
Abstract
Increased understanding of the genetic aetiology of advanced non-small-cell lung cancer (aNSCLC) has facilitated personalised therapies that target specific molecular aberrations associated with the disease. Biopsy samples for mutation testing may be taken from primary or metastatic sites, depending on which sample is most accessible, and upon differing diagnostic practices between territories. However, the mutation status concordance between primary tumours and corresponding metastases is the subject of debate. This review aims to ascertain whether molecular diagnostic testing of either the primary or metastatic tumours is equally suitable to determine patient eligibility for targeted therapies. A literature search was performed to identify articles reporting studies of mutations in matched primary and metastatic aNSCLC tumour samples. Clinical results of mutation status concordance between matched primary and metastatic tumour samples from patients with aNSCLC were collated. Articles included in this review (N =26) all reported mutation status data from matched primary and metastatic tumour samples obtained from adult patients with aNSCLC. Generally, substantial concordance was observed between primary and metastatic tumours in terms of EGFR, KRAS, BRAF, p16 and p53 mutations. However, some level of discordance was seen in most studies; mutation testing methodologies appeared to play a key role in this, along with underlying tumour heterogeneity. Substantial concordance in mutation status observed between primary and metastatic tumour sites suggests that diagnostic testing of either tumour type may be suitable to determine a patient’s eligibility for personalised therapies. As with all diagnostic testing, highly sensitive and appropriately validated mutation analysis methodologies are desirable to ensure accuracy. Additional work is also required to define how much discordance is clinically significant given natural tumour heterogeneity. The ability of both primary and metastatic tumour sites to accurately reflect the tumour mutation status will allow more patients to receive therapies personalised to their disease.
Similar content being viewed by others
Introduction
Lung cancer is the leading cause of cancer mortality [1], with non-small-cell lung cancer (NSCLC) accounting for ~85 % of primary lung cancers [2]. Metastatic spread of the disease is a complication of advanced NSCLC (aNSCLC) [3], which usually precedes the fatal stages by a few months. Unfortunately, many patients present with metastases at diagnosis [4] due to the relatively asymptomatic earlier stages of the disease.
Increased understanding of the genetic aetiology of aNSCLC over the past decade has provided the opportunity for personalised treatment for some patients, targeting several of the key molecular aberrations now known to be associated with aNSCLC [5, 6]. A recent meta-analysis of the incidence and coincidence of mutations in aNSCLC reported that three genes – tumour protein p53 (TP53), epidermal growth factor receptor (EGFR) and v-Ki-ras2 Kirsten rat sarcoma viral oncogenes homolog (KRAS) – were commonly mutated in NSCLC of adenocarcinoma (ADC) histology [7], one of the most common histological subtypes of NSCLC [2, 8]. The functional pathways associated with these genes are well-documented [9–11], however in brief: mutations in EGFR are known to activate the MAPK/ERK pathway [10, 11]; mutations in KRAS, BRAF and PIK3CA are known to alter MAPK/ERK activation [10, 11]; and mutations in TP53 are known to lead to loss of function of this tumor suppressor [9].
Molecular diagnostic testing is now recommended by several clinical guidelines [12–14] for patients with NSCLC to determine eligibility for targeted therapies. For example, EGFR tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, are approved for patients with mutations in the EGFR gene; it is now widely accepted that response to EGFR TKIs is greater in patients with tumours harbouring EGFR mutations compared with wild-type EGFR oncogenes [15]. Similarly, translocations involving the anaplastic lymphoma kinase gene predict patients who will respond to the TKI crizotinib [16]. Further to this, new targeted therapies are being developed for patients with other molecular aberrations; for example, selumetinib, cobimetinib and trametinib are being developed for patients with KRAS mutation-positive tumours [17].
As a result of the availability of targeted therapies, determining tumour mutation status in patients with aNSCLC is now a key component of diagnosis in many countries, with the hope, where possible, of optimising treatment outcomes [18, 19]. Currently, most aNSCLC cases are diagnosed by a histological analysis of the tumour tissue; for example, around 77 % of patients in England and Wales (UK) are diagnosed in this manner [20]. However, depending on patient ability and/or willingness to undergo sampling, whether samples are available or evaluable, and differing diagnostic practices between countries [12, 13, 21–25], either the primary tumour or a metastatic lesion may be biopsied [26]. However, the concordance in mutation status between matched primary and metastatic tumours is the subject of debate [3, 27–33], with limited understanding as to whether discordance reflects actual heterogeneity in mutations, or is an artifact of technical/sensitivity limitations in testing methodology [34–43]. Nevertheless, given that the collection of multiple invasive samples from a patient with NSCLC is undesirable, it is important to ascertain whether the mutation status of an individual patient’s NSCLC can be accurately characterised from biopsies of either the primary or metastatic sites.
Although research into the concordance of EGFR and KRAS mutation status between matched primary and metastatic tumours exists [33], to the authors’ knowledge, no review to date has systematically assessed the currently available data regarding whether metastatic samples are representative of primary tumour samples in patients with aNSCLC in terms of multiple mutations, and included consideration of the mutation testing methodologies employed. To address this knowledge gap, we describe in this review the level of mutation status concordance between matched primary and metastatic tumour samples, considering EGFR, KRAS and any other molecular aberrations noted in the included literature, as well as describing the mutation testing methodologies used.
Methods
Literature search
Literature searches of the MEDLINE® and PubMed® databases were carried out to identify journal articles published before 8 September 2014, which reported studies of mutations in aNSCLC tumour samples of primary or metastatic origin. The following search criteria were used: [NSCLC OR Lung] AND [mutation] AND [Primary] AND [Metas*]. The following were excluded: [non-English papers] AND [Editorials] AND [Commentaries]. Case reports, reviews and meta-analyses were also excluded, due to the variability in mutation testing methodologies used, which would limit the conclusions that could be drawn from such studies.
Articles were reviewed to identify those reporting results of clinical studies of mutation status concordance between matched primary and metastatic tumour samples from patients with aNSCLC. Where possible, the following parameters were recorded: study population demographics (age, gender, ethnicity and smoking status); number of matched tumour samples; description of matched tumour samples; molecular marker assessed; molecular marker assessment technique; mutation frequency in primary compared with metastatic tumour samples; and mutation status concordance rate between primary and metastatic tumour samples. Gene expression data, where reported, were not included.
Results
Included studies
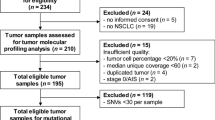
The literature search yielded 370 relevant abstracts, of which 26 were considered relevant for inclusion based upon the fact that they reported mutation data (any gene) for both primary and corresponding metastatic samples from patients with aNSCLC (Fig. 1). One study included data from small-cell lung cancers [44], and another included data from paired synchronous double tumours [45]; the remaining 24 studies are listed in Tables 1, 2, 3 and 4.

Study selection flow diagram. Literature searches carried out on 25 July 2013, 3 January 2014 and 8 September 2014
Patient demographics
Key demographic data, where available/applicable, are presented in Tables 1, 2, 3 and 4.
EGFR mutation status concordance
In total, 14 reports of EGFR mutation status concordance between matched primary and metastatic tumours were identified, of which four were in Caucasian patients: Kalikaki et al. [46] (Table 3) analysed 25 primary tumours and corresponding lung (n = 9), thoracic wall (n = 5), adrenal gland (n = 4), brain (n = 3), bone (n = 2), skin (n = 1) and liver (n = 1) metastases, and determined the EGFR mutation status concordance to be 72 % (18/25). Schmid et al. [47] (Table 3) analysed 96 primary tumours and corresponding lymph node metastases of Austrian patients, and found EGFR mutation status concordance to be 94 % (90/96). In a US study, Munfus-McCray et al. [48] (Table 3) assessed 9 primary tumours and corresponding metastatic tumours of the brain (n = 3), lymph node (n = 3) or pleura/knee/lung (n = 1 each), and found EGFR mutation status concordance to be 89 % (8/9). Lastly, Mansuet-Lupo et al. [30] (Table 1) analysed 10 primary tumours and corresponding lymph node (n = 8) or pleural metastases (n = 2) of French patients, and found EGFR mutation status concordance to be 90 % (9/10); the discordant result was obtained from a metastatic lymph node containing <15 % tumour cells, and an EGFR mutation corresponding to the primary tumour was subsequently identified in another lymph node from this patient, resulting in 100 % concordance.
Other studies of lymph node metastases, a commonly assessed metastatic site, were in Asian patients. Sun et al. [32] (Table 3) analysed 80 primary tumours and corresponding lymph node metastases of Chinese patients, and found EGFR mutation status concordance to be 91 % (73/80). Two of the discordant cases resulted from a different EGFR mutation being present in the primary versus metastatic tumour (E746-A750 vs L747-T751 and L747-P753insS vs R748-P752, respectively). In two more recent studies of Chinese patients, Han et al. [49] (Table 3) and Wei et al. [50] (Table 1) analysed, respectively, 22 and 50 primary tumours and corresponding lymph node metastases, and found EGFR mutation status concordance to be 95 % (21/22) and 94 % (47/50). However, quantitative analysis in Wei et al’s study indicated that EGFR mutation ratios (amount of mutant EGFR:all EGFR present) were significantly lower in metastatic compared with primary tumour samples (Wilcoxon matched-pair test; P < 0.01), suggesting a more moderate mutation ratio concordance of 84 %. In two studies of Japanese patients, Yatabe et al. [51] (Table 1) and Shimizu et al. [52] (Table 1) analysed, respectively, 77 and 70 primary tumours and corresponding lymph node metastases, and found EGFR mutation status concordance to be 100 % (77/77) and 86 % (60/70). Park et al. [31] (Table 1) analysed 101 primary tumours and corresponding lymph node metastases of Korean patients. EGFR mutation status concordance was found to be 88 % (89/101) via direct sequencing, with 11 discordant cases EGFR mutation-positive in the primary tumour only and one discordant case EGFR mutation-positive in the metastatic tumour only; however, retesting with a more sensitive heteroduplex analysis decreased the concordance to 83 % (84/101).
Another commonly studied metastatic site was the brain. Matsumoto et al. [53] (Table 1) analysed 8 primary tumours and corresponding brain metastases of Japanese patients, with an EGFR mutation status concordance of 100 % (8/8) detected via direct sequencing. Luo et al. [54] (Table 1) implemented the amplification-refractory mutation system (ARMS) method in their retrospective study of EGFR mutations in 15 primary tumours and corresponding brain metastases obtained from Chinese patients, which yielded a concordance of 93 % (14/15).
Other studies included additional metastatic sites as well as the brain. Gow et al. [28] (Table 1) analysed 67 primary tumours and corresponding metastases of the following sites obtained from Taiwanese patients: brain (n = 25); bone (n = 20); and pleura/skin/soft tissue, distant lymph node, gastrointestinal system, metastatic lung tumour or adrenal gland (n = 22). EGFR mutation status concordance was found to be 61 % (41/67) via direct sequencing. The 26 discordant results were EGFR mutation-positive in their metastatic tumour only; these were reanalysed using the ARMS method, which indicated that 10/26 (38 %) of these were in fact concordant. Combining the ARMS and direct sequencing results yielded an overall concordance of 73 % (49/67). Han et al. [29] (Table 3) analysed 37 primary tumours and corresponding metastases of the following sites obtained from Korean patients: pleural effusion (n = 12), pleura (n = 9), brain (n = 5), lymph node (n = 3), lung (n = 2), soft tissue (n = 2), adrenal gland (n = 1), pericardial effusion (n = 1), pericardium (n = 1) and ovary (n = 1); EGFR mutation status concordance was found to be 81 % (30/37).
KRAS mutation status concordance
Overall, 10 reports of KRAS mutation status concordance between matched primary and metastatic tumours were identified, of which three analysed lymph node metastases. Schmid et al. [47] (Table 3, previously described) found KRAS mutation status concordance to be 74 % (71/96), including one discordant result of a KRAS mutation that was different in the primary (G12C) versus the metastatic (G12R) tumour, and another with unknown mutation status in the corresponding primary tumour of a KRAS mutation-positive metastasis. Unusually, two patients with an EGFR mutation (one with the mutation in their primary tumour and one with the mutation in a lymph node metastasis) had an additional KRAS mutation in the corresponding metastases. Sun et al. [32] (Table 3, previously described) found a KRAS mutation status concordance of 93 % (74/80). Alsdorf et al. [55] (Table 2) analysed 19 primary tumours and corresponding lymph node metastases of German patients. Direct sequencing yielded 4 discordant results; however, re-evaluation of mutation status using a combination of the ARMS method and enrichment of tumour cells by laser capture microdissection found identical KRAS mutations in all 19 matched pairs (100 % concordance).
Other studies assessed mixed/non-lymph node metastatic sites, mostly in Caucasian patients apart from Han et al’s study [29] in Korean patients (Table 3, previously described), which found a KRAS mutation status concordance of 97 % (36/37).
The remaining studies were as follows: Li et al. [56] (Table 2) analysed 15 primary tumours and corresponding metastases of various origin obtained from Spanish patients (with multiple sites tested per patient), and found a KRAS mutation status concordance rate of 100 % (15/15). A retrospective study by Badalian et al. [57] (Table 2) analysed 11 primary tumours and corresponding bone metastases of Hungarian patients using a restriction fragment length polymorphism-polymerase chain reaction (RFLP-PCR) method, and found a KRAS mutation status concordance of 64 % (7/11). Another retrospective study by Kalikaki et al. [46] (Table 3, previously described) found KRAS mutation status concordance to be 76 % (19/25). Cortot et al. [3] (Table 2) analysed 21 primary tumours and corresponding metachronous/synchronous metastases of the brain (n = 13), lung (n = 4) or bone/soft tissue (n = 2 each), obtained from French patients. Direct sequencing found KRAS mutation status concordance to be 71 % (15/21); however, re-testing using a mutant-enriched PCR analysis increased the concordance to 81 % (17/21). In a study of US patients, Holst et al. [58] (Table 2) analysed 10 primary tumours and corresponding intrathoracic metastases of patients with the bronchioloalveolar adenocarcinoma NSCLC subtype, and found a KRAS mutation status concordance of 100 % (10/10). In a further study of US patients, Munfus-McCray et al. [48] (Table 3, previously described) found a KRAS mutation status concordance of 89 % (8/9).
Concordance of other mutations
In total, four studies included analysis of other mutations as well as/instead of those in KRAS and EGFR.
Reichel et al. [44] examined the pattern of p53 mutations in 26 primary lung tumours and 60 corresponding metastases obtained from Swiss patients. A total of 7/9 patients with p53 mutation in the primary tumour had identical mutations in all corresponding metastases. In one patient with discordant results, a p53 mutation was found in one metastatic site (liver), but wild-type p53 was detected in the primary tumour and in a metastatic lesion of the kidney. In the other patient, a p53 mutation was detected in the primary tumour and one metastatic site (kidney), whereas wild-type p53 was detected in a metastatic lesion of the liver. Further to this, Holst et al. [58] (Table 2, previously described) found that when p53 loss of heterozygosity was detected in the primary tumour, it was also detected in the corresponding metastases.
An Italian study by Marchetti et al. [59] (Table 4) assessed 30 primary tumours and corresponding lymph node metastases, and found a p16 mutation status concordance of 100 % (30/30).
Schmid et al. [47] (Table 3, previously described) observed novel BRAF exon 15 mutations in 2 primary tumours and not in corresponding metastases in Austrian patients. However, KRAS/BRAF and EGFR/BRAF mutations were found to be mutually exclusive.
Allelic patterns between primary and metastatic tumours
Two studies included in this review presented results related to EGFR mutation heterogeneity in matched primary and metastatic tumour samples.
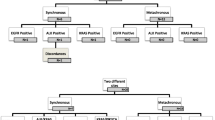
Tang et al. [60] (Table 1) assessed EGFR mutation heterogeneity in primary tumours and corresponding lymph node metastases from 9 EGFR mutation-positive patients, by taking multiple samples from non-contiguous sites of both primary tumours and metastases. Overall, 54/56 (96 %) and 25/30 (83 %) primary and metastatic tumour sites were EGFR mutation-positive, respectively. A total of 5/9 patients had identical EGFR mutations at multiple sites within the primary tumour and corresponding metastases; however, 2/9 patients presented with two different variants of Exon 19 deletions within the primary tumour: 1/9 patients had a mixture of wild-type EGFR and EGFR Exon 19 deletions; and 1/9 patients carried both L858R mutations and Exon 19 deletions. However, metastases were non-heterogeneous, with only a single type of mutation detected in each which was always present in at least one site of the primary tumour (Fig. 2).

EGFR mutation pattern in 56 primary tumour and 30 lymph node metastasis sites obtained from nine patients with EGFR-mutant lung adenocarcinomas. A homogeneous mutation pattern was detected in five primary tumours (cases 2, 3, 4, 7 and 9) and all but one (case 6) metastasis case. Case 6 had mixed wild-type and mutant sites in both primary tumour sites and corresponding metastases. EGFR, epidermal growth factor receptor. Reprinted from Cancer Prev Res (Phila), 2008, 1, 192–200, Tang et al., Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas, with permission from AACR [60]
Further to this, Takahashi et al. [61] (Table 3) compared whole-genome allelic imbalance of 8 primary tumours and corresponding brain (n = 7), lymph node (n = 3), liver (n = 2), pulmonary (n = 1) and pleural (n = 1) metastases obtained from Japanese patients. p53 and EGFR mutations were detected in 7/8 (88 %) and 3/8 (38 %) primary tumours and corresponding metastases, respectively. Genetic alterations were similar between the majority of cases (>67 %); however, there were genetic alterations (specifically, chromosomal regions of allelic imbalance indicated by imbalance of allele homo-/heterozygosity) in the metastatic but not primary tumour in 7/8 cases; and in the primary but not metastatic tumour in 4/8 cases.
Mutation patterns in multiple primary tumours
Two papers included in our review included analysis of mutation status concordance between multiple primary tumours.
A Japanese study by Matsuzoe et al. [45] investigated p53 mutations in 20 paired synchronous double tumours; p53 mutations occurred in 7/20 samples, with three different distributions: (i) only one tumour had the mutation (4/7); (ii) each tumour had a different mutation (2/7); and (iii) the same mutation was found in both tumours (1/7). In addition, 3/7 patients had metastatic lymph nodes in which p53 mutations were found that were identical to those found in the corresponding primary tumour. The third pattern (iii) was suggested to represent metastatic lung cancer.
Chen et al. [27] (Table 1) analysed 180 primary tumours and corresponding metastases of Asian patients. They found the following concordance rates for EGFR mutations: paired pulmonary primary nodules, 76 % (31/41); paired primary lung tumours and distant metastases, 86 % (30/35); paired primary lung tumours and metastatic lymph nodes, 90 % (44/49); and paired metachronous primary tumours (i.e. diagnosed at different times), 91 % (50/55). Overall concordance was estimated at 86 % (155/180) using a high-resolution melting method.
Somatic versus passenger mutations
Most studies included in this review were restricted to analysis of a small set of biomarkers. Interestingly, a French study by Vignot et al. [62] (Table 4) investigated the presence of multiple somatic alterations in 15 primary tumours and corresponding metastases (locoregional [n = 73], central nervous system [n = 3], distant adenopathy [n = 2], adrenal [n = 1], cutaneous [n = 1], parietal [n = 1]) and determined which alterations were likely to be driving recurrent mutations (defined as mutations that occur in ≥5 % of NSCLC samples in the Catalogue of Somatic Mutations in Cancer or are amplified/deleted in ≥5 % of NSCLC samples in the literature) or passenger mutations (all other mutations). A total of 161 and 190 somatic alterations were identified in the primary and metastatic tumours, respectively. Of these, 159 of these were classed as likely to be passenger mutations; the concordance rate between mutations found in primary compared with metastatic tumours was 94 % for recurrent mutations and 63 % for those considered likely to be passenger mutations.
Discussion
This literature review aimed to describe the level of mutation status concordance between primary and corresponding metastatic tumours, considering EGFR, KRAS and any other molecular aberrations noted. Various factors could contribute to mutation status discordance, such as differences in the sensitivity of mutation testing methods [63], mutation heterogeneity within the samples themselves [60, 61], or evolution of the mutation status of the primary and metastatic tumours [61]. Understanding these factors is important to learn how mutation testing may be improved, in order to ensure that as many patients as possible can access therapies personalised to the mutation status of their NSCLC tumours.
Most studies in this review focussed on EGFR and KRAS mutations, and in general, substantial mutation status concordance was found in terms of both. Furthermore, there was limited evidence to suggest, as other studies have [64], that KRAS mutation status concordance was lower than EGFR mutation status concordance in studies that analysed both mutations [29, 32, 46, 48, 49]. EGFR mutation frequency was found to be higher than KRAS mutation frequency in most [29, 48, 49, 61], but not all [46, 47], studies. A minority of studies included analysis of other molecular aberrations (BRAF, p53 and p16); results of these also indicated substantial mutation status concordance.
Where reported, discordance appeared to be partly related to the mutation testing methodology utilised. Many studies used direct sequencing to assess mutation status, which is known to be relatively insensitive [65], with mutations needing to be present in around 20 % of alleles interrogated [42, 43] to avoid false-negative results [33]. Retesting and confirmation of mutation results with more sensitive techniques was commonly employed by studies in this review. Park et al. [31] found EGFR mutation concordance was 83 % upon retesting with heteroduplex analysis versus 88 % with direct sequencing; 8 EGFR mutations were detected by heteroduplex analysis that were not picked up by direct sequencing, indicating the presence of false negatives. Similarly, the ARMS methodology was employed by some studies, which increased the concordance rates found by direct sequencing [28, 55].
The number of neoplastic versus non-neoplastic cells in tumour samples can also affect detection of mutations [66]. For example, Mansuet-Lupo et al. [30] detected an EGFR mutation in all metastatic lymph node samples of a patient aside from one sample which contained just 15 % tumour cells. Most studies included in this review used samples with >30 % tumour cells, and some employed laser-capture microdissection [67] to enrich tumour cell content in their samples [53, 55, 61].
Another methodological limitation of most studies in this review was the use of formalin-fixed paraffin-embedded (FFPE) tumour tissues; DNA breakages can occur during formalin fixation [68], with corrupt DNA linked to false-negative or -positive results. Other sample types may be more appropriate for mutation testing, including fresh/archived cytologic samples [69] and blood serum/plasma samples [26, 70] (not included in this review). There are data that have shown EGFR concordance rates are higher when analysing archival smear slides compared with FFPE tissues; Sun et al. [4] compared primary tumour FFPE histological material with fresh-frozen metastatic material, and found a higher rate of mutation in the fresh samples. In addition to this, a relatively large direct sequencing PCR amplicon was used (292 base pairs), which could have resulted in a much decreased sensitivity in fragmented FFPE-derived material compared with fresh-frozen tissue.
Another potential cause of mutation status discordance was the site of the metastatic sample; only one study included in our review specifically included mutation analysis comparing primary lung tumours and both corresponding lymph node and distant metastases [27], which yielded concordance rates of 90 and 86 %, respectively. This study also compared discordance rates between metachronous and synchronous tumours, which were 16 and 8 %, respectively. There was a lack of similar data from other studies, with most not reporting whether primary and metastatic samples were taken simultaneously.
However, although mutation test methodologies contribute to discordance, natural intratumoural heterogeneity cannot be excluded as a factor, which was observed in some studies in this review. For example, two studies investigated allelic patterns in tumours [60, 61], and found differences in heterozygosity and mutation subtype between primary tumours and their corresponding metastases. When Takahashi et al. [61] investigated the nature of each case where different genetic alterations were observed between matched primary and metastatic tumours, the process of metastasis was found to vary, suggesting multiple models of tumour progression and metastatic origins may apply in lung cancer; larger studies are needed to investigate this further. It is also interesting to note the findings of the study by Vignot et al. [62], who found that the global level of discordance could at least be partly attributed to passenger mutations.
Although not the focus of this review, limited data were found regarding the association between clinicopathological characteristics and mutation status concordance. Where data were available, as with previous studies, EGFR mutations were associated with female sex [32], non-smoker status [32, 47] and ADC histology [32]; one study found KRAS was associated with current smokers [47].
EGFR mutations in both primary and metastatic tumours were also found to be linked to response to EGFR TKI therapy. For example, Shimizu et al. [52] found the disease control rate to be significantly higher in patients with EGFR mutation-positive primary and metastatic tumours versus patients with EGFR mutation-positive primary tumours only (P = 0.062). Furthermore, Kalikaki et al. [46] found that of two patients who developed metachronous metastasis following EGFR TKI therapy, one had acquired resistance to the TKI therapy due to a metastatic tumour with a T790M EGFR mutation. The T790M EGFR mutation is one of the most common mechanisms leading to resistance to TKI therapy, of which there are several [71]. This suggests that consideration of alterations in EGFR mutation status during tumour progression is important, and repeat mutation testing may, therefore, be appropriate during clinical management of patients.
Most, but not all, studies included in this review reported that where both primary and corresponding metastatic tumours were EGFR mutation-positive prior to any therapy, the same mutation subtype was observed. However, further work is needed to determine the impact of both TKI therapies and chemotherapies on the mutation status of both primary and metastatic tumours (including the emergence of new mutation subtypes), and the resulting need for repeat biopsy.
Furthermore, there is a need for further data reporting on the concordance of the presentation of other biomarkers between primary and metastatic tumours; for example, although mutation expression profiles were beyond the scope of this review, there are ongoing and important developments in personalised therapies targeting proteins differentially expressed in tumours [72].
This review is limited by the heterogeneous patient populations included in the studies, with sample size, patient demographics, disease characteristics, tumour sampling methods and treatment histories differing substantially, and not always reported. This review was also limited to English papers only. Furthermore, until optimum testing techniques have been further researched and defined, the effects of the different mutation testing techniques discussed in this review cannot be fully evaluated.
Conclusions
The high level of concordance in mutation status between matched primary and metastatic tumours reported in studies here suggest that both sample types are equally viable options for informing treatment decisions based on mutation status. Robust mutation testing must be carried out to ensure accuracy of analysis; key components of robust mutation testing include the sensitivity of the assay and the quality and quantity of the tumour sample used. Furthermore, additional work is required to describe and define how much discordance is clinically relevant given natural tumour heterogeneity. The opportunity, therefore, exists for patients whose primary tumours are not available and/or evaluable to receive personalised therapy following mutation analysis of a metastatic lesion.
Abbreviations
- aNSCLC:
-
Advanced non-small-cell lung cancer
- ADC:
-
Adenocarcinoma
- ARMS:
-
Amplification‐refractory mutation system
- BAC:
-
Bronchioloalveolar carcinoma
- BRAF:
-
Murine sarcoma viral oncogene homolog B1
- CNS:
-
Central nervous system
- DNA:
-
Deoxyribonucleic acid
- EGFR:
-
Epidermal growth factor receptor
- FFPE:
-
Formalin-fixed paraffin-embedded
- GCC:
-
Giant cell carcinoma
- KRAS:
-
Kirsten rat sarcoma viral oncogenes homolog
- LCC:
-
Large cell carcinoma
- N/A:
-
Not available
- PCR:
-
Polymerase chain reaction
- PCR-SSCP:
-
Polymerase chain reaction-single-strand conformation polymorphism
- PNA-LNA:
-
Peptide nucleic acid-locked nucleic acid
- RFLP-PCR:
-
Restriction fragment length polymorphism-polymerase chain reaction
- SCC:
-
Small cell carcinoma
- SNP:
-
Single nucleotide polymorphism
- SSC:
-
Squamous cell carcinoma
- TKI:
-
Tyrosine kinase inhibitor
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29.
Youlden DR, Cramb SM, Baade PD. The International Epidemiology of Lung Cancer: geographical distribution and secular trends. J Thorac Oncol. 2008;3:819–31.
Cortot AB, Italiano A, Burel-Vandenbos F, Martel-Planche G, Hainaut P. KRAS mutation status in primary nonsmall cell lung cancer and matched metastases. Cancer. 2010;116:2682–7.
Sun PL, Jin Y, Kim H, Lee CT, Jheon S, Chung JH. High concordance of EGFR mutation status between histologic and corresponding cytologic specimens of lung adenocarcinomas. Cancer Cytopathol. 2013;121:311–9.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8.
Davies H, Hunter C, Smith R, Stephens P, Greenman C, Bignell G, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–5.
Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24:2371–6.
Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung cancer in never smokers–a review. Eur J Cancer. 2012;48:1299–311.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908.
Berg M, Soreide K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer: implications for targeted therapy. Discov Med. 2012;14:207–14.
Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 1802;2010:396–405.
Keedy VL, Temin S, Somerfield MR, Beasley MB, Johnson DH, McShane LM, et al. American Society of Clinical Oncology Provisional Clinical Opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol. 2011;29:2121–7.
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8:823–59.
Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244–85.
Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;12:7232–41.
Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94.
Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47.
Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616–24.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67.
Health and Social Care Information Centre. National Lung Cancer Audit Report 2013. http://www.hscic.gov.uk/catalogue/PUB12719/clin-audi-supp-prog-lung-nlca-2013-rep.pdf (2013). Accessed 19 May 2015.
National Comprehensive Cancer Network. Practice guidelines in oncology – version V.3.2012 (non-small-cell lung cancer). http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (2012). Accessed 19 May 2015.
Peters S, Adjei AA, Gridelli C, Reck M, Kerr K, Felip E. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23 Suppl 7:vii56–64.
Ellison G, Zhu G, Moulis A, Dearden S, Speake G, McCormack R. EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol. 2013;66:79–89.
Choi YL, Sun JM, Cho J, Rampal S, Han J, Parasuraman B, et al. EGFR mutation testing in patients with advanced non-small cell lung cancer: a comprehensive evaluation of real-world practice in an East Asian tertiary hospital. PLoS ONE. 2013;8:e56011.
National Collaborating Centre for Cancer (UK). http://www.nice.org.uk/guidance/cg121/resources/guidance-lung-cancer-pdf (2011). Accessed 19 May 2015.
Marchetti A, Normanno N. Recommendations for mutational analysis of EGFR in lung carcinoma. Pathologica. 2010;102:119–26.
Chen ZY, Zhong WZ, Zhang XC, Su J, Yang XN, Chen ZH, et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist. 2012;17:978–85.
Gow CH, Chang YL, Hsu YC, Tsai MF, Wu CT, Yu CJ, et al. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol. 2009;20:696–702.
Han HS, Eom DW, Kim JH, Kim KH, Shin HM, An JY, et al. EGFR mutation status in primary lung adenocarcinomas and corresponding metastatic lesions: discordance in pleural metastases. Clin Lung Cancer. 2011;12:380–6.
Mansuet-Lupo A, Zouiti F, Alifano M, Tallet A, Charpentier MC, Ducruit V, et al. Intratumoral distribution of EGFR mutations and copy number in metastatic lung cancer, what impact on the initial molecular diagnosis? J Transl Med. 2014;12:131.
Park S, Holmes-Tisch AJ, Cho EY, Shim YM, Kim J, Kim HS, et al. Discordance of molecular biomarkers associated with epidermal growth factor receptor pathway between primary tumors and lymph node metastasis in non-small cell lung cancer. J Thorac Oncol. 2009;4:809–15.
Sun L, Zhang Q, Luan H, Zhan Z, Wang C, Sun B. Comparison of KRAS and EGFR gene status between primary non-small cell lung cancer and local lymph node metastases: implications for clinical practice. J Exp Clin Cancer Res. 2011;30:30.
Wang F, Fang P, Hou DY, Leng ZJ, Cao LJ. Comparison of epidermal growth factor receptor mutations between primary tumors and lymph nodes in non-small cell lung cancer: a review and meta-analysis of published data. Asian Pac J Cancer Prev. 2014;15:4493–7.
Liu X, Lu Y, Zhu G, Lei Y, Zheng L, Qin H, et al. The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J Clin Pathol. 2013;66:1065–9.
Hasanovic A, Ang D, Moreira AL, Zakowski MF. Use of mutation specific antibodies to detect EGFR status in small biopsy and cytology specimens of lung adenocarcinoma. Lung Cancer. 2012;77:299–305.
Wu SG, Chang YL, Lin JW, Wu CT, Chen HY, Tsai MF, et al. Including total EGFR staining in scoring improves EGFR mutations detection by mutation-specific antibodies and EGFR TKIs response prediction. PLoS ONE. 2011;6:e23303.
Angulo B, Conde E, Suárez-Gauthier A, Plaza C, Martinez R, Redondo P, et al. A comparison of EGFR mutation testing methods in lung carcinoma: direct sequencing, real-time PCR and immunohistochemistry. PLoS ONE. 2012;7:e43842.
Goto K, Satouchi M, Ishii G, Nishio K, Hagiwara K, Mitsudomi T, et al. An evaluation study of EGFR mutation tests utilized for non-small-cell lung cancer in the diagnostic setting. Ann Oncol. 2012;23:2914–9.
Tsai T-H, Wu S-G, Chang Y-L, Wu C-T, Tsai M-F, Wei P-F, et al. Effusion immunocytochemistry as an alternative approach for the selection of first-line targeted therapy in advanced lung adenocarcinoma. J Thorac Oncol. 2012;7:993–1000.
Ilie MI, Hofman V, Bonnetaud C, Havet K, Lespinet-Fabre V, Coëlle C, et al. Usefulness of tissue microarrays for assessment of protein expression, gene copy number and mutational status of EGFR in lung adenocarcinoma. Virchows Arch. 2010;457:483–95.
Kozu Y, Tsuta K, Kohno T, Sekine I, Yoshida A, Watanabe S, et al. The usefulness of mutation-specific antibodies in detecting epidermal growth factor receptor mutations and in predicting response to tyrosine kinase inhibitor therapy in lung adenocarcinoma. Lung Cancer. 2011;73:45–50.
Li J, Wang L, Mamon H, Kulke MH, Berbeco R, Makrigiorgos GM. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14:579–84.
Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–21.
Reichel MB, Ohgaki H, Petersen I, Kleihues P. p53 mutations in primary human lung tumors and their metastases. Mol Carcinog. 1994;9:105–9.
Matsuzoe D, Hideshima T, Ohshima K, Kawahara K, Shirakusa T, Kimura A. Discrimination of double primary lung cancer from intrapulmonary metastasis by p53 gene mutation. Br J Cancer. 1999;79:1549–52.
Kalikaki A, Koutsopoulos A, Trypaki M, Souglakos J, Stathopoulos E, Georgoulias V, et al. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer. 2008;99:923–9.
Schmid K, Oehl N, Wrba F, Pirker R, Pirker C, Filipits M. EGFR/KRAS/BRAF mutations in primary lung adenocarcinomas and corresponding locoregional lymph node metastases. Clin Cancer Res. 2009;15:4554–60.
Munfus-McCray D, Harada S, Adams C, Askin F, Clark D, Gabrielson E, et al. EGFR and KRAS mutations in metastatic lung adenocarcinomas. Hum Pathol. 2011;42:1447–53.
Han CB, Ma JT, Li F, Zhao JZ, Jing W, Zhou Y, et al. EGFR and KRAS mutations and altered c-Met gene copy numbers in primary non-small cell lung cancer and associated stage N2 lymph node-metastasis. Cancer Lett. 2012;314:63–72.
Wei B, Yang K, Zhao J, Chang Y, Ma Z, Dong B, et al. Quantification of EGFR mutations in primary and metastatic tumors in non-small cell lung cancer. J Exp Clin Cancer Res. 2014;33:5.
Yatabe Y, Matsuo K, Mitsudomi T. Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J Clin Oncol. 2011;29:2972–7.
Shimizu K, Yukawa T, Hirami Y, Okita R, Saisho S, Maeda A, et al. Heterogeneity of the EGFR mutation status between the primary tumor and metastatic lymph node and the sensitivity to EGFR tyrosine kinase inhibitor in non-small cell lung cancer. Target Oncol. 2013;8:237–42.
Matsumoto S, Takahashi K, Iwakawa R, Matsuno Y, Nakanishi Y, Kohno T, et al. Frequent EGFR mutations in brain metastases of lung adenocarcinoma. Int J Cancer. 2006;119:1491–4.
Luo D, Ye X, Hu Z, Peng K, Song Y, Yin X, et al. EGFR mutation status and its impact on survival of Chinese non-small cell lung cancer patients with brain metastases. Tumour Biol. 2014;35:2437–44.
Alsdorf WH, Clauditz TS, Hoenig T, Quaas A, Sirma H, Koenig AM, et al. Intratumoral heterogeneity of KRAS mutation is rare in non-small-cell lung cancer. Exp Mol Pathol. 2013;94:155–9.
Li S, Rosell R, Urban A, Font A, Ariza A, Armengol P, et al. K-ras gene point mutation: a stable tumor marker in non-small cell lung carcinoma. Lung Cancer. 1994;11:19–27.
Badalian G, Barbai T, Rásó E, Derecskei K, Szendrôi M, Tímár J. Phenotype of bone metastases of non-small cell lung cancer: epidermal growth factor receptor expression and K-RAS mutational status. Pathol Oncol Res. 2007;13:99–104.
Holst VA, Finkelstein S, Yousem SA. Bronchioloalveolar adenocarcinoma of lung: monoclonal origin for multifocal disease. Am J Surg Pathol. 1998;22:1343–50.
Marchetti A, Buttitta F, Pellegrini S, Bertacca G, Chella A, Carnicelli V, et al. Alterations of P16 (MTS1) in node-positive non-small cell lung carcinomas. J Pathol. 1997;181:178–82.
Tang X, Varella-Garcia M, Xavier AC, Massarelli E, Ozburn N, Moran C, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Cancer Prev Res (Phila). 2008;1:192–200.
Takahashi K, Kohno T, Matsumoto S, Nakanishi Y, Arai Y, Yamamoto S, et al. Clonal and parallel evolution of primary lung cancers and their metastases revealed by molecular dissection of cancer cells. Clin Cancer Res. 2007;13:111–20.
Vignot S, Frampton GM, Soria JC, Yelensky R, Commo F, Brambilla C, et al. Next-generation sequencing reveals high concordance of recurrent somatic alterations between primary tumor and metastases from patients with non-small-cell lung cancer. J Clin Oncol. 2013;31:2167–72.
Ellison G, Donald E, McWalter G, Knight L, Fletcher L, Sherwood J, et al. A comparison of ARMS and DNA sequencing for mutation analysis in clinical biopsy samples. J Exp Clin Cancer Res. 2010;29:132.
Han B, Zhou X, Zhang RX, Zang WF, Chen ZY, Song HD, et al. Mutations of the epidermal growth factor receptor gene in NSCLC patients. Oncol Lett. 2011;2:1233–7.
Jancik S, Drabek J, Berkovcova J, Xu YZ, Stankova M, Klein J, et al. A comparison of Direct sequencing, Pyrosequencing, High resolution melting analysis, TheraScreen DxS, and the K-ras StripAssay for detecting KRAS mutations in non small cell lung carcinomas. J Exp Clin Cancer Res. 2012;31:79.
Dufort S, Richard MJ, Lantuejoul S. de FF. Pyrosequencing, a method approved to detect the two major EGFR mutations for anti EGFR therapy in NSCLC. J Exp Clin Cancer Res. 2011;30:57.
Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, et al. Laser-capture microdissection. Nat Protoc. 2006;1:586–603.
Jakobsen JN, Sørensen JB. Intratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;69:289–99.
da Cunha SG, Saieg MA, Geddie W, Leighl N. EGFR gene status in cytological samples of nonsmall cell lung carcinoma: controversies and opportunities. Cancer Cytopathol. 2011;119:80–91.
Pirker R, Herth FJ, Kerr KM, Filipits M, Taron M, Gandara D, et al. Consensus for EGFR mutation testing in non-small cell lung cancer: results from a European workshop. J Thorac Oncol. 2010;5:1706–13.
Tartarone A, Lazzari C, Lerose R, Conteduca V, Improta G, Zupa A, et al. Mechanisms of resistance to EGFR tyrosine kinase inhibitors gefitinib/erlotinib and to ALK inhibitor crizotinib. Lung Cancer. 2013;81:328–36.
D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer. 2015;112:95–102.
Acknowledgements
We thank Louise Brown from Complete Medical Communications, who provided medical writing support, funded by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
This work was supported by AstraZeneca. JS, SD, MR and JW are employees of AstraZeneca and hold shares in AstraZeneca.
Authors’ contributions
All authors contributed to the study design and interpretation of literature review data. JS and SD performed the literature review. All authors contributed to the content of the manuscript, reviewed and approved the final version, and contributed to the decision to publish the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sherwood, J., Dearden, S., Ratcliffe, M. et al. Mutation status concordance between primary lesions and metastatic sites of advanced non-small-cell lung cancer and the impact of mutation testing methodologies: a literature review. J Exp Clin Cancer Res 34, 92 (2015). https://doi.org/10.1186/s13046-015-0207-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-015-0207-9