 and RhoC resulted in the abolition of actin stress fiber formation and fibronectin expression, the two important changes associated with EMT [56]. The Rho proteins regulate cytoskeletal organization, and true to its nature, RhoC has been shown to regulate actin organization in tumors resulting in enhanced migration, invasion and metastasis [21, 24, 57,58,59,60]. Significantly, using stable benign breast epithelial cell lines with inducible RhoA and RhoC expression, Sarah Lang et al. have shown, that RhoC and not RhoA, is indispensable for invasion [61].
The close association between TGF-β1 and RhoC has been reported in several tumors. RhoC, known to play an important role in rearrangement of the cytoskeleton, has been implicated in EMT, invasion and metastasis of lung adenocarcinoma cells when induced by TGF-β1. Moreover, the down-regulation of RhoC using shRNA abolished TGF-β1-mediated EMT induction [62]. Similarly, in ovarian epithelial carcinoma cells, RhoC has been shown to mediate EMT that is stimulated by TGF-β1 and VEGF [63]. A similar study performed on the cervical carcinoma model demonstrated that RhoC is necessary for TGF-β1 driven EMT [64]. Notably, it has been postulated that tumor cells disseminate either as single cells or move collectively. Using intravital imaging, Giampieri and group showed that TGF-β switches breast cancer cells from cohesive to single cell motility, which is essential for intravasation, by transcriptionally reprograming tumor cells, thereby leading to alteration in the expression of several genes, including RhoC [65].
In cervical cancer, Notch1 has been proven to regulate RhoC leading to changes in migration and invasion [56]. Similarly, stromal cell-derived factor-1 (SDF-1) was seen to modulate Jurkat cell migration through the RhoC-ROS pathway [66]. Using the SUM-149 Inflammatory Breast Cancer (IBC) cell line, Joglekar et al., have reported that caveolin-1 regulates RhoC-mediated invasion by activation of Akt-1 [67]. In the colon cancer model, HOXD10 and RhoC were shown to negatively correlate with each other in both patient specimens and cell lines. Further analysis revealed that increased HOXD10 led to suppression of the MAPK and AKT pathways known to regulate RhoC [68]. The interaction of FMNL-3 with RhoC was seen to lead to increased MMP2, MMP9 and VEGF, consequently leading to increased invasion in colon cancer cell lines [69]. The knockdown of RhoC in cholangiocellular carcinoma cells on the other hand, resulted in the suppression of invasion and migration [70]. On similar lines, YMO1, a protein belonging to the Yurt and mosaic family, was seen to reduce the invasion and metastatic ability of hepatocellular carcinoma cells, by targeting RhoC [71].
There are several pathways, which are regulated by RhoC, that contribute to carcinoma progression and maintenance. RhoC alters the Mitogen Activated Protein Kinase (MAPK) and Phosphoinositide 3 kinase/AKT Serine Threonine Kinase (PI3K/AKT) pathways to regulate invasion [72, 73]. Interestingly, while RhoC is an important player in the motility of inflammatory breast cancer (IBC) and melanoma, it does not contribute to motility in prostate cancer cell lines, such as PC-3. However, RhoC does regulate the invasion of PC-3 [74]. RhoC has also been shown to activate the Protein-Tyrosine Kinase 2 (PYK2) pathway in prostate cancer, consequently leading to metastasis in prostate cancer [75]. Immunohistochemical analysis of RhoC expression in this study showed a significant correlation between both lymph node and distant metastases and the activation of Matrix Metalloprotease 2 (MMP2) and Matrix Metalloprotease 9 (MMP9). Further, antibody array analysis showed that RhoC activated several kinases, including MAPK, Focal Adhesion Kinase (FAK), AKT and PYK2. RhoC also regulates Formin-like 3 (FMNL3) mediated cell migration and invasion as it is involved in polarized migration [19]. In another study, RhoC has been shown to stimulate alpha5 integrin expression and the Src-dependent activation of p130 Crk-associated substrate/Ras-Related C3 Botulinum Toxin Substrate 1 (Cas/Rac1) signaling [76]. RhoC also controls cofilin activity to modulate actin organization, resultantly affecting invasion and invadopodia formation [12, 77]. Table 1 summarizes the pathways in which RhoC has been implicated. These and several more studies elucidate the mechanisms of RhoC-mediated regulation of cancer phenotypes.
Not only does RhoC regulate tumor growth, EMT, migration, invasion, and metastasis, it also regulates angiogenesis in tumors. Vasculogenesis and angiogenesis are controlled by angiogenic factors, such as VEGF-A [78]. In the physiological context, RhoC stimulates the proliferation of human umbilical vein endothelial cells (HUVECs) by stabilizing beta-catenin, which in turn enhances cyclin D1 expression. Cyclin-D1 subsequently drives cell-cycle progression [79]. Apart from proliferation, RhoC also regulates various angiogenic features like pseudopod formation and migration ability in HUVECs and MVECs (myeloma vascular endothelial cells) via ROCK and MAPK signaling [80]. RhoC has been shown to regulate angiogenesis in breast cancer [73, 81], where it modulates the expression of VEGF, fibroblast growth factor-basic (bFGF), interleukin-6 and interleukin-8, which are important in angiogenesis [81]. Similarly, RhoC may promote VEGF expression in oesophageal squamous cell carcinoma, thus regulating angiogenesis [82]. In cervical cancer, conditioned media from SiHa cells stably over-expressing the RhoC gene resulted in increased in vitro tube formation by HUVEC cells. Additionally, immunohistochemical analysis of clinical specimens revealed that RhoC and VEGF were expressed in the same areas of tumor sections [56]. Cancers promote the development of an alternate vascular system (known as vasculogenic mimicry) to support its growth and proliferation. RhoC and its effector ROCK2 have been shown to play important roles in this context by activation of the ERK and MMP pathways in the hepatocellular carcinoma model [83]. The importance of RhoC to carcinoma progression was exemplified by Hakem et al. Using a mouse model, they showed that RhoC was dispensable for post-natal development, however it’s depletion reduced metastasis [21]. The disseminated tumor cells survived in blood vessels until they relocated to a distant site and formed metastases. The ability to survive in these conditions is an important attribute of metastatic tumor cells. We have earlier published that RhoC is also involved in anoikis resistance [56], which may aid the cells to survive for prolonged durations in suspension in blood. As summarized in Fig. 2, RhoC therefore plays an important role at various stages of tumor progression.

An illustration depicting the diverse roles of RhoC in various aspects of cancer progression: RhoC significantly contributes to cancer initiation, proliferation, stemness maintenance, angiogenesis, invasion, intravasation, and metastasis across numerous tumor models, as shown
Intriguingly, while RhoC has been highly implicated in several aspects of carcinoma progression, there is no report of a mutation associated with this gene [84]. Analysis of the COSMIC database suggests that mutations of RhoC in cancer are very rare. Only 60 unique samples out of 47,923 showed mutations. In total, there were only 17 missense mutations, 7 silent mutations and 1 deletion mutation included in the database. Interestingly, these mutations are scattered across the protein domains, indicating that they are unlikely to be driver mutations and are most likely passenger mutations.
RhoC in Cancer stem cells
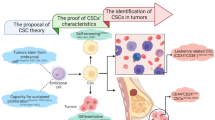
The phenotypic and functional heterogeneity observed among cells within the same tumor represents one of the greatest challenges in cancer therapeutics and has caused confounding clinical outcomes, as it results in heterogeneous therapy response. The plasticity of tumor cells enables them to adapt and survive at different stages of tumor progression in a dynamically changing microenvironment, beginning from the site of tumor initiation and ending at a distant metastatic site. Such plastic tumor cells exhibit several stem-like characteristics, such as self-renewal, high drug efflux capacity and better DNA repair, and are thus termed cancer stem cells (CSCs) [85, 86].
Tumor formation is broadly believed to adhere to the stochastic/clonal evolution model or the hierarchical/classical CSC model [87]. The clonal evolution model attributes cancer initiation to genetic abnormalities within a normal cell. According to the clonal evolution theory, these aberrations lead to a heterogenous tumor pool consisting of multiple clones, each of them being equally proficient at giving rise to a tumor. The classical CSC model on the other hand, entrusts tumor induction capability solely to the CSC population. This theory believes that a cancer stem cell gives rise to transit amplifying cells, which further give rise to the differentiated tumor bulk. Recent findings have led scientists to believe that this model is not unidirectional, but is in fact highly dynamic and plastic, allowing for interconversion of these states via differentiation and dedifferentiation resulting in a complex, heterogenous tumor [87]. An outline of these models has been illustrated in Fig. 3. Several reports cumulatively suggest that RhoC regulates numerous steps of tumor progression, including proliferation [56, 88, 89], EMT [62, 63], invasion [61, 90, 91], intravasation [56, 92], extravasation [92], anoikis resistance [56], angiogenesis [56, 92] and metastasis [58, 61, 93]. Resultantly, it is apt to believe that RhoC may be involved in regulating or maintaining tumor plasticity, which endows adaptability at every stage of tumor progression. Plasticity is known to be an inherent feature of stem cells and in line with this, recent research has shown that RhoC is involved in maintenance of the stemness phenotype.

Tumor induction models and the possible role of RhoC: The clonal evolution theory postulates that genetic abnormalities lead to tumor formation, with every clone of cells thus produced being equally capable of regenerating the tumor (a). On the contrary, the cancer stem cell theory proposes the presence of a minute sub-population known as cancer stem cells (CSCs), which alone hold the potential for resurgence of the various populations that constitute the tumor. This includes the differentiated tumor bulk, transit amplifying cells which are mildly pluripotent and proliferative and an intermediate substantially pluripotent “plastic” state (b). These cellular states are highly dynamic with cells being capable of constantly moving from one state to another. We propose that RhoC, with it’s involvement in multiple tumor phenotypes could play a pivotal role in regulating this “switch” via it’s downstream effectors
An important finding by Rosenthal et al. indicates a strong correlation between RhoC and ALDH, a breast cancer stem cell (BCSC) marker [93]. Using the aggressive BCSC cell line SUM149, Rosenthal et al. show that cells with active ALDH (ALDH+) have higher levels of RhoC than those with inactive ALDH (ALDH−). Tumorigenicity studies utilizing a limiting number of 50 cells in mice resulted in no induction of tumors in mice injected with ALDH+/shRhoC cells, whereas 5 out of 9 mice with ALDH+/scrambled cells formed tumors. Moreover, incidences of lung metastases were found to be around five times higher in mice injected with ALDH+/scrambled cells compared to those injected with ALDH+/shRhoC cells, indicating the stem-like property of cells containing RhoC. Finally, a tissue microarray of breast cancer samples from 136 patients indicated a high correlation between RhoC and ALDH1, further supporting RhoC’s association with ALDH.
The role of RhoC in CSC maintenance has also been illustrated in head and neck squamous cell carcinoma (HNSCC) by Islam et al. [94]. Using UM-SCC-1 and UM-SCC-47 cell lines, they show that the siRNA mediated inhibition of RhoC led to decreased expression of ALDH, CD44, Oct3/4, Sox2, and Nanog, in addition to a diminished formation of tumorspheres. Further, Islam et al. determine that tumorspheres have increased levels of RhoC and genes associated with stemness compared to cells grown as monolayers, whereas the inhibition of RhoC leads to a reduction in the expression of stemness genes, which points towards RhoC’s possible role in CSC induction. Islam et al. then demonstrate that RhoC leads to stemness induction in head and neck cancer by activation of STAT3 via IL-6. In a study by Sang et al., ovarian cancer stem cells (OCSCs) were sorted using the CD117 marker from A2780-PM and A2780-PTX-PM, two drug-resistant and invasive ovarian cancer cell lines [90]. These OCSCs were found to have elevated expressions of RhoC. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay revealed that cells inhibited for RhoC had decreased cell proliferation and drug resistance. Further, inhibition of RhoC by RhoC-specific siRNA led to decreased expression of stemness markers like CD133 and CD117, as observed by real-time quantitative PCR, suggesting a possible role of RhoC in the formation of OCSCs. It may be thus appropriate to deduce that overexpression of RhoC results in enhanced plasticity/stemness of cancer cells (Fig. 3).
RhoC and resistance to Cancer therapy
Therapy resistance occurs when the tumor stops responding to treatments, such as radiation, chemotherapy and other forms of targeted therapy. Current approaches primarily kill treatment-sensitive cells, while the therapy-resistant cells survive and expand over time to cause recurrence. In order to ensure a complete cure, it is important to eliminate all cancer cells, including the therapy-resistant ones, which have now come to be known as CSCs. Indeed, recent reports suggest a vast number of molecular mechanisms that regulate and contribute to resistance. RhoC has also been shown to contribute to therapy resistance in some tumor models. Interestingly, a seminal article by Mani et al., which reported that EMT induces stemness in cancer cells, opened up a new understanding of CSCs and their targeting [95]. This study showed that transformed human mammary epithelial cells that had undergone EMT formed tumors much more efficiently and had stemness properties. Therefore, this study opens up avenues to explore opportunities to target pathways that regulate EMT and resultantly eliminate CSCs, the culprit for recurrence and metastasis.
RhoC is one such protein whose role in EMT induction and regulation is well documented. It has been demonstrated that RhoC along with LIM Domain Kinase 2 (LIMK2) is a direct target of p53 during chemotherapy [96]. Kawata et al. demonstrated that RhoC may have a role to play in endocrine therapy resistance, a significant barrier to successful prostate cancer treatment [97]. Two years later, the same group reported that RhoC was up-regulated in breast cancer samples post chemotherapy treatment, indicating increased RhoC levels in the chemoresistant population. They also showed a correlation between RhoC expression and reduced E-cadherin levels, pointing towards a possible role for RhoC in EMT, thereby leading to therapy resistance in breast cancer [98]. Research from our laboratory (unpublished data) shows that RhoC and its downstream effector, ROCK2 regulates the radioresistance in cervical cancer.
Several groups have reported that RhoC inhibitors have a profound effect on carcinoma phenotypes in vitro, using both cell lines and tumor biopsy-derived cells. Wenandy et al. attempted to understand RhoC’s clinical application and found that RhoC has a Human Leukocyte Antigen-A3 (HLA-A3) restricted epitope, which is recognized by cytotoxic T cells. Wenandy et al. propose that RhoC may serve as a target for anti-cancer immunotherapy [99]. Inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG-CoA reductase), commonly known as statins, have been widely used to understand the function of this molecule. The HMG-CoA pathway produces geranylgeranyl pyrophosphate (GGPP) and Farnesyl pyrophosphate (FPP) as intermediate products, which are important for activation of RhoGTPases [100]. Interestingly, the use of farnesyltransferase inhibitors (FTI) has been shown to have a profound effect on tumor phenotype. Treatment of IBC cells with FTI showed a reversal of RhoC-induced phenotypes like anoikis resistance, motility, and invasion [73]. Collisson EA et al. proposed statins as a primary prophylaxis for melanoma, demonstrating a reduction in RhoC activation upon treatment with atorvastatin, consequently leading to inhibition of invasion and metastasis [100]. Another study reported that a combinatorial use of atorvastatin and celecoxib, in vitro, resulted in an induction of cell cycle arrest and apoptosis in colon cancer cells [101]. Atorvastatin-mediated inhibition of RhoC also blocked metastasis in head and neck cancer cells, in vitro [102]. Encouraging reports indicate that use of the statin group of drugs reduces incidence of esophageal cancer [103]. A study by Kaushal et al. showed that antiRhoC siRNA led to decreased invasion, motility, and migration of the breast cancer cell lines SUM149 and MDA-MB-231, suggesting that RhoC is a potential therapeutic target [104]. This group went on to further design “smart” nanoparticles that delivered anti-RhoC siRNA into breast cancer cells, thereby successfully impeding migration and invasion [105].
Despite a series of convincing reports on RhoC’s role in various tumor phenotypes, it has not been developed further as a prognostic marker or therapeutic target. There have been attempts to use inhibitors, such as atorvastatin, to understand its function, but further development has not been reported. Considering its extensive contribution to carcinomas and their progression, it is important to initiate studies to define RhoC as a potential therapeutic target.
Conclusion
CSCs have the ability to evade therapy, repair and survive under stressful conditions, such as hypoxia. These cells also have EMT properties, coupled with the ability to invade and migrate. Resistance to therapy has also been attributed to CSCs in several tumors. Given that CSCs are an important subset of the tumor and can elicit various tumor phenotypes, it is important to develop targets against these cells for better cancer care. The ideal target for such adaptive and plastic cells would be a molecular pathway that is important for CSC maintenance and regulates several functional attributes of these cells. The available literature suggests that RhoC has a major contribution in CSC maintenance. The role of RhoC in carcinoma progression has been well studied and reported. This molecule has a central role in most of the reported tumor phenotypes, with recent reports pointing to its possible role in the stemness of cancer cells. Given the evidence implicating RhoC in various aspects of tumor progression, this molecule seems to be an ideal druggable target. However, the three Rho GTPases: RhoA, RhoB, and RhoC show an 85% amino acid sequence identity, leaving little room for the development of a specific inhibitor to RhoC alone [15]. Nevertheless, considering that the role of RhoC in tumor progression is overwhelming, efforts must be steered towards developing siRNA, antibodies, or small molecule-based inhibitors of RhoC. It is, therefore, of prime importance to thoroughly explore the application of this molecule in cancer prognostication, in order to efficiently tackle the disease.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- BCSC:
-
Breast cancer stem cell
- bFGF:
-
Fibroblast growth factor-basic
- Cas/Rac1:
-
Crk-associated substrate/Ras-Related C3 Botulinum Toxin Substrate 1
- CSCs:
-
Cancer stem cells
- EGFR:
-
Epidermal Growth Factor Receptor
- EMT:
-
Epithelial to mesenchymal transition
- FAK:
-
Focal Adhesion Kinase
- FMNL3:
-
Formin-like 3
- FPP:
-
Farnesyl pyrophosphate
- GAPs:
-
GTPase Activating Proteins
- GDIs:
-
Guanine Dissociation Inhibitors
- GDP:
-
Guanosine Diphosphate
- GEFs:
-
Guanine Nucleotide Exchange Factors
- GGPP:
-
Geranylgeranyl pyrophosphate
- GTPases:
-
Guanosine Triphosphatases
- HLA-A3:
-
Human Leukocyte Antigen-A3
- HNSCC:
-
Head and neck squamous cell carcinoma
- HUVECs:
-
Human umbilical vein endothelial cells
- IBC:
-
Inflammatory breast cancer
- LIMK2:
-
LIM Domain Kinase 2
- MAPK:
-
Mitogen Activated Protein Kinase
- mDia:
-
Diaphanous Related Formin
- miRNAs:
-
MicroRNAs
- MMP9:
-
Matrix Metalloprotease 9
- MRTF:
-
Myocardin-related transcription factors
- MVECs:
-
Myeloma vascular endothelial cells
- NFκB:
-
Nuclear Factor kappa-light-chain-enhancer of activated B cells
- OCSCs:
-
Ovarian cancer stem cells
- PI3K/AKT:
-
Phosphoinositide 3 kinase/AKT Serine Threonine Kinase
- PYK2:
-
Protein-Tyrosine Kinase 2
- RhoC:
-
Ras homolog gene family member C
- ROCK:
-
Rho Associated Coiled-Coil Containing Protein Kinase
- TGF-beta:
-
Tumor Growth Factor-beta
- VEGF:
-
Vascular Endothelial Growth Factor
References
Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16(10):522–9.
Ridley AJ. The GTP-binding protein Rho. Int J Biochem Cell Biol. 1997;29(11):1225–9.
Hall A. Ras-related GTPases and the cytoskeleton. Mol Biol Cell. 1992;3(5):475–9.
Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279(5350):509–14.
Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33(Pt 5):891–5.
Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13(1):13–22.
Lamarche N, Hall A. GAPs for rho-related GTPases. Trends Genet. 1994;10(12):436–40.
Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26(12):724–32.
Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 1999;11(8):545–54.
Hall A, Paterson HF, Adamson P, Ridley AJ. Cellular responses regulated by rho-related small GTP-binding proteins. Philos Trans R Soc Lond Ser B Biol Sci. 1993;340(1293):267–71.
Healy KD, Hodgson L, Kim TY, Shutes A, Maddileti S, Juliano RL, et al. DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol Carcinog. 2008;47(5):326–37.
Bravo-Cordero JJ, Sharma VP, Roh-Johnson M, Chen X, Eddy R, Condeelis J, et al. Spatial regulation of RhoC activity defines protrusion formation in migrating cells. J Cell Sci. 2013;126(Pt 15):3356–69.
Mohl M, Winkler S, Wieland T, Lutz S. Gef10--the third member of a rho-specific guanine nucleotide exchange factor subfamily with unusual protein architecture. Naunyn Schmiedebergs Arch Pharmacol. 2006;373(5):333–41.
Shi GX, Yang WS, Jin L, Matter ML, Ramos JW. RSK2 drives cell motility by serine phosphorylation of LARG and activation of Rho GTPases. Proc Natl Acad Sci U S A. 2017;115(2):E190–E9.
Wheeler AP, Ridley AJ. Why three rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301(1):43–9.
Heasman SJ, Ridley AJ. Mammalian rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701.
Aepfelbacher M, Essler M, Huber E, Sugai M, Weber PC. Bacterial toxins block endothelial wound repair. Evidence that rho GTPases control cytoskeletal rearrangements in migrating endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17(9):1623–9.
Melendez J, Stengel K, Zhou X, Chauhan BK, Debidda M, Andreassen P, et al. RhoA GTPase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J Biol Chem. 2011;286(17):15132–7.
Vega FM, Fruhwirth G, Ng T, Ridley AJ. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol. 2011;193(4):655–65.
Durkin CH, Leite F, Cordeiro JV, Handa Y, Arakawa Y, Valderrama F, et al. RhoD inhibits RhoC-ROCK-dependent cell contraction via PAK6. Dev Cell. 2017;41(3):315–29 e7.
Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19(17):1974–9.
Liu AX, Rane N, Liu JP, Prendergast GC. RhoB is dispensable for mouse development, but it modifies susceptibility to tumor formation as well as cell adhesion and growth factor signaling in transformed cells. Mol Cell Biol. 2001;21(20):6906–12.
Wang HB, Liu XP, Liang J, Yang K, Sui AH, Liu YJ. Expression of RhoA and RhoC in colorectal carcinoma and its relations with clinicopathological parameters. Clin Chem Lab Med. 2009;47(7):811–7.
Bellovin DI, Simpson KJ, Danilov T, Maynard E, Rimm DL, Oettgen P, et al. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 2006;25(52):6959–67.
Simpson KJ, Dugan AS, Mercurio AM. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 2004;64(23):8694–701.
Chardin P, Boquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 1989;8(4):1087–92.
Suwa H, Ohshio G, Imamura T, Watanabe G, Arii S, Imamura M, et al. Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br J Cancer. 1998;77(1):147–52.
Carr KM, Bittner M, Trent JM. Gene-expression profiling in human cutaneous melanoma. Oncogene. 2003;22(20):3076–80.
Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, et al. Significant association of rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9(7):2632–41.
Shikada Y, Yoshino I, Okamoto T, Fukuyama S, Kameyama T, Maehara Y. Higher expression of RhoC is related to invasiveness in non-small cell lung carcinoma. Clin Cancer Res. 2003;9(14):5282–6.
Shinto E, Tsuda H, Matsubara O, Mochizuki H. Significance of RhoC expression in terms of invasion and metastasis of colorectal cancer. Nihon Rinsho. 2003;61(Suppl 7):215–9.
van Golen KL, Wu ZF, Qiao XT, Bao LW, Merajver SD. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000;60(20):5832–8.
Wang W, Yang LY, Yang ZL, Huang GW, Lu WQ. Expression and significance of RhoC gene in hepatocellular carcinoma. World J Gastroenterol. 2003;9(9):1950–3.
Wu D, Asiedu M, Wei Q. Myosin-interacting guanine exchange factor (MyoGEF) regulates the invasion activity of MDA-MB-231 breast cancer cells through activation of RhoA and RhoC. Oncogene. 2009;28(22):2219–30.
Shen CH, Chen HY, Lin MS, Li FY, Chang CC, Kuo ML, et al. Breast tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res. 2008;68(19):7779–87.
Kusama T, Mukai M, Endo H, Ishikawa O, Tatsuta M, Nakamura H, et al. Inactivation of rho GTPases by p190 RhoGAP reduces human pancreatic cancer cell invasion and metastasis. Cancer Sci. 2006;97(9):848–53.
Xie S, Zhu M, Lv G, Geng Y, Chen G, Ma J, et al. Overexpression of Ras homologous C (RhoC) induces malignant transformation of hepatocytes in vitro and in nude mouse xenografts. PLoS One. 2013;8(1):e54493.
Zhao Y, Zheng HC, Chen S, Gou WF, Xiao LJ, Niu ZF. The role of RhoC in ovarian epithelial carcinoma: a marker for carcinogenesis, progression, prognosis, and target therapy. Gynecol Oncol. 2013;130(3):570–8.
Chen S, Chen X, Xiu YL, Sun KX, Zhao Y. Inhibition of ovarian epithelial carcinoma tumorigenesis and progression by microRNA 106b mediated through the RhoC pathway. PLoS One. 2015;10(5):e0125714.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–8.
Chen X, Chen S, Xiu YL, Sun KX, Zong ZH, Zhao Y. RhoC is a major target of microRNA-93-5P in epithelial ovarian carcinoma tumorigenesis and progression. Mol Cancer. 2015;14:31.
Chen S, Wang LL, Sun KX, Xiu YL, Zong ZH, Chen X, et al. The role of the long non-coding RNA TDRG1 in epithelial ovarian carcinoma tumorigenesis and progression through miR-93/RhoC pathway. Mol Carcinog. 2018;57(2):225–34.
Wu DD, Chen X, Sun KX, Wang LL, Chen S, Zhao Y. Role of the lncRNA ABHD11-AS1 in the tumorigenesis and progression of epithelial ovarian cancer through targeted regulation of RhoC. Mol Cancer. 2017;16(1):138.
He H, Wei Z, Du F, Meng C, Zheng D, Lai Y, et al. Transcription of HOTAIR is regulated by RhoC-MRTF-A-SRF signaling pathway in human breast cancer cells. Cell Signal. 2017;31:87–95.
Zhou W, Zhang C, Jiang H, Zhang Z, Xie L, He X. MiR-493 suppresses the proliferation and invasion of gastric cancer cells by targeting RhoC. Iran J Basic Med Sci. 2015;18(10):1027–33.
Liu BL, Sun KX, Zong ZH, Chen S, Zhao Y. MicroRNA-372 inhibits endometrial carcinoma development by targeting the expression of the Ras homolog gene family member C (RhoC). Oncotarget. 2016;7(6):6649–64.
Bailey JM, Singh PK, Hollingsworth MA. Cancer metastasis facilitated by developmental pathways: sonic hedgehog, notch, and bone morphogenic proteins. J Cell Biochem. 2007;102(4):829–39.
Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Vincent-Salomon A, Thiery JP. Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003;5(2):101–6.
Wallerand H, Cai Y, Wainberg ZA, Garraway I, Lascombe I, Nicolle G, et al. Phospho-Akt pathway activation and inhibition depends on N-cadherin or phospho-EGFR expression in invasive human bladder cancer cell lines. Urol Oncol. 2010;28(2):180–8.
Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–5.
Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11(3):257–68.
Haak AJ, Appleton KM, Lisabeth EM, Misek SA, Ji Y, Wade SM, et al. Pharmacological inhibition of Myocardin-related transcription factor pathway blocks lung metastases of RhoC-overexpressing melanoma. Mol Cancer Ther. 2017;16(1):193–204.
Zhou X, Guo X, Chen M, Xie C, Jiang J. HIF-3alpha promotes metastatic phenotypes in pancreatic Cancer by transcriptional regulation of the RhoC-ROCK1 signaling pathway. Mol Cancer Res. 2017;16(1):124–34.
Srivastava S, Ramdass B, Nagarajan S, Rehman M, Mukherjee G, Krishna S. Notch1 regulates the functional contribution of RhoC to cervical carcinoma progression. Br J Cancer. 2010;102(1):196–205.
Faried A, Faried LS, Kimura H, Nakajima M, Sohda M, Miyazaki T, et al. RhoA and RhoC proteins promote both cell proliferation and cell invasion of human oesophageal squamous cell carcinoma cell lines in vitro and in vivo. Eur J Cancer. 2006;42(10):1455–65.
Ikoma T, Takahashi T, Nagano S, Li YM, Ohno Y, Ando K, et al. A definitive role of RhoC in metastasis of orthotopic lung cancer in mice. Clin Cancer Res. 2004;10(3):1192–200.
Lin M, DiVito MM, Merajver SD, Boyanapalli M, van Golen KL. Regulation of pancreatic cancer cell migration and invasion by RhoC GTPase and caveolin-1. Mol Cancer. 2005;4(1):21.
Liu N, Zhang G, Bi F, Pan Y, Xue Y, Shi Y, et al. RhoC is essential for the metastasis of gastric cancer. J Mol Med (Berl). 2007;85(10):1149–56.
Lang S, Busch H, Boerries M, Brummer T, Timme S, Lassmann S, et al. Specific role of RhoC in tumor invasion and metastasis. Oncotarget. 2017;8(50):87364–78.
Lu X, Guo H, Chen X, Xiao J, Zou Y, Wang W, et al. Effect of RhoC on the epithelial-mesenchymal transition process induced by TGF-beta1 in lung adenocarcinoma cells. Oncol Rep. 2016;36(6):3105–12.
Gou WF, Zhao Y, Lu H, Yang XF, Xiu YL, Zhao S, et al. The role of RhoC in epithelial-to-mesenchymal transition of ovarian carcinoma cells. BMC Cancer. 2014;14:477.
He X, Qian Y, Cai H, Yang S, Cai J, Wang Z. RhoC is essential in TGF-beta1 induced epithelial-mesenchymal transition in cervical cancer cells. Oncol Lett. 2015;10(2):985–9.
Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11(11):1287–96.
Luo J, Li D, Wei D, Wang X, Wang L, Zeng X. RhoA and RhoC are involved in stromal cell-derived factor-1-induced cell migration by regulating F-actin redistribution and assembly. Mol Cell Biochem. 2017;436(1–2):13–21.
Joglekar M, Elbazanti WO, Weitzman MD, Lehman HL, van Golen KL. Caveolin-1 mediates inflammatory breast Cancer cell invasion via the Akt1 pathway and RhoC GTPase. J Cell Biochem. 2017;118(5):1273.
Yuan YH, Wang HY, Lai Y, Zhong W, Liang WL, Yan FD, et al. Epigenetic inactivation of HOXD10 is associated with human colon cancer via inhibiting the RHOC/AKT/MAPK signaling pathway. Cell Commun Signal. 2019;17(1):9.
Zeng YF, Xiao YS, Liu Y, Luo XJ, Wen LD, Liu Q, et al. Formin-like 3 regulates RhoC/FAK pathway and actin assembly to promote cell invasion in colorectal carcinoma. World J Gastroenterol. 2018;24(34):3884–97.
Yang H, Liang J, Zhou J, Mi J, Ma K, Fan Y, et al. Knockdown of RHOC by shRNA suppresses invasion and migration of cholangiocellular carcinoma cells via inhibition of MMP2, MMP3, MMP9 and epithelial-mesenchymal transition. Mol Med Rep. 2016;13(6):5255–61.
Chang RM, Pei L, Fang F, Xu JF, Yang H, Zuo CH, et al. YMO1 suppresses invasion and metastasis by inhibiting RhoC signaling and predicts favorable prognosis in hepatocellular carcinoma. Oncotarget. 2016;7(34):55585–600.
Ruth MC, Xu Y, Maxwell IH, Ahn NG, Norris DA, Shellman YG. RhoC promotes human melanoma invasion in a PI3K/Akt-dependent pathway. J Invest Dermatol. 2006;126(4):862–8.
van Golen KL, Bao LW, Pan Q, Miller FR, Wu ZF, Merajver SD. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin Exp Metastasis. 2002;19(4):301–11.
Yao H, Dashner EJ, van Golen CM, van Golen KL. RhoC GTPase is required for PC-3 prostate cancer cell invasion but not motility. Oncogene. 2006;25(16):2285–96.
Iiizumi M, Bandyopadhyay S, Pai SK, Watabe M, Hirota S, Hosobe S, et al. RhoC promotes metastasis via activation of the Pyk2 pathway in prostate cancer. Cancer Res. 2008;68(18):7613–20.
Arpaia E, Blaser H, Quintela-Fandino M, Duncan G, Leong HS, Ablack A, et al. The interaction between caveolin-1 and Rho-GTPases promotes metastasis by controlling the expression of alpha5-integrin and the activation of Src, Ras and Erk. Oncogene. 2011;31(7):884–96.
Bravo-Cordero JJ, Oser M, Chen X, Eddy R, Hodgson L, Condeelis J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr Biol. 2011;21(8):635–44.
Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273(2):114–27.
Hoeppner LH, Sinha S, Wang Y, Bhattacharya R, Dutta S, Gong X, et al. RhoC maintains vascular homeostasis by regulating VEGF-induced signaling in endothelial cells. J Cell Sci. 2015;128(19):3556–68.
Zhao Z, Liu K, Tian X, Sun M, Wei N, Zhu X, et al. Effects of RhoC downregulation on the angiogenesis characteristics of myeloma vascular endothelial cells. Cancer Med. 2019;8(7):3502–3510.
van Golen KL, Wu ZF, Qiao XT, Bao L, Merajver SD. RhoC GTPase overexpression modulates induction of angiogenic factors in breast cells. Neoplasia. 2000;2(5):418–25.
Zhao ZH, Tian Y, Yang JP, Zhou J, Chen KS. RhoC, vascular endothelial growth factor and microvascular density in esophageal squamous cell carcinoma. World J Gastroenterol. 2015;21(3):905–12.
Zhang JG, Zhang DD, Liu Y, Hu JN, Zhang X, Li L, et al. RhoC/ROCK2 promotes vasculogenic mimicry formation primarily through ERK/MMPs in hepatocellular carcinoma. Biochim Biophys Acta Mol basis Dis. 2019;1865(6):1113–25.
Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer. 2002;87(6):635–44.
Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101(39):14228–33.
Srivastava S, Krishna S. Cancer stem cells: an approach to identify newer therapeutic targets. J Stem Cells. 2009;4(2):123–31.
Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59(1):168–79.
Wu Y, Tao Y, Chen Y, Xu W. RhoC regulates the proliferation of gastric cancer cells through interaction with IQGAP1. PLoS One. 2012;7(11):e48917.
Xu XD, Shen HB, Zhu L, Lu JQ, Zhang L, Luo ZY, et al. Anti-RhoC siRNAs inhibit the proliferation and invasiveness of breast cancer cells via modulating the KAI1, MMP9, and CXCR4 expression. Onco Targets Ther. 2017;10:1827–34.
Sang XB, Sun KX, Wang LL, Chen S, Wu DD, Zong ZH, et al. Effects and mechanism of RhoC downregulation in suppressing ovarian cancer stem cell proliferation, drug resistance, invasion and metastasis. Oncol Rep. 2016;36(6):3267–74.
Stoletov K, Montel V, Lester RD, Gonias SL, Klemke R. High-resolution imaging of the dynamic tumor cell vascular interface in transparent zebrafish. Proc Natl Acad Sci U S A. 2007;104(44):17406–11.
Reymond N, Im JH, Garg R, Cox S, Soyer M, Riou P, et al. RhoC and ROCKs regulate cancer cell interactions with endothelial cells. Mol Oncol. 2015;9(6):1043–55.
Rosenthal DT, Zhang J, Bao L, Zhu L, Wu Z, Toy K, et al. RhoC impacts the metastatic potential and abundance of breast cancer stem cells. PLoS One. 2012;7(7):e40979.
Islam M, Sharma S, Teknos TN. RhoC regulates cancer stem cells in head and neck squamous cell carcinoma by overexpressing IL-6 and phosphorylation of STAT3. PLoS One. 2014;9(2):e88527.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15.
Croft DR, Crighton D, Samuel MS, Lourenco FC, Munro J, Wood J, et al. p53-mediated transcriptional regulation and activation of the actin cytoskeleton regulatory RhoC to LIMK2 signaling pathway promotes cell survival. Cell Res. 2010;21(4):666–82.
Kawata H, Shimada N, Kamiakito T, Komatsu K, Morita T, Ota T, et al. RhoC and guanine nucleotide exchange factor Net1 in androgen-unresponsive mouse mammary carcinoma SC-4 cells and human prostate cancer after short-term endocrine therapy. Prostate. 2012;72(10):1071–9.
Kawata H, Kamiakito T, Omoto Y, Miyazaki C, Hozumi Y, Tanaka A. RhoC upregulation is correlated with reduced E-cadherin in human breast cancer specimens after chemotherapy and in human breast cancer MCF-7 cells. Horm Cancer. 2014;5(6):414–23.
Wenandy L, Sorensen RB, Svane IM, Thor Straten P, Andersen MH. RhoC a new target for therapeutic vaccination against metastatic cancer. Cancer Immunol Immunother. 2008;57(12):1871–8.
Collisson EA, Kleer C, Wu M, De A, Gambhir SS, Merajver SD, et al. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol Cancer Ther. 2003;2(10):941–8.
Xiao H, Zhang Q, Lin Y, Reddy BS, Yang CS. Combination of atorvastatin and celecoxib synergistically induces cell cycle arrest and apoptosis in colon cancer cells. Int J Cancer. 2008;122(9):2115–24.
Islam M, Sharma S, Kumar B, Teknos TN. Atorvastatin inhibits RhoC function and limits head and neck cancer metastasis. Oral Oncol. 2013;49(8):778–86.
Beales IL, Hensley A, Loke Y. Reduced esophageal cancer incidence in statin users, particularly with cyclo-oxygenase inhibition. World J Gastrointest Pharmacol Ther. 2013;4(3):69–79.
Kaushal N, Durmaz YY, Bao L, Merajver SD, ElSayed ME. “Smart” nanoparticles enhance the cytoplasmic delivery of anti-RhoC silencing RNA and inhibit the migration and invasion of aggressive breast Cancer cells. Mol Pharm. 2015;12(7):2406–17.
Kaushal N, Tiruchinapally G, Durmaz YY, Bao L, Gilani R, Merajver SD, et al. Synergistic inhibition of aggressive breast cancer cell migration and invasion by cytoplasmic delivery of anti-RhoC silencing RNA and presentation of EPPT1 peptide on “smart” particles. J Control Release. 2018;289:79–93.
Acknowledgements
Not applicable
Funding
We would like to thank the Department of Science and Technology for financial support. Pavana Thomas and Annapurna Pranatharthi have received fellowships from CSIR and CSIR-UGC respectively. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Author information
Authors and Affiliations
Contributions
PT and SS are major contributors towards the writing, making of figures and proofreading of the article. AP and CR contributed partly in manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Thomas, P., Pranatharthi, A., Ross, C. et al. RhoC: a fascinating journey from a cytoskeletal organizer to a Cancer stem cell therapeutic target. J Exp Clin Cancer Res 38, 328 (2019). https://doi.org/10.1186/s13046-019-1327-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1327-4