
Abstract
RNA sensors detect foreign and endogenous RNAs to protect the host by initiating innate and adaptive immune response. In tumor microenvironment (TME), activation of RNA sensors induces tumor-inhibitory cytotoxic T lymphocyte responses and inhibits the activity of immunosuppressive cells though stimulating type I IFN signaling pathway. These characteristics allow RNA sensors to be prospective targets in tumor immunotherapy. Therefore, a comprehensive understanding of the roles of RNA sensors in TME could provide new insight into the antitumor immunotherapy. Moreover, RNA sensors could be prominent triggering targets to synergize with immunotherapies. In this review, we highlight the diverse mechanisms of RNA sensors in cancer immunity and their emerging contributions in cancer immunotherapy, including monotherapy with RNA sensor agonists, as well as combination with chemotherapy, radiotherapy, immune checkpoint blockade or cancer vaccine.
Similar content being viewed by others
Introduction
Activation of pattern recognition receptors (PRRs), a kind of germline-encoded host sensors, produces type I interferon and interleukin-1β (IL-1β) in the innate immune system [1]. The PRRs sense nucleic acids are called nucleic acid sensors, and can be divided into two categories: one is sensors that detect nucleic acids in endosomes, such as Toll-like receptor (TLR) family members, another group is represented by sensors that detect nucleic acids in cytosol, such as retinoic-acid inducible gene-I (RIG-I)-like receptors (RLRs) and cyclic GMP-AMP synthase (cGAS) [2,3,4,5]. According to the sensed nucleic acid types, there exist two types of nucleic acid sensors, defined as DNA sensors and RNA sensors [2]. These sensors detect exogenous and endogenous nucleic acids not only responding to cellular stress, damage and destruction of homeostasis, but also mediating innate immunity and antitumor immunity.
TLRs and RLRs are two classical receptor families that act as RNA sensors. TLRs family members localize to cell surface and endosomes, among which TLR3, TLR7 and TLR8 function as RNA sensors. TLR3 recognizes double-stranded RNA (dsRNA) of various viral genome or replication intermediates, and boosts host immune response to viral infection [6, 7], whereas TLR7 and TLR8 recognize guanosine and uridine-containing single-stranded RNA (ssRNA) motifs in viral RNA [8]. When combined with RNA, the RNA sensors of TLRs switch through conformational changes, recruit adaptor protein TRIF for TLR3 and MyD88 for TLR7/TLR8, causing IRF3/7 phosphorylation and IFN-β secretion [9]. RLRs predominately localize to the cytoplasm and consist of three members: RIG-I, melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) [10,11,12]. RIG-I recognizes 5’-triphosphate RNA (3pRNA) in the cytoplasm of virus-infected cells, and initiates antiviral immune response by activating MAVS and TBK1 signals [13,14,15]. MDA5 recognizes longer dsRNA, while LGP2 facilitates the interaction between MDA5 and dsRNA [5]. After binding to RNA, RIG-I and MDA5 undergo conformational modifications, induce type I IFN and immune related genes through direct activation and recruitment domains CARD-CARD mediated interactions with mitochondrial associated adaptor protein MAV, and activate IRF3 and NF-κB-mediated signal cascades [16,17,18]. The characteristics of these RNA sensors are listed in Table 1. Although RNA sensors are important for rapid detection of viral RNAs and restrain of viral replication and transmission, they have the risk of potential recognition of self-RNAs, which might generate autoimmune diseases. Therefore, the dynamic regulation of RNA sensors is essential to prevent inappropriate recognition of endogenous RNA [19].
As RNA sensors can recognize pathogen-associated molecular patterns (PAMPs) and induce protective immunity to protect host from foreign intruders, more and more researchers have paid attention to their roles in cancer immunity. Accumulating evidences have suggested that RNA sensors within human cancer cells can respond to cytosolic RNA to induce type I IFN signals, and trigger antitumor immunity and tumor clearance [20]. Nowadays, RNA sensors have been widely used in cancer immunotherapy attributed to their anti-tumor potentiality. Herein, we summarize the roles of RNA sensors in cancer immunity, especially their expression and interaction with immune cells in tumor microenvironment (TME), and describe the insights and emerging cancer immunotherapy strategies based on RNA sensors.
RNA sensors contribute to cancer immunity
Toll-like receptors
TLRs are primarily expressed on immune cells and stimulate innate and adaptive immune responses against pathogens by activating various downstream signaling cascades to induce cytokines and chemokines secretion [21]. Besides, TLRs also express on malignant epithelial cells and mediate apoptosis in several tumors [22]. However, TLR RNA sensors have different cellular expression profiles and intracellular signal pathways, raising the possibility that distinct TLRs differentially influence the TME. Similar to immune cells, cancer cells respond to TLR ligands by secreting cytokines and chemokines, which enhance the recruitment and activation of immune cells (Fig. 1).

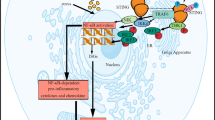
Signaling pathways of RNA sensors in normal and cancer cells. RNA derived from virus infection, intracellular uptake, mitochondrial stress, chromosomal instability can be sensed by RNA sensors. RNA-sensing TLRs, including TLR3, TLR7 and TLR8, predominately localize to the endosome. All RNA-sensing TLRs form homodimers upon activation. TLR3 recruits TRIF to activate the kinases TBK1 and IKKE via activation of TRAF, resulting in phosphorylation and activation of the transcription factor IRF3 to drive type I IFN expression. TLR7 and TLR8 recruit MyD88. MyD88 then recruits the kinases IRAK4 and IRAK1, activates TRAF6 and TAK1, resulting in activation of IRF7 and NF-kB which drives IFN-I expression. RIG-I, LGP2, MDA5 and NOD2 recognize intracellular RNA, bind to the mitochondrial located adaptor protein MAVS, and trigger the activation of TRAF3 to activate TBK1 and drive type I IFN expression. RIG-I senses 3pRNA though MAVS signaling pathway and MAD5 induces IFN responses via TBK1-IRF3 pathway
TLR3 is one of the TLR targets that represents the potential of anti-tumor activity, and its expression is significantly increased in tumor tissues [23]. Studies have shown that the responsive tumors are characterized by up-regulation of STAT1 and TLR3 signaling, down-regulation of IL-10 signaling, and more infiltrating-activated natural killer (NK) cells [24]. TLR3 in TME is mainly expressed in Batf3-positive dendritic cells (DCs) (CD141+ DCs in human, CD8a+ and CD103+ DCs in mouse) and tumor associated macrophages (TAMs) [25,26,27]. In human malignant tumors, CD141+ DCs (cDC1) were essential to induce tumor-inhibitory cytotoxic T lymphocyte (CTL) responses [28,29,30]. TLR3 stimulation by poly-IC has been found to expand and activate cDC1 by inducing IFN-λ1 production, and recruit cytotoxic effector cells in TME [31, 32]. In non-small-cell lung cancer (NSCLC), the expression of TLR3 on cancer cells contributed to stimulated CD103+ lung dendritic cell subset, activated caspase-3 and induced apoptosis [33]. TAMs are the main component of TME, and known to release tumor necrosis factor-α (TNF-α) in response to poly-IC to induce cell death [26, 31, 32]. Vidyarthi et al. [34] reported that TLR3 stimulation reverted macrophages phenotype from M2 type to M1 type and regressed tumor growth via IFN-α/β signaling pathway. In melanoma, TLR3 agonists could induce antitumor immunity by activating NK cells to hinder B16 melanoma lung metastasis [35, 36], whereas the activation of NK cells in lung was mediated by alveolar macrophages and shapes macrophage behavior [37]. Moreover, TLR3 activation decreased the expression of PD-L1, ablated FoxP3 positive CD4+ T cells and elicited a distinct CD8+ T cell activation phenotype in TME [38]. However, TLR3 activation have also been reported to promote cancer progression. For example, lung cancer microparticles (L-MPs) activated TLR3 and NLRP3 inflammasome, induced macrophages to release IL-1β, and thus promoted lung cancer development [39]. In addition, cancer cells induced chemotactic signaling pathway in endothelial cells by activating TLR3-SLIT2 axis [40].
TLR7 is usually expressed in endosomes of different immune cells, including B lymphocytes, plasmacytoid and conventional dendritic cells (pDCs and cDCs) and macrophages [41]. High expression of TLR7 is reported to be associated with poor prognosis of cancerous patients [42,43,44,45,46]. TLR7 not only played an immunosurveillance role on activation of innate and adaptive immune effectors, but also exhibited a dual regulatory effect on tumor progression [47]. On the one hand, TLR7 activation recruited immunosuppressive cells to facilitate tumor-immune escape. For example, TLR7 stimulation in cancer cells favored tumor progression through increasing the secretion of C–C motif chemokine ligand 2 (CCL2) and granulocyte–macrophage colony stimulating factor (GM-CSF) in TME and eliciting the recruitment of myeloid derived suppressor cells (MDSCs) into the tumor [42]. On the other hand, TLR7 stimulation promoted immune cell infiltration in TME, which functioned as a tumor suppressor. For instance, increased TLR7 expression indicated poor prognosis and was positively correlated with immune cell infiltration (such as T cells, macrophages, neutrophils and DCs) and immune checkpoint expression in stomach adenocarcinoma [46]. Additionally, TLR7 also has the potential to induce CD4+ T cells and CD8+ T cell infiltration into TME [48].
TLR8 is expressed in the endosomal compartment and is significantly enriched in monocytes, macrophage and DCs [49]. TLR8 signaling could reverse the suppressive functions of tumor-derived CD4+ T cells, CD8+ T cells and γδ regulatory T (Treg) cells resulting in enhanced anti-tumor immunity [50,51,52,53,54,55]. Activation of TLR8 in cancer cells is found to prevent the induction of senescence in responder T cells and DCs [56], stimulate glucose uptake and glycolysis in CD4+ T cells [57], and induce apoptosis of MDSCs to enhance the anti-tumor effects of adaptive immune response [58]. TLR8 activation also stimulated the release of distinct inflammatory mediators, such as Th1-polarizing cytokines and chemokines, skewed monocytes toward an M1 phenotype and reversed MDSC-mediated suppression of T cell proliferation [59, 60]. Moreover, Safarzadeh et al. [61] reported that TLR7/8 agonist reduced the immunosuppressive activity of patient-derived MDSCs on T cells via promoting MDSCs repolarization into mature myeloid cells.
RIG-I-like receptors
RLRs are cytosolic PRRs which can sense cytosolic RNA, and have been found to be expressed in several human normal and cancer cells [62,63,64,65,66,67]. RIG-I signaling activation promotes immune activation in TME, drives transcriptional activation of pro-inflammatory genes involving type I IFNs and pro-inflammatory cytokines and results in immunogenic cell death [68]. Previous studies demonstrated that RIG-I sensitized cancer cells to irradiation treatment by interacting with XRCC4 to compromise virus integration and DNA repair [66]. Activation of MDA5 and RIG-I induced apoptosis in colorectal cancer through mitochondrial pathway [67]. Recently, the role of RLR RNA sensors on antitumor immunity has also been revealed (Fig. 1).
In melanoma, RIG-I signaling triggered surface expression of membrane-bound TNF-related apoptosis-inducing ligand (TRAIL) in naïve NK cells, and induced a TRAIL-dependent cytotoxic NK cell response [69]. In humanized lung cancer model, RIG-I activated by MAPK/IRF1 triggered an interferon and a pro-apoptotic response, resulting in the reduction of exhausted CD8+ T cells and tumor shrinkage [70]. Overexpression of RIG-I in hepatocellular carcinoma (HCC) promoted the polarization of M1 macrophages in vitro and increased cancer cell apoptosis in vivo through RIG-I/MAVS/NF-κB pathway [71]. In a hypoxic murine melanoma model, RIG-I was activated and has been found to provoke melanocyte antigen-specific CD8+ T cells and NK cells attack, and enhance 3pRNA antitumor efficacy [72]. However, RIG-I also contributed to tumor immune escape. For example, high expression of RIG-I predicted worse clinical outcome in ovarian cancer, and was correlated with immune-regulatory signatures involving checkpoint molecules (PD-L1/PD-1) [73]. In nasopharyngeal carcinoma, Epstein-Barr virus (EBV)-encoded circBART2.2 induced PD-L1 transcription via binding the helicase domain of RIG-I and activating NF-κB and IRF3 cascades, leading to immune escape [74].
MDA5 is another important RNA sensor of RLR family which recognizes longer dsRNA in the cytosol [13]. Activation of MDA5 generated type-I IFN in various DC subsets, and enhanced cytotoxic T cell expansion [75]. Recently, Brown et al. [76] reported that MDA5 could also orchestrate TBK1-IRF3 signaling and sustain type-I/III IFN release, helping Th1 differentiated antitumor T cell phenotypes in TME.
LGP2 poses dual regulating effect of RNA sensing. In neuroblastoma cells, ectopic expression of LGP2 significantly promoted poly-IC‐induced cell death and was associated with downregulation of RIG‐I, MDA5 and MAVS [77]. In breast cancer patients who received radiotherapy, DCs in TME were correlated with LGP2 expression and linked to the clinical outcome. The absence of LGP2 in DCs inhibited the production of type-I IFN and the priming capacity of DCs, and impaired the function of tumor infiltrating CD8+ T cells [78].
Other RNA sensors
Besides, some emerging RNA sensors have been revealed and defined, including NOD-like receptors (NLRs), heterogeneous nuclear ribonucleoproteins (hnRNPs), DEAD-box or DEAH-box RNA helicases and ZBP1 [5, 10]. These RNA sensors are found to sense RNA and interact with TLRs and RLRs in innate immunity. However, few studies have reported their relationship with antitumor immune response.
NOD2, a member of NLRs family, has been demonstrated to function as an RNA sensor by recognizing viral genomic ssRNA and regulate IRF3-dependent antiviral immunity responses via MAVS pathway in both hematopoietic and non-hematopoietic cells [79]. Dysregulation of NOD2 has also been reported in tumorigenesis. In lung adenocarcinoma, cancer cells induced decreased NOD2 expression, resulting in the phenotypic polarization of macrophages through NF-κB signalling pathway [80]. Recently, cGAS-like receptors (cGLRs) are shown to recognize distinct molecular patterns and catalyze synthesis of different nucleotide second messenger signals. In drosophila, cGLRs could sense dsRNA and induce an enhanced antiviral response by synthesizing 3′2′-cGAMP [81]. A study found that chicken Asp-Glu-Ala-Asp (DEAD)-box helicase 1 (DDX1) was an RNA sensor in antiviral innate immunity and mediated IRF7-IFN-β signaling pathway [82]. Poly (ADP-ribose) polymerase 9 (PARP9), a member of PARP family, served as a non-canonical sensor for dsRNA in human or mouse DCs and macrophages to produce type I IFN via activation of the phosphoinositide 3-kinase (PI3K) and AKT3 pathway [83].
To sum up, RNA sensors have been found to contribute to cancer immunity across multiple cancer types (Table 2). The activation of RNA sensors in TME plays positive and negative regulatory roles, and interacts with immune cells, making them attractive targets in cancer immunotherapy (Fig. 2).

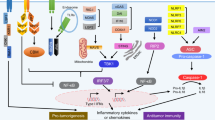
Model for RNA Sensing in the context of anti-tumor immunity. RNA sensors can induce anti-tumor efficacy through multiple mechanisms. Tumor-derived Type I IFN and antigen which contain RNA activates antigen-presenting cells (APC), which mainly including DCs and macrophages. RNA sensors in APCs sense RNA and promote DCs mature and macrophage M1 polarization. Then these cells trigger production of type I IFN and other proinflammatory factors to promote an antitumor immune microenvironment by activating T cells, NK cells and inhibiting Treg and MDSC cells
RNA sensors in cancer immunotherapy
As most RNA sensors have function on anti-tumor immunity based on their induction of IFN signaling [84], many drugs targeting RNA sensors are under clinical evaluation for cancer treatment (data from https://clinicaltrials.gov/) (Table 3). Not only local delivery of the single use of RNA sensor agonists, but also the combination with other cancer immunotherapy strategies give great contribution to cancer immunotherapy (Fig. 3).

Contribution of RNA sensors in cancer immunotherapy. RNA sensor agonists singly use benefit to cancer immunotherapy by boosting anti-cancer immune response. When loaded into some vehicles such as liposomes, nanoparticle and some novel compounds, they get more effective drug concentration in tumor bed. RNA sensor agonists can also act as adjuvant for other cancer immunotherapy strategies including cancer vaccines, immune cells engineering, immune checkpoint therapy and chemoradiotherapy to induce antitumor immune microenvironment, ameliorate the treatment efficacy and reduce the side effects
RNA sensor agonists single use contributes to cancer immunotherapy
Classical RNA sensor agonists
TLR agonists have received considerable attention as promising targets for cancer immunotherapy owing to their ability to convert immunosuppressive TME towards a T cell inflamed phenotype. Poly-IC, an agonist of TLR3, MDA5 and RIG-I, is usually used as an immune adjuvant. It can directly target tumor-resident DCs, mimic natural infection of dsRNA virus, and initiate a strong inflammatory response by recruiting and activating CD8+ T cells [85, 86]. Poly-L-lysine stabilized (Poly-ICLC), a synthetic dsRNA and an agonist of TLR3 and MDA5, stabilizes with poly-lysine and carboxymethylcellulose. Poly-ICLC has been evaluated in a large number of clinical trials with the goal to exert anti-tumor immune effect and can be safely applied to patients [86]. Resiquimod (R848), a TLR7/8 agonist, is usually used as an immune adjuvant, which specifically binds to the PAMPs of DCs and promotes the maturation and activation of DCs to improve anti-tumoral immune response [87,88,89]. In a mice model of head and neck cancer, R848 could recruit immune cells to the tumor and inhibit tumor growth [87]. TLR7/8 agonist imiquimod was proven to activate NK cells to kill tumor cells and induct tumor-specific CD4+ T cells, resulting in a strong regression of low MHC-I tumors [90]. Additionally, Wiedemann et al. [91] reported that a small molecule TLR7-agonist SC-1 could activate NK cell responses and restore NK cell-mediated tumor killing effect in vivo. SC-1 also exerted antitumor effect through releasing type I IFN, activating plasmacytoid DCs, polarizing macrophages to M1 phenotype and decreasing MDSC [92]. Motolimod (VTX-2337), a selective small-molecule TLR8 agonist, could alter lymphocyte differentiation and function, stimulate Th1 polarizing cytokines, enhance antibody-dependent cellular cytotoxicity, and promote innate and adaptive antitumor immunity [93,94,95]. In a clinical trial of head and neck squamous cell carcinoma (HNSCC), motolimod significantly improved the prognosis of patients with HPV-positive oropharyngeal cancer [96].
Two recent studies demonstrated that RIG-I activated by short 5′-triphosphate-modified RNA (ppp-RNA) reduced tumor burden in melanoma and acute myeloid leukemia (AML) model, which was dependent on CD4+ T cells, CD8+ T cells and the intact MAVS/IFN signaling in the host [97, 98]. Jiang et al. [99] tested the antitumor activity of stem loop RNA 14 (SLR14), a RIG-I agonist, in immunogenic tumor models. They found that tumor growth was delayed and survival was extended in SLR14-treated mice. The numbers of CD8+ T lymphocytes, CD11b+ cells and NK cells were observed to be increased in the model. Moreover, SLR14 significantly inhibited the growth of nonimmunogenic B16 tumor, and the cured mice developed immunologic memory. Another study in AML showed that RIG-I activation overcame the intrinsic T cell resistance of IFN-sensitive/resistant melanoma and enhanced the clinical effect of immunotherapy [100].
Novel RNA sensor agonists
Nowadays, more emerging RNA sensor agonists have been developed and used for safe and effective cancer immunotherapy, including some RNA-based agonists and small-molecule agonists (Table 4). ARNAX, a TLR3-specific RNA agonist, not only established Th1 immunity in TME, but also upregulated genes involved in the recruitment and function of T cells, NK cells and DCs [101,102,103]. Single-stranded RNA origami (RNA-OG) based on nucleic acid nanotechnology can stimulate a strong immune response via TLR3 signaling pathway. In a mice peritoneal metastatic colon cancer model, RNA-OG was found to induce obvious tumor growth arrest by activating CD8+ T cells and NK cells and antagonize the peritoneal immunosuppressive TME. Unlike poly-IC, RNA-OG administration did not significantly produce high level of type-I IFN in blood, nor did it cause apparent toxicity in the animal model, which make it a potential safe and effective RNA sensor agonist for cancer immunotherapy [104]. In a melanoma model, Liu et al. [105] reported that a ssRNA-Pim-3-small hairpin RNA (shRNA) dual-function vector could activate TLR7 by ssRNA fragments to stimulate antitumor immune response, such as activation of CD8+ T cells and NK cells and reduction of intratumoral Treg and MDSCs. In addition, several noncoding RNAs and even recombinant virus have been identified as RNA senor inducers. MiR-574-5p derived from small extracellular vesicles activated TLR7/8, thereby decreased PGE2-levels in lung cancer [106]. CircNDUFB2 is reported to be recognized by RIG-I to activate RIG-I-MAVS signaling cascades and recruit immune cells into the TME in NSCLC [107]. PVSRIPO, a neuro-attenuated recombinant poliovirus, shows strong cytotoxicity in infected tumor cells expressing poliovirus receptor CD155. In malignant glioma, PVSRIPO could induce IFN response and elicit antitumor T cell immunity through MDA5-TBK1-IRF3 signaling [76, 108]. As for small-molecule agonists, 17e (CU-CPT17e), a newly discovered small molecule capable of activating TLRs 3, 8 and 9 simultaneously, could induce THP-1 cells to produce various cytokines, such as IL-6, IL-8 and TNF-α, and inhibit the proliferation of HeLa cancer cells [109]. 1V270, a small molecule TLR7-specific ligand conjugated to a phospholipid moiety, is another RNA sensor agonist. It has been shown to induce tumor-specific adaptive immune responses and suppress primary tumor growth in HNSCC and melanoma mice models [110, 111]. Additionally, local 1V270 treatment could activate TAM and convert an immune-suppressive TME to a tumoricidal environment without inducing systemic cytokine [111]. The 1V270 therapy also inhibited tumor colonization in an NK cell dependent manner, which exhibited a suppression role of lung metastasis by inducing tumor-specific adaptive immune responses [112]. MEDI9197 (3M-052), a novel designed lipophilic TLR7/8 agonist, is found to regulate the enrichment and activation of CD8+ T cells and NK cells, polarization of Th1 cells and inhibition of tumor growth in multiple syngeneic models [113].
RNA sensor agonist delivery vehicles ameliorate cancer immunotherapy efficacy
As it is difficult to achieve effective drug concentration in tumors for systemic administration of agonists, some delivery vehicles such as liposomes, nanoparticle and novel compounds for delivering RNA sensor agonists to the target tumor region have been developed.
Liposomes
R848 delivered by complement C3-targeted liposomes triggered various signal cascades to increase the expression of cytokines and factors (such as TNF-α, IL-1β, IL-6 and IL-12), leading to the delay of tumor growth in 4T1 tumor-bearing mice [114]. Zhang et al. [115] developed an intravenously-injectable formulation with R848 by using thermosensitive liposomes (TSLs) as a delivery vehicle (R848-TSLs). Combined with local hyperthermia and αPD-1, systemic administration of R848-TSLs could significantly inhibit tumor growth. Local injection of R848-TSLs combined with αPD-1 also showed superior anti-tumor efficacy. They observed that complete regression of neu deletion (NDL) tumors in both treated and abscopal sites was achieved in 8 of 11 tumor bearing mice with enhanced infiltration and accumulation of CD8+ T cells in tumors. In another recent study, Wan et al. [116] conjugated the small molecule TLR7 agonist 1V209 with cholesterol (1V209-Cho) and prepared into liposomes (1V209-Cho-Lip). Compared with 1V209, 1V209-Cho-Lip exerted less toxic effect and enhanced transport capacity of lymph nodes (LN). Subsequent in vivo experiments showed that 1V209-Cho-Lip treatment could inhibit the progression of Pan02 pancreatic ductal cancer, 4T1 breast cancer and CT26 colorectal cancer models by eliciting CD8+ T cell responses and inducing effective DC activation. In addition, cholesterol conjugation with 1V209 also induced tumor-specific memory immunity to reduce tumor relapse and metastasis. Cationic liposomes loaded with tumor-specific synthetic long peptides (SLPs) and TLR3 ligands as adjuvants could also induce cytotoxicity against target cells in vivo by strongly activating functional antigen-specific CD4+ T cells and CD8+ T cells [117].
Nanoparticle
Nanoparticle is a class of microscopic particle and has been found to possess anti-tumor therapeutic potential by inducing pro-inflammatory TME. It was also used as a vector to deliver cancer vaccines. When synergized with the poly-IC, nanoparticles such as Ferumoxytol and BO-112, were shown to exert anti-tumor therapeutic potential by inducing macrophage activation and enhancing tumor antigen-specific CTLs in TME [118, 119]. In a mouse model, combination of CHP-NY-ESO-1, a nanoparticle complex of cholesteryl pullulan (CHP) and NY-ESO-1 antigen protein, with anti-PD-1 antibody suppressed the growth of NY-ESO-1-expressing tumors. Further phase 1 clinical trial reported that CHP-NY-ESO-1 could induce higher antibody responses in patients with advanced or recurrent esophageal cancer when combined with poly-ICLC [120]. Nanoparticle that delivers TLR7 ligand to tumor-draining lymph nodes can induce a local cytotoxic T cell response [121], leading to the proliferation of tumor antigen-specific CD8+ T cells and potent activation of DCs in the sentinel lymph nodes [89, 122]. When co-delivers TLR‑7 agonists with anti-CD47 antibodies, nanoparticles can induce systematic immune responses and superb antitumor efficacy [123]. R848-loaded nanoparticles were proven to effectively deliver drugs to TAMs, induce the shift of macrophages from M2 to M1 phenotype, activate DCs and increase cytotoxic T cells [124, 125]. Moreover, a self-assembling vehicle-free multi-component antitumor nanovaccine (SVMAV) loaded with R848 and STAT3 inhibitors could effectively migrate into lymph nodes, promote CD8+ T cell response and DC function not only in primary melanoma, but also in lung metastasis. It is worth noting that neoantigen-specific SVMAV showed stronger antitumor activity than aPD-1 in an orthotopic HCC model [126]. In addition, a semiconducting polymer nanoadjuvant (SPNIIR) can effectively generate heat not only to induce immunogenic cell death and ablate tumors, but also release TLR agonists, which promotes the maturation of DCs and enhances anti-tumor immune response [127]. The application of a nanoparticles/bacteria complex (Ec-PR848) composed of Escherichia coli, DOX-loaded PLGA and R848 was shown to polarize macrophages from M2 to M1 phenotype, impair the immunosuppression of TME, and significantly improve the efficacy of immunotherapy [128]. Delivery of 5’-triphosphate RNA together with endosomolytic nanoparticles could induce immunogenic cell death, trigger the expression of type I IFN and proinflammatory cytokines, and increase the infiltration of CD8+ T cells through activating RIG-I pathway in CT26 tumor model [129]. Nanoparticle delivery of RIG-I agonist dsRNA also strongly induced the level of pro-inflammatory Th1 cytokines, further increased the proportion of M1 over M2 macrophages, CD8+ T cells over regulatory T cells, and reduced the levels of plasma cells and immunosuppressive B regulatory in pancreatic cancer [130]. These findings provide a promising nanoparticle-based immunotherapy approaches for malignant tumors.
Novel compounds
Recent years, some novel compounds for the treatment of malignant tumors have been developed and synthesized. RNA sensor agonists can also be loaded in these compounds as adjuvants to promote their antitumor immune responses. CH-NPs, an ionic complex ovalbumin as a model antigen and poly-IC as the adjuvant, could increase intracellular delivery and maturation of DCs, resulting in the activation of antigen-specific cytotoxic CD8+ T cells in vivo [131]. Liu et al. [132] developed a galactose-functionalized zinc protoporphyrin IX (ZnPP) grafted poly(l-lysine)-b-poly(ethylene glycol) polypeptide micelles (ZnPP PM) for TAM-targeted immunopotentiator delivery. ZnPP PM loaded by poly-IC could activate T lymphocytes and NK cells, and effectively repolarize TAM in B16-F10 melanoma tumor model. JOC-x is a conjugatable tumor tight junction opener, when conjugated with poly-IC, it could not only recruit and activate of CD8+ T cells by targeting DCs, but also play a tumor killing role by initiating apoptosis in tumor cells [133].
RNA sensor agonists combined with other cancer immunotherapy strategies
Cancer vaccines
Cancer vaccines, produced by tumor-derived antigens (such as microparticles, proteins, peptides and mRNA), can deliver tumor antigens to local tumor region, trigger strong antitumor immune response in situ. Protein or peptide vaccine combined with RNA sensor agonists have been demonstrated to induce activation of antigen-specific CD8+ T cells that translates into potent antitumor immunity [134,135,136,137]. In a murine melanoma model, tumor antigen vaccination based on anti-CD40 and poly-IC increased the number of CD8+ T cells in tumor tissue and delayed tumor growth [138]. In cervical cancer, lung cancer and melanoma models, tumor antigens adjuvant with nanoemulsion (NE) loaded with TLR7/8 agonists showed enhanced infiltration of lymphocytes, polarization of tumor-associated M2 macrophages, strong local and systemic anti-tumor immune response, resulting in inhibited tumor growth and prolonged survival [139, 140]. Additionally, studies have reported that TLR7 agonist imiquimod augmented the immunogenicity of peptide vaccine by activating the strong and durable response of CD4+ T cells and CD8+ T cells in melanoma [141]. Moreover, tumor cell-derived microparticles (TMPs) by oral vaccination activated NOD2 leading to subsequent antitumor T cell responses, inhibited the tumor growth of CT26 colon cancer and B16 melanoma in mice [142]. Koerner et al. [143] recently reported that biodegradable poly (lactic-co-glycolic acid) (PLGA) particles carrying TLR3/RIG-I ligand Riboxxim could potently activate murine and human DCs and elevate tumor-specific CD8+ T cell responses, showing effective anti-cancer effect in preclinical tumor models.
Immune cell engineering
The combination of RNA sensor agonists and engineered immune cells provides a new immunotherapeutic strategy for solid tumors. In the immunotherapy of chimeric antigen receptor T cells (CAR-T), Di et al. [144] found that poly-IC significantly promoted the higher lytic activity of CAR-T, enhanced the tumor growth inhibition from CAR-T cells after systemic administration in vivo. Moreover, poly-IC reduced the number of MDSC in peripheral blood and spleen, and weakened the immunosuppressive activity of MDSC on proliferation and cytolytic function of CAR-T cells. In one recent reported study, researchers delivered RN7SL1, an endogenous RNA that activates RIG-I/MDA5 signal, through engineered CAR-T cells to promote the expansion and effector memory differentiation of CAR-T cells. They found that when RN7SL1 was deployed in extracellular vesicles, it could selectively transfer to immune cells to restrict the development of MDSC and reduce TGF-β in myeloid cells. Even when heterogenous CAR antigen tumors lack sufficient neoantigens, CAR-T cells still could co-deploy peptide antigens with RN7SL1 to improve efficacy [145]. In addition, Li et al. [54] found that TLR8 could reverse Treg suppression by selectively inhibiting glucose uptake and glycolysis in Treg cells, and then enhance antitumor immunity in a melanoma adoptive transfer T cell therapy model. Antigen sensitized DCs has found to induce antigen-specific CD8+ T cell response in vivo, making them as attractive targets for cancer immunotherapies [28, 29, 146]. When cultured in presence of poly-IC, DCs can more effectively enhance T cell responses [147]. Two previous studies found that the combination of a DC-based vaccination and poly-ICLC was well-tolerated in glioblastoma and glioma [148, 149]. Another clinical trial of pancreatic cancer demonstrated that the combination of peptide pulsed DCs and poly-ICLC was safe and could induce a measurable tumor specific T cell population [150]. Additionally, reovirus-activated NK cells combined with cetuximab could synergistically enhance their antitumor cytotoxicity in colorectal cancer, which was dependent on TLR3 and its downstream signals [151].
Immune checkpoint therapy
Application of monoclonal antibodies targeting immune checkpoints, such as programmed cell death 1 ligand 1 (PD-L1), integrin-associated protein (CD47) and cytotoxic T lymphocyte associated antigen 4 (CTLA-4) has been found to improve the survival rate of patients with several cancer types [152,153,154]. However, there are still a large proportion of patients who cannot benefit from immune checkpoint blockade (ICB) therapy, whereas some cancer types even seem to be less sensitive to ICB [155, 156]. In order to achieve effective outcome, some researches have focused on the effect of RNA sensor agonists on ICB [157]. One study found that pretreatment of IFN-γ, TLR3 ligand poly-IC and anti-IL-10 antibody could sensitize tumors to ICB by increasing infiltrating-activated NK cells [24]. In an HNSCC model, Sato-Kaneko et al. [111] demonstrated that the combined treatment of intravenous TLR agonist and PD-1 blockade activated TAMs, induced tumor specific adaptive immune response, and inhibited primary tumor growth and metastasis. In two breast cancer models, PD-1 blockade combined with poly-IC efficiently modulates immune cell profiles, such as increase in CD8+ T cells, type-1 conventional DCs, immunogenic M1 macrophages and CD169+ macrophages, and reduction in MDSC, plasmacytoid DCs, regulatory T cells and immunotolerant M2 macrophages, which in turn eliminates not only the primary tumor, but also metastasis [158]. TLR3-specific RNA agonist ARNAX could activate tumor-specific CTLs, and overcome anti-PD-1 resistance without cytokinemia when combined with anti-PD-L1 antibody and a tumor-associated antigen [101]. Application of ppp-RNA also induced the expression of PD-L1 on AML cells and established therapeutic sensitivity against PD-1 checkpoint blocking in vivo [97]. Another RIG-I agonist SLR14 could improve the antitumor efficacy of anti-PD1 antibody over monotherapy [100]. In CT26 colon cancer model, nanoparticle conjugated TLR7 agonists could potentiate the efficiency of checkpoint inhibitors targeting PD-1 and CTLA-4, and even promote a long-term specific immunological memory [159]. Another TLR7/8 agonist-based nanovaccine combined with sunitinib and PD-L1 antibody treatment was proven to upregulate activation of CD8+ T cells and reduce MDSCs and PD-L1high M2 macrophages in the tumor, leading to enhanced antitumor efficacy in B16F10 and MB49 mice models [160]. Moreover, combining irreversible electroporation (IRE) with intratumoral TLR7 agonist 1V270 and systemic anti-PD-1 blockade not only improved treatment responses, but also eliminated untreated concomitant distant tumors [161]. In addition, anti-CTLA-4 and its combined immunotherapy with anti-PD-1 have also been found to be dependent on the activation of RIG-I, which could induce cross-presentation of tumor-associated antigen by CD103+ DCs, caspase-3-mediated tumor cell death and expansion of tumor antigen-specific CD8+ T cells [162, 163].
Combination immunotherapy
A major challenge of cancer immunotherapy is to develop a durable, effective and tumor-specific immune response without systemic toxicity. The applications of immune adjuvants combined with chemotherapy, radiotherapy and targeting therapy are able to improve clinical efficacy. Emerging studies have revealed that some RNA sensor agonists used as adjuvants can help to boost the antitumor immune microenvironment, so as to ameliorate the treatment efficacy and reduce the side effects.
In formyl peptide receptor 1 (FPR1)-deficient mice, immunotherapy with Poly-IC has been found to restore the deficient chemotherapeutic responses by improving DC and T lymphocyte-mediated anticancer immunity [164]. Wei et al. [165] developed two targeted polymer micelles to deliver immunomodulator imiquimod (R837) and anticancer drug doxorubicin (DOX) to TAMs and tumor cells through intravenous and intratumoral injection. They found that R837 could stimulate the maturation of TAM, induce the anti-tumor immune response in TME. Meanwhile, the release of DOX in the cytoplasm of tumor cells by the chemotherapeutic micelles could also directly induce cancer cell death. Administration of R848 combined with oxaliplatin reversed the function of MDSCs and strengthened antitumor effect of oxaliplatin in colorectal cancer [166], whereas oxaliplatin-based platinum prodrug bearing TLR7 agonist SZU101 enhanced activation of cytotoxic T cells in tumors and contributed to the high anticancer efficiency in breast cancer model [167]. Ringgaard et al. [168] revealed that combining a liposomal oxaliplatin formulation (PCL8-U75) with R848 induced immunological rejection of established tumors by increasing infiltration of Foxp3-T helper cells and cancer antigen-specific cytotoxic T cells. Moreover, the therapeutic effect of radiotherapy in combination with poly-IC was shown to enhance radiation-sensitivity via TNF-α produced by intra-tumor macrophages and CTL induced by TLR3-positive DC [169]. In EBV-positive nasopharyngeal cancer, Poly-ICLC can strengthen cetuximab-based immunotherapy through enhancing NK-mediated IFN-ɣ expression, antibody-dependent cellular cytotoxicity (ADCC) and DC maturation [170]. In syngeneic human CD20 (hCD20)-expressing models of lymphoma, the combination of R848 and obinutuzumab improved the clearance of lymphoma and produced long-term antitumor immune response [171]. In HNSC, encouraging antitumor activity and strong pharmacodynamic response were also observed when TLR8 agonist Motolimod combined with cetuximab [172].
Conclusion and perspectives
RNA sensors are important for recognizing PAMPs and could help to protect the host from both exogenous and endogenous RNA. Intriguingly, recent studies show that PRR-mediated RNA sensing also occurs in the nucleus and mitochondrion, highlighting an orchestrated multi-compartmental RNA-sensing paradigm [173,174,175,176]. The principle has subsequently been used to develop new treatment strategies, making RNA sensors as an important target for cancer immunotherapy. Deep understanding of the mechanism into the activation and regulation of RNA sensors in cancer immunity is necessary for exploring their applications in antitumor immunotherapy. The accepted notion so far is that activation of RNA sensing pathways is able to suppress tumors. Conversely, in some cases, triggering of RNA sensors by their ligands does not result in antitumor immunity but favors tumor progress and immune escape instead [39, 42, 73, 74]. Therefore, more in-depth exploration of their function is needed in future researches.
Immunotherapy has achieved remarkable success in the treatment of malignancy. Recent progresses in the understanding of how RNA sensing signals affect cancer immunity of agonists, antagonists and novel treatment strategies. As we mentioned above, single or combined application of RNA sensor agonists is becoming a potential effective treatment of cancers, and more burgeoning agonists have been found and developed. It is reported that even recombinant viruses and cellular noncoding RNAs are expected to be agonists [74, 76, 106,107,108]. Although the primary focus lies on RNA sensor agonists in combination with other conventional treatments or as components of cancer vaccines, systematic treatment strategies based on RNA sensors, novel drug delivery methods and innovative combination with other immunotherapies will continue to promote progress in this field.
Availability of data and materials
Not applicable.
Abbreviations
- PRR:
-
Pattern recognition receptor
- IL-1β:
-
Interleukin-1β
- TLR:
-
Toll-like receptor
- RIG-I:
-
Retinoic-acid inducible gene-I
- cGAS:
-
Cyclic GMP-AMP synthase
- dsDNA:
-
Double-stranded RNA
- ssRNA:
-
Single-stranded RNA
- MDA5:
-
Melanoma differentiation-associated protein 5
- LGP2:
-
Laboratory of genetics and physiology 2
- PAMP:
-
Pathogen-associated molecular pattern
- TME:
-
Tumor microenvironment
- NK:
-
Natural killer
- DC:
-
Dendritic cell
- TAM:
-
Tumor associated macrophage
- CTL:
-
Cytotoxic T lymphocyte
- NSCLC:
-
Non-small-cell lung cancer
- TNF-α:
-
Tumor necrosis factor-α
- GM-CSF:
-
Granulocyte-macrophage colony stimulating factor
- MDSC:
-
Myeloid derived suppressor cell
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- HCC:
-
Hepatocellular carcinoma
- NLR:
-
NOD-like receptor
- cGLR:
-
cGAS-like receptor
- PARP9:
-
Poly (ADP-ribose) polymerase 9
- PI3K:
-
Phosphoinositide 3-kinase
- SLR14:
-
Stem loop RNA 14
- TSL:
-
Thermosensitive liposome
- FPR1:
-
Formyl peptide receptor 1
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- SLP:
-
Synthetic long peptide
- TMP:
-
Tumor cell-derived microparticle
- CAR-T:
-
Chimeric antigen receptor T cell
- PD-1:
-
Programmed death receptor 1
- CD47:
-
Integrin-associated protein
- CTLA4:
-
Cytotoxic T lymphocyte associated antigen 4
- ICB:
-
Immune checkpoint blockade
References
Hoffmann J, Akira S. Innate immunity. Curr Opin Immunol. 2013;25(1):1–3.
Qiao Y, Zhu S, Deng S, et al. Human cancer cells sense cytosolic nucleic acids through the RIG-I-MAVS pathway and cGAS-STING pathway. Front Cell Dev Biol. 2021;8:606001.
Sparrer KM, Gack MU. Intracellular detection of viral nucleic acids. Curr Opin Microbiol. 2015;26:1–9.
Cui J, Chen YJ, Wang HY, et al. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccine Immunother. 2014;10(11):3270–85.
Liu GQ, Gack MU. Distinct and orchestrated functions of RNA sensors in innate immunity. Immunity. 2020;53(1):26–42.
Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–8.
Huang YL, Huang MT, Sung PS, et al. Endosomal TLR3 co-receptor CLEC18A enhances host immune response to viral infection. Commun Biol. 2021;4(1):229.
Zhang ZK, Ohto U, Shibata T, et al. Structural analysis reveals that Toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity. 2016;45(4):737–48.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
Chan CP, Jin DY. Cytoplasmic RNA sensors and their interplay with RNA-binding partners in innate antiviral response: theme and variations. RNA. 2022;28(4):449–77.
Andrejeva J, Childs KS, Young DF, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101(49):17264–9.
Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–7.
Kato H, Takeuchi O, Mikamo-Satoh E, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(7):1601–10.
Hornung V, Ellegast J, Kim S, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–7.
Schlee M, Roth A, Hornung V, et al. Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31(1):25–34.
Chow KT, Gale M, Loo YM. RIG-I and other RNA sensors in antiviral immunity. Annu Rev Immunol. 2018;36:667–94.
Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175(5):2851–8.
Seth RB, Sun LJ, Ea CK, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell. 2005;122(5):669–82.
Maurano M, Snyder JM, Connelly C, et al. Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity. 2021;54(9):1948-1960.e5.
Zitvogel L, Galluzzi L, Kepp O, et al. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–14.
Tatematsu M, Seya T, Matsumoto M. Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem J. 2014;458(2):195–201.
Estornes Y, Toscano F, Virard F, et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012;19(9):1482–94.
Lomphithak T, Choksi S, Mutirangura A, et al. Receptor-interacting protein kinase 1 is a key mediator in TLR3 ligand and Smac mimetic-induced cell death and suppresses TLR3 ligand-promoted invasion in cholangiocarcinoma. Cell Commun Signal. 2020;18(1):161.
Zemek RM, Jong ED, Chin WL, et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Transl Med. 2019;11(501):eaav7816.
Azuma M, Takeda Y, Nakajima H, et al. Biphasic function of TLR3 adjuvant on tumor and spleen dendritic cells promotes tumor T cell infiltration and regression in a vaccine therapy. Oncoimmunology. 2016;5(8):e1188244.
Shime H, Matsumoto M, Oshiumi H, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A. 2012;109(6):2066–71.
Jongbloed SL, Kassianos AJ, McDonald KJ, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207(6):1247–60.
Spranger S, Dai D, Horton B, et al. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711-723.e4.
Böttcher JP, Bonavita E, Chakravarty P, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022-1037.e14.
Kline DE, MacNabb BW, Chen XF, et al. CD8alpha(+) dendritic cells dictate leukemia-specific CD8(+) T cell fates. J Immunol. 2018;201(12):3759–69.
Hubert M, Gobbini E, Couillault C, et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci Immunol. 2020;5(46):eaav3942.
Lee YS, O'Brien LJ, Walpole CM, et al. Human CD141(+) dendritic cells (cDC1) are impaired in patients with advanced melanoma but can be targeted to enhance anti-PD-1 in a humanized mouse model. J Immunother Cancer. 2021;9(3):e001963.
Bianchi F, Alexiadis S, Camisaschi C, et al. TLR3 expression induces apoptosis in human non-small-cell lung cancer. Int J Mol Sci. 2020;21(4):1440.
Vidyarthi A, Khan N, Agnihotri T, et al. TLR-3 stimulation skews M2 macrophages to M1 through IFN-alphabeta signaling and restricts tumor progression. Front Immunol. 2018;9:1650.
Le Noci V, Tortoreto M, Gulino A, et al. Poly(I:C) and CpG-ODN combined aerosolization to treat lung metastases and counter the immunosuppressive microenvironment. Oncoimmunology. 2015;4(10):e1040214.
Le Noci V, Sommariva M, Tortoreto M, et al. Reprogramming the lung microenvironment by inhaled immunotherapy fosters immune destruction of tumor. Oncoimmunology. 2016;5(11):e1234571.
Sommariva M, Le Noci V, Storti C, et al. Activation of NK cell cytotoxicity by aerosolized CpG-ODN/poly(I:C) against lung melanoma metastases is mediated by alveolar macrophages. Cell Immunol. 2017;313:52–8.
Thomas G, Micci L, Yang W, et al. Intra-tumoral activation of endosomal TLR pathways reveals a distinct role for TLR3 agonist dependent type-1 interferons in shaping the tumor immune microenvironment. Front Oncol. 2021;11:711673.
Chen J, Sun W, Zhang H, et al. Macrophages reprogrammed by lung cancer microparticles promote tumor development via release of IL-1beta. Cell Mol Immunol. 2020;17(12):1233–44.
Tavora B, Mederer T, Wessel KJ, et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature. 2020;586(7828):299–304.
Barr TA, Brown S, Ryan G, et al. TLR-mediated stimulation of APC: distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37(11):3040–53.
Dajon M, Iribarren K, Petitprez F, et al. Toll like receptor 7 expressed by malignant cells promotes tumor progression and metastasis through the recruitment of myeloid derived suppressor cells. Oncoimmunology. 2019;8(1):e1505174.
Michaelis KA, Norgard MA, Zhu X, et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat Commun. 2019;10(1):4682.
Grimmig T, Matthes N, Hoeland K, et al. TLR7 and TLR8 expression increases tumor cell proliferation and promotes chemoresistance in human pancreatic cancer. Int J Oncol. 2015;47(3):857–66.
Chatterjee S, Crozet L, Damotte D, et al. TLR7 promotes tumor progression, chemotherapy resistance, and poor clinical outcomes in non-small cell lung cancer. Cancer Res. 2014;74(18):5008–18.
Yuan Q, Zhou Q, Ren J, et al. WGCNA identification of TLR7 as a novel diagnostic biomarker, progression and prognostic indicator, and immunotherapeutic target for stomach adenocarcinoma. Cancer Med. 2021;10(12):4004–16.
Dajon M, Iribarren K, Cremer I. Dual roles of TLR7 in the lung cancer microenvironment. Oncoimmunology. 2015;4(3):e991615.
Fotin-Mleczek M, Duchardt KM, Lorenz C, et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother. 2011;34(1):1–15.
Roychowdhury A, Jondhale M, Saldanha E, et al. Landscape of toll-like receptors expression in tumor microenvironment of triple negative breast cancer (TNBC): distinct roles of TLR4 and TLR8. Gene. 2021;792:145728.
Kiniwa Y, Miyahara Y, Wang HY, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13(23):6947–58.
Peng G, Guo Z, Kiniwa Y, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309(5739):1380–4.
Peng G, Wang HY, Peng WY, et al. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity. 2007;27(2):334–48.
Ye J, Ma CL, Hsueh EC, et al. Tumor-derived γδ regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol. 2013;190(5):2403–14.
Li L, Liu X, Sanders KL, et al. TLR8-mediated metabolic control of human Treg function: a mechanistic target for cancer immunotherapy. Cell Metab. 2019;29(1):103-123 e105.
Xu R, Wu M, Liu S, et al. Glucose metabolism characteristics and TLR8-mediated metabolic control of CD4(+) Treg cells in ovarian cancer cells microenvironment. Cell Death Dis. 2021;12(1):22.
Ye J, Ma C, Hsueh EC, et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med. 2014;6(10):1294–311.
Shang WW, Xu R, Xu T, et al. Ovarian cancer cells promote glycolysis metabolism and TLR8-mediated metabolic control of human CD4(+) T cells. Front Oncol. 2020;10:570899.
Dang Y, Rutnam ZJ, Dietsch G, et al. TLR8 ligation induces apoptosis of monocytic myeloid-derived suppressor cells. J Leukoc Biol. 2018;103(1):157–64.
Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180(6):1044–66.
Shayan G, Kansy BA, Gibson SP, et al. Phase Ib study of immune biomarker modulation with neoadjuvant Cetuximab and TLR8 stimulation in head and neck cancer to overcome suppressive myeloid signals. Clin Cancer Res. 2018;24(1):62–72.
Safarzadeh E, Mohammadi A, Mansoori B, et al. STAT3 silencing and TLR7/8 pathway activation repolarize and suppress myeloid-derived suppressor cells from breast cancer patients. Front Immunol. 2020;11:613215.
Hirata Y, Broquet AH, Menchén L, et al. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J Immunol. 2007;179(8):5425–32.
Furr SR, Moerdyk-Schauwecker M, Grdzelishvili VZ, et al. RIG-I mediates nonsegmented negative-sense RNA virus-induced inflammatory immune responses of primary human astrocytes. Glia. 2010;58(13):1620–9.
Broquet AH, Hirata Y, McAllister CS, et al. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol. 2011;186(3):1618–26.
Glas M, Coch C, Trageser D, et al. Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem Cells. 2013;31(6):1064–74.
Guo G, Gao M, Gao X, et al. Reciprocal regulation of RIG-I and XRCC4 connects DNA repair with RIG-I immune signaling. Nat Commun. 2021;12(1):2187.
Wang SQ, Yang XY, Yu XF, et al. Knockdown of IGF-1R triggers viral RNA sensor MDA5- and RIG-I-mediated mitochondrial apoptosis in colonic cancer cells. Mol Ther Nucleic Acids. 2019;16:105–17.
Iurescia S, Fioretti D, Rinaldi M. the innate immune signalling pathways: turning RIG-I sensor activation against cancer. Cancers (Basel). 2020;12(11):3158.
Daßler-Plenker J, Paschen A, Putschli B, et al. Direct RIG-I activation in human NK cells induces TRAIL-dependent cytotoxicity toward autologous melanoma cells. Int J Cancer. 2019;144(7):1645–56.
Brägelmann J, Lorenz C, Borchmann S, et al. MAPK-pathway inhibition mediates inflammatory reprogramming and sensitizes tumors to targeted activation of innate immunity sensor RIG-I. Nat Commun. 2021;12(1):5505.
Zhou B, Li C, Yang Y, et al. RIG-I promotes cell death in hepatocellular carcinoma by inducing M1 polarization of perineal macrophages through the RIG-I/MAVS/NF-kappaB pathway. Onco Targets Ther. 2020;13:8783–94.
Engel C, Brugmann G, Lambing S, et al. RIG-I resists hypoxia-induced immunosuppression and dedifferentiation. Cancer Immunol Res. 2017;5(6):455–67.
Wolf D, Fiegl H, Zeimet AG, et al. High RIG-I expression in ovarian cancer associates with an immune-escape signature and poor clinical outcome. Int J Cancer. 2020;146(7):2007–18.
Ge J, Wang J, Xiong F, et al. Epstein–Barr virus-encoded circular RNA CircBART2.2 promotes immune escape of nasopharyngeal carcinoma by regulating PD-L1. Cancer Res. 2021;81(19):5074–88.
Sultan H, Wu J, Kumai T, et al. Role of MDA5 and interferon-I in dendritic cells for T cell expansion by anti-tumor peptide vaccines in mice. Cancer Immunol Immunother. 2018;67(7):1091–103.
Brown MC, Mosaheb MM, Mohme M, et al. Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling. Nat Commun. 2021;12(1):1858.
Lin LL, Huang CC, Wu MT, et al. Innate immune sensor laboratory of genetics and physiology 2 suppresses tumor cell growth and functions as a prognostic marker in neuroblastoma. Cancer Sci. 2018;109(11):3494–502.
Zheng W, Ranoa DRE, Huang X, et al. RIG-I-like receptor LGP2 is required for tumor control by radiotherapy. Cancer Res. 2020;80(24):5633–41.
Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–80.
Wang Y, Miao Z, Qin X, et al. NOD2 deficiency confers a pro-tumorigenic macrophage phenotype to promote lung adenocarcinoma progression. J Cell Mol Med. 2021;25(15):7545–58.
Slavik KM, Morehouse BR, Ragucci AE, et al. cGAS-like receptors sense RNA and control 3’2’-cGAMP signalling in Drosophila. Nature. 2021;597(7874):109–13.
Lin Z, Wang J, Zhu W, et al. Chicken DDX1 acts as an RNA sensor to mediate IFN-β signaling pathway activation in antiviral innate immunity. Front Immunol. 2021;12:7474.
Xing J, Zhang A, Du Y, et al. Identification of poly(ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat Commun. 2021;12(1):2681.
Khodarev NN, Intracellular RNA. Sensing in mammalian cells: role in stress response and cancer therapies. Int Rev Cell Mol Biol. 2019;344:31–89.
Ammi R, Waele JD, Willemen Y, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. 2015;146:120–31.
Martins KA, Bavari S, Salazar AM. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev Vaccines. 2015;14(3):447–59.
Lu R, Groer C, Kleindl PA, et al. Formulation and preclinical evaluation of a toll-like receptor 7/8 agonist as an anti-tumoral immunomodulator. J Control Release. 2019;306:165–76.
Kanzler H, Barrat FJ, Hessel EM, et al. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13(5):552–9.
Nuhn L, Koker SD, Lint SV, et al. Nanoparticle-conjugate TLR7/8 agonist localized immunotherapy provokes safe antitumoral responses. Adv Mater. 2018;30(45):e1803397.
Doorduijn EM, Sluijter M, Salvatori DC, et al. CD4(+) T cell and NK cell interplay key to regression of MHC class i(low) tumors upon TLR7/8 agonist therapy. Cancer Immunol Res. 2017;5(8):642–53.
Wiedemann GM, Jacobi SJ, Chaloupka M, et al. A novel TLR7 agonist reverses NK cell anergy and cures RMA-S lymphoma-bearing mice. Oncoimmunology. 2016;5(7):e1189051.
Vascotto F, Petschenka J, Walzer KC, et al. Intravenous delivery of the toll-like receptor 7 agonist SC1 confers tumor control by inducing a CD8+ T cell response. Oncoimmunology. 2019;8(7):1601480.
Dietsch GN, Randall TD, Gottardo R, et al. Late-stage cancer patients remain highly responsive to immune activation by the selective TLR8 agonist motolimod (VTX-2337). Clin Cancer Res. 2015;21(24):5445–52.
Stephenson RM, Lim CM, Matthews M, et al. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol Immunother. 2013;62(8):1347–57.
Northfelt DW, Ramanathan RK, Cohen PA, et al. A phase I dose-finding study of the novel Toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin Cancer Res. 2014;20(14):3683–91.
Ferris RL, Saba NF, Gitlitz BJ, et al. Effect of adding motolimod to standard combination chemotherapy and cetuximab treatment of patients with squamous cell carcinoma of the head and neck: the active8 randomized clinical trial. JAMA Oncol. 2018;4(11):1583–8.
Ruzicka M, Koenig LM, Formisano S, et al. RIG-I-based immunotherapy enhances survival in preclinical AML models and sensitizes AML cells to checkpoint blockade. Leukemia. 2020;34(4):1017–26.
Helms MW, Jahn-Hofmann K, Gnerlich F, et al. Utility of the RIG-I agonist triphosphate RNA for melanoma therapy. Mol Cancer Ther. 2019;18(12):2343–56.
Jiang X, Muthusamy V, Fedorova O, et al. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J Exp Med. 2019;216(12):2854–68.
Such L, Zhao F, Liu D, et al. Targeting the innate immunoreceptor RIG-I overcomes melanoma-intrinsic resistance to T cell immunotherapy. J Clin Investig. 2020;130(8):4266–81.
Takeda Y, Kataoka K, Yamagishi J, et al. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. 2017;19(9):1874–87.
Takeda Y, Yoshida S, Takashima K, et al. Vaccine immunotherapy with ARNAX induces tumor-specific memory T cells and durable anti-tumor immunity in mouse models. Cancer Sci. 2018;109(7):2119–29.
Matsumoto M, Takeda Y, Seya T. Targeting Toll-like receptor 3 in dendritic cells for cancer immunotherapy. Expert Opin Biol Ther. 2020;20(8):937–46.
Qi X, Liu X, Matiski L, et al. RNA origami nanostructures for potent and safe anticancer immunotherapy. ACS Nano. 2020;14(4):4727–40.
Liu J, Hu Y, Guo Q, et al. Enhanced anti-melanoma efficacy of a Pim-3-targeting bifunctional small hairpin RNA via single-stranded RNA-mediated activation of plasmacytoid dendritic cells. Front Immunol. 2019;10:2721.
Donzelli J, Proestler E, Riedel A, et al. Small extracellular vesicle-derived miR-574-5p regulates PGE2-biosynthesis via TLR7/8 in lung cancer. J Extracell Vesicles. 2021;10(12):e12143.
Li B, Zhu L, Lu C, et al. circNDUFB2 inhibits non-small cell lung cancer progression via destabilizing IGF2BPs and activating anti-tumor immunity. Nat Commun. 2021;12(1):295.
Walton RW, Brown MC, Sacco MT, et al. Engineered oncolytic poliovirus PVSRIPO subverts MDA5-dependent innate immune responses in cancer cells. J Virol. 2018;92(19):e00879-e918.
Zhang L, Dewan V, Yin H. Discovery of small molecules as multi-toll-like receptor agonists with proinflammatory and anticancer activities. J Med Chem. 2017;60(12):5029–44.
Hayashi T, Chan M, Norton JT, et al. Additive melanoma suppression with intralesional phospholipid-conjugated TLR7 agonists and systemic IL-2. Melanoma Res. 2011;21(1):66–75.
Sato-Kaneko F, Yao S, Ahmadi A, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. 2017;2(18):e93397.
Hosoya T, Sato-Kaneko F, Ahmadi A, et al. Induction of oligoclonal CD8 T cell responses against pulmonary metastatic cancer by a phospholipid-conjugated TLR7 agonist. Proc Natl Acad Sci USA. 2018;115(29):E6836–44.
Mullins SR, Vasilakos JP, Deschler K, et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer. 2019;7(1):244.
Francian A, Widmer A, Olsson T, et al. Delivery of toll-like receptor agonists by complement C3-targeted liposomes activates immune cells and reduces tumour growth. J Drug Target. 2021;29(7):754–60.
Zhang H, Tang WL, Kheirolomoom A, et al. Development of thermosensitive resiquimod-loaded liposomes for enhanced cancer immunotherapy. J Control Release. 2021;330:1080–94.
Wan D, Que H, Chen L, et al. Lymph-node-targeted cholesterolized TLR7 agonist liposomes provoke a safe and durable antitumor response. Nano Lett. 2021;21(19):7960–9.
Varypataki EM, Benne N, Bouwstra J, et al. Efficient eradication of established tumors in mice with cationic liposome-based synthetic long-peptide vaccines. Cancer Immunol Res. 2017;5(3):222–33.
Zhao J, Zhang Z, Xue Y, et al. Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I : C) promote melanoma regression. Theranostics. 2018;8(22):6307–21.
Kotting C, Hofmann L, Lotfi R, et al. Immune-stimulatory effects of curcumin on the tumor microenvironment in head and neck squamous cell carcinoma. Cancers (Basel). 2021;13(6):1335.
Ishikawa T, Kageyama S, Miyahara Y, et al. Safety and antibody immune response of CHP-NY-ESO-1 vaccine combined with poly-ICLC in advanced or recurrent esophageal cancer patients. Cancer Immunol Immunother. 2021;70(11):3081–91.
Mottas I, Bekdemir A, Cereghetti A, et al. Amphiphilic nanoparticle delivery enhances the anticancer efficacy of a TLR7 ligand via local immune activation. Biomaterials. 2019;190–191:111–20.
Smith AAA, Gale EC, Roth GA, et al. Nanoparticles presenting potent TLR7/8 agonists enhance anti-PD-L1 immunotherapy in cancer treatment. Biomacromol. 2020;21(9):3704–12.
Ni K, Luo T, Culbert A, et al. Nanoscale metal-organic framework co-delivers TLR-7 agonists and anti-CD47 antibodies to modulate macrophages and orchestrate cancer immunotherapy. J Am Chem Soc. 2020;142(29):12579–84.
Figueiredo P, Lepland A, Scodeller P, et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomater. 2021;133:231–43.
Rodell CB, Arlauckas SP, Cuccarese MF, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578–88.
Zhang L, Huang J, Chen X, et al. Self-assembly nanovaccine containing TLR7/8 agonist and STAT3 inhibitor enhances tumor immunotherapy by augmenting tumor-specific immune response. J Immunother Cancer. 2021;9(8):e003132.
Li J, Yu X, Jiang Y, et al. Second near-infrared photothermal semiconducting polymer nanoadjuvant for enhanced cancer immunotherapy. Adv Mater. 2021;33(4):e2003458.
Wei B, Pan J, Yuan R, et al. Polarization of Tumor-Associated Macrophages by Nanoparticle-Loaded Escherichia coli Combined with Immunogenic Cell Death for Cancer Immunotherapy. Nano Lett. 2021;21(10):4231–40.
Jacobson ME, Wang-Bishop L, Becker KW, et al. Delivery of 5’-triphosphate RNA with endosomolytic nanoparticles potently activates RIG-I to improve cancer immunotherapy. Biomater Sci. 2019;7(2):547–59.
Das M, Shen L, Liu Q, et al. Nanoparticle delivery of RIG-I agonist enables effective and safe adjuvant therapy in pancreatic cancer. Mol Ther. 2019;27(3):507–17.
Han HD, Byeon Y, Jang JH, et al. In vivo stepwise immunomodulation using chitosan nanoparticles as a platform nanotechnology for cancer immunotherapy. Sci Rep. 2016;6:38348.
Liu L, He H, Liang R, et al. ROS-inducing micelles sensitize tumor-associated macrophages to TLR3 stimulation for potent immunotherapy. Biomacromol. 2018;19(6):2146–55.
Pitner R, Kim J, Davis-Bergthold J, et al. Structure-based design of JOC-x, a conjugatable tumor tight junction opener to enhance cancer therapy. Sci Rep. 2019;9(1):6169.
Melssen MM, Petroni GR, Chianese-Bullock KA, et al. A multipeptide vaccine plus toll-like receptor agonists LPS or polyICLC in combination with incomplete Freund’s adjuvant in melanoma patients. J Immunother Cancer. 2019;7(1):163.
Patel SP, Petroni GR, Roszik J, et al. Phase I/II trial of a long peptide vaccine (LPV7) plus toll-like receptor (TLR) agonists with or without incomplete Freund’s adjuvant (IFA) for resected high-risk melanoma. J Immunother Cancer. 2021;9(8):e003220.
Dillon PM, Petroni GR, Smolkin ME, et al. A pilot study of the immunogenicity of a 9-peptide breast cancer vaccine plus poly-ICLC in early stage breast cancer. J Immunother Cancer. 2017;5(1):92.
Akache B, Agbayani G, Stark FC, et al. Sulfated lactosyl archaeol archaeosomes synergize with poly(I:C) to enhance the immunogenicity and efficacy of a synthetic long peptide-based vaccine in a melanoma tumor model. Pharmaceutics. 2021;13(2):257.
Stevens AD, Bullock TNJ. Therapeutic vaccination targeting CD40 and TLR3 controls melanoma growth through existing intratumoral CD8 T cells without new T cell infiltration. Cancer Immunol Immunother. 2021;70(8):2139–50.
Kim SY, Kim S, Kim JE, et al. Lyophilizable and multifaceted Toll-like receptor 7/8 agonist-loaded nanoemulsion for the reprogramming of tumor microenvironments and enhanced cancer immunotherapy. ACS Nano. 2019;13(11):12671–86.
Koh J, Kim S, Lee SN, et al. Therapeutic efficacy of cancer vaccine adjuvanted with nanoemulsion loaded with TLR7/8 agonist in lung cancer model. Nanomedicine. 2021;37:102415.
Meneveau MO, Petroni GR, Salerno EP, et al. Immunogenicity in humans of a transdermal multipeptide melanoma vaccine administered with or without a TLR7 agonist. J Immunother Cancer. 2021;9(5):e002214.
Dong W, Zhang H, Yin X, et al. Oral delivery of tumor microparticle vaccines activates NOD2 signaling pathway in ileac epithelium rendering potent antitumor T cell immunity. Oncoimmunology. 2017;6(3):e1282589.
Koerner J, Horvath D, Herrmann VL, et al. PLGA-particle vaccine carrying TLR3/RIG-I ligand Riboxxim synergizes with immune checkpoint blockade for effective anti-cancer immunotherapy. Nat Commun. 2021;12(1):2935.
Di S, Zhou M, Pan Z, et al. Combined adjuvant of poly I: C improves antitumor effects of CAR-T cells. Front Oncol. 2019;9:241.
Johnson LR, Lee DY, Eacret JS, et al. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell. 2021;184(19):4981-4995 e4914.
Barry KC, Hsu J, Broz ML, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24(8):1178–91.
Salem ML, Díaz-Montero CM, Al-Khami AA, et al. Recovery from cyclophosphamide-induced lymphopenia results in expansion of immature dendritic cells which can mediate enhanced prime-boost vaccination antitumor responses in vivo when stimulated with the TLR3 agonist poly(I:C). J Immunol. 2009;182(4):2030–40.
Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–15.
Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–6.
Mehrotra S, Britten CD, Chin S, et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J Hematol Oncol. 2017;10(1):82.
Long S, Gu Y, An Y, et al. Reovirus enhances cytotoxicity of natural killer cells against colorectal cancer via TLR3 pathway. J Transl Med. 2021;19(1):185.
Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33.
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33.
Lesterhuis WJ, Bosco A, Millward MJ, et al. Dynamic versus static biomarkers in cancer immune checkpoint blockade: unravelling complexity. Nat Rev Drug Discov. 2017;16(4):264–72.
The Lancet Oncology. Calling time on the immunotherapy gold rush. Lancet Oncol. 2017;18(8):981.
Chen M, Hu S, Li Y, Jiang TT, Jin H, Feng L. Targeting nuclear acid-mediated immunity in cancer immune checkpoint inhibitor therapies. Signal Transduct Target Ther. 2020;5(1):270.
Babikr F, Wan J, Xu A, et al. Distinct roles but cooperative effect of TLR3/9 agonists and PD-1 blockade in converting the immunotolerant microenvironment of irreversible electroporation-ablated tumors. Cell Mol Immunol. 2021;18(12):2632–47.
Huang CH, Mendez N, Echeagaray OH, et al. Immunostimulatory TLR7 agonist-nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Adv Ther (Weinh). 2020;3(6):1900200.
Kim H, Khanna V, Kucaba TA, et al. Combination of Sunitinib and PD-L1 blockade enhances anticancer efficacy of TLR7/8 agonist-based nanovaccine. Mol Pharm. 2019;16(3):1200–10.
Narayanan JSS, Ray P, Hayashi T, et al. Irreversible electroporation combined with checkpoint blockade and TLR7 stimulation induces antitumor immunity in a murine pancreatic cancer model. Cancer Immunol Res. 2019;7(10):1714–26.
Heidegger S, Wintges A, Stritzke F, et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci Immunol. 2019;4(39):eaau8943.
Poeck H, Wintges A, Dahl S, et al. Tumor cell-intrinsic RIG-I signaling governs synergistic effects of immunogenic cancer therapies and checkpoint inhibitors in mice. Eur J Immunol. 2021;51(6):1531–4.
Le Naour J, Liu P, Zhao L, et al. A TLR3 ligand reestablishes chemotherapeutic responses in the context of FPR1 deficiency. Cancer Discov. 2021;11(2):408–23.
Wei X, Liu L, Li X, et al. Selectively targeting tumor-associated macrophages and tumor cells with polymeric micelles for enhanced cancer chemo-immunotherapy. J Control Release. 2019;313:42–53.
Liu Z, Xie Y, Xiong Y, et al. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett. 2020;469:173–85.
Tang L, Cai D, Qin M, et al. Oxaliplatin-based platinum(IV) prodrug bearing toll-like receptor 7 agonist for enhanced immunochemotherapy. ACS Omega. 2020;5(1):726–34.
Ringgaard L, Melander F, Eliasen R, et al. Tumor repolarization by an advanced liposomal drug delivery system provides a potent new approach for chemo-immunotherapy. Sci Adv. 2020;6(36):eaba5628.
Yoshida S, Shime H, Takeda Y, et al. Toll-like receptor 3 signal augments radiation-induced tumor growth retardation in a murine model. Cancer Sci. 2018;109(4):956–65.
Tan LSY, Wong B, Gangodu NR, et al. Enhancing the immune stimulatory effects of cetuximab therapy through TLR3 signalling in Epstein–Barr virus (EBV) positive nasopharyngeal carcinoma. Oncoimmunology. 2018;7(11):e1500109.
Cheadle EJ, Lipowska-Bhalla G, Dovedi SJ, et al. A TLR7 agonist enhances the antitumor efficacy of obinutuzumab in murine lymphoma models via NK cells and CD4 T cells. Leukemia. 2016;31(7):1611–21.
Chow LQM, Morishima C, Eaton KD, et al. Phase Ib trial of the toll-like receptor 8 agonist, motolimod (VTX-2337), combined with cetuximab in patients with recurrent or metastatic SCCHN. Clin Cancer Res. 2017;23(10):2442–50.
Cao L, Liu S, Li Y, et al. The nuclear matrix protein SAFA surveils viral RNA and facilitates immunity by activating antiviral enhancers and super-enhancers. Cell Host Microbe. 2019;26(3):369-384.e8.
Liu GQ, Lu Y, Thulasi Raman SN, et al. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat Commun. 2018;9(1):3199.
Wang Y, Yuan S, Jia X, et al. Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS. Nat Cell Biol. 2019;21(11):1346–56.
Zhang T, Yin C, Boyd DF, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180(6):1115-1129.e13.
Acknowledgements
Not applicable.
Funding
This study was supported by Grants from Natural Science Foundation of Shanghai [22ZR1450700] and National Natural Science Foundation of China [81971340 and 81502230].
Author information
Authors and Affiliations
Contributions
R.Y. and S.H.Y. collected the related papers and drafted the manuscript; R.Y., S.H.Y., T.H.X. and J.W.Z. revised the manuscript and drafted the figures; J.W.Z. and S.F.W. participated in the design of the review and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
All authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, R., Yu, S., Xu, T. et al. Emerging role of RNA sensors in tumor microenvironment and immunotherapy. J Hematol Oncol 15, 43 (2022). https://doi.org/10.1186/s13045-022-01261-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01261-z