
Abstract
High-grade B-cell lymphoma with translocations involving MYC and BCL2 or BCL6, usually referred to as double hit lymphoma (DHL), is an aggressive hematological malignance with distinct genetic features and poor clinical prognosis. Current standard chemoimmunotherapy fails to confer satisfying outcomes and few targeted therapeutics are available for the treatment against DHL. Recently, the delineating of the genetic landscape in tumors has provided insight into both biology and targeted therapies. Therefore, it is essential to understand the altered signaling pathways of DHL to develop treatment strategies with better clinical benefits. Herein, we summarized the genetic alterations in the two DHL subtypes (DHL-BCL2 and DHL-BCL6). We further elucidate their implications on cellular processes, including anti-apoptosis, epigenetic regulations, B-cell receptor signaling, and immune escape. Ongoing and potential therapeutic strategies and targeted drugs steered by these alterations were reviewed accordingly. Based on these findings, we also discuss the therapeutic vulnerabilities that coincide with these genetic changes. We believe that the understanding of the DHL studies will provide insight into this disease and capacitate the finding of more effective treatment strategies.
Similar content being viewed by others


Background
Diffuse large B cell lymphomas (DLBCLs) are aggressive, heterogeneous neoplasms with distinct biological, pathological and clinical characteristics, representing ~ 25% of non-Hodgkin lymphomas (NHL) [1]. DLBCLs harboring translocation of MYC(8q24) and BCL2(18q21) or/and BCL6(3q27) displays a highly aggressive profile, including high incidence of advanced disease at diagnosis, and poor response to up-front R-CHOP therapy (5-year overall survival [OS] and progression-free survival [PFS] rates of 27% and 18%, versus 71% and 65% in other DLBCLs) [2]. Owing to their unique genomic features, biological behaviors, and prognosis, the subtypes were classified as a new category in the 2016 revision of the World Health Organization (WHO) classification for lymphoma. They were termed high-grade B-cell lymphoma (HGBL) with translocations involving MYC and BCL2 and/or BCL6, also called double hit lymphoma (DHL) or triple hit lymphoma (THL) [3]. DHL involves the gene translocation of MYC and BCL2 (DHL-BCL2) or MYC and BCL6 (DHL-BCL6), while THL involves the translocation of all these three genes.
Given inferior outcomes with current therapies in DHL patients, further understanding disease pathology is of great importance to developing better regimens. Herein, we reviewed current clinical management, genetic alterations, and their physiological functions, as well as novel agents in development, hoping to enlighten future exploration in DHL. Since the majority of histological subgroups of DHLs are DLBCLs [3], this review focuses on DHLs fulfilling DLBCL histology. These DHLs were termed as DHL-BCL2 (MYC and BCL2 translocation) and DHL-BCL6 (MYC and BCL6 translocation) in this review.
Diagnosis, prognosis, and current treatment
Diagnosis of DHLs
DHLs are identified in 5–15% DLBCLs [3]. Diagnosis of DHLs requires identification of translocations of MYC(8q24) and BCL2(18q21) or/and BCL6(3q27), which is usually accomplished by fluorescent in-situ hybridization (FISH) [4]. Up to now, no consensus has been established as to which patients should have FISH tests. Some institutions recommend FISH to all DLBCL patients (routine testing), whereas others only recommend it to certain patients (selective testing) due to high cost and low prevalence [5]. In the centers performing selective testing, decisions are made based on patients’ pathologic characteristics, including MYC and BCL2 protein expression, Ki-67 percentage and cell-of-origin (COO) subtype, etc. [6, 7].
Based on COO, DLBCL could be classified into GCB and non-GCB via immunohistochemistry test (IHC), which is the most common technique with low cost used in clinic. In academic institutions, gene expression profiling (GEP) could also be used to further classify DLBCL into GCB, ABC and unclassified subtypes. Due to different prevalence of DHL-BCL2 and DHL-BCL6 in these COO subtypes, COO diagnosis could help the decision of selective testing. However, the consistency between IHC and GEP is not 100%. Sriram Balasubramanian and colleagues reviewd 910 patients’ IHC and GEP results from PHOENIX study. IHC result was 82.7% concordant with that of GEP. Due to the cost issue, IHC is currently more accessible.
Prognosis and risk stratification
Even within the same molecular group, the clinical outcome can be very different. Though conflicting results were yielded from different studies when associating MYC partner to prognosis [8,9,10,11], IG translocation partner of MYC likely indicates inferior outcome, especially in DHLs [12]. Additionally, DLBCL co-expressing MYC and BCL2 (double expressor lymphomas (DEL)) are associated with significantly shorter OS and PFS in both DHL and non-DHL [2, 12,13,14]. Interestingly, DHL cases without concurrently expressing MYC and BCL2 had more favorable outcomes than DEL after ASCT [13]. Sha et al. defined a molecular high-grade (MHG) group in DLBCL patients based on gene expression profile [15]. The 3-year PFS of the MHG group and others treated with bortezomib plus R-CHOP were 37% and 72%, respectively. Intriguingly, only half of the MHG group was diagnosed with DHL, whereas the other half demonstrated inferior outcomes compared to the non-MHG group, irrespective of their translocation status. The majority of DHLs were classified as MHG. However, no signs of worse outcomes were observed in DHLs lacking MHG features.
When compared to DHL-BCL2/THL, DHL-BCL6, the minority of all DHL, possess distinct features in many aspects. In contrast to DHL-BCL2/THL which are distinct GCB subtypes, DHL-BCL6 can be found in lymphoma with various COO including GCB, ABC, and unclassified. DHL-BCL6 has a lower proportion of MYC + by IHC compared to DHL-BCL2/THL, whereas DHL-BCL2/THL share similar morphology (high-grade with a starry sky appearance) and immunophenotype (CD10+, MCY + BCL2+, and MUM1) [16, 17]. Also, DHL-BCL6 tends to have a less cytogenetic complexity [18]. Recent studies indicate that compared to MYC/BCL2 lymphoma, lymphoma with BCL6 translocation shas a distinct molecular profile [19,20,21,22,23], suggesting a distinct molecular group from the DHL-BCL2/THL, the majority of DHL. Therefore, more study should be done to investigate the molecular mechanism and category of these two subtypes.
In terms of prognosis, DHL-BCL2 and THL have similar OS [17, 24]. As for DHL-BCL6, some studies suggest that this subtype is more aggressive, frequently involving extranodal sites, and has worse OS compared to DHL-BCL2 [17, 18, 24]. However, there are contradicting results indicating that DHL-BCL6 has a similar, or even better prognosis than DHL-BCL2 [24, 25]. The conflicting conclusion may be the result of a limited sample population (~ 10 in most studies).
The prognosis is different within the DHL-BCL2 group. Parallelly, a 104-gene double-hit signature (DHITsig) developed by Ennishi et al. distinguished a cohort of GCB-DLBCLs (DHITsig positive) from other GCB-DLBCLs (DHITsig negative) [26]. DHITsig positive group was composed of 48% non-DHL/THL-BCL2s and 52% DHL/THL-BCL2s (88% of all DHL/THL-BCL2s). DHITsig positive patients had dismal outcomes after being treated with R-CHOP in terms of DSS (BioElectric Massage) (pos vs neg, 63% vs 90%), 5-year PFS (pos vs neg, 53% vs 79%), and OS (pos vs neg, 60% vs 83%). Several literature highlighted the importance of risk stratification depending on molecular features like GEP (gene expression profile), instead of merely genetic translocation status, to avoid missing out on a cluster of high-risk patients while exposing low-risk ones to excessive therapies [27, 28].
Current chemotherapy
The standard regimen for DHL has not been established yet. When clinical trials are not available, chemotherapy is the first choice. Regimens, like R-CHOP, R-DA-EPOCH, R-Hyper-CVAD, and R-CODOX-M/IVAC, have been clinically evaluated in Table 1 [29].
When treated with R-CHOP, DHLs were found to be associated with inferior OS compared to other DLBCLs (median OS: 13 months vs 95 months; 3-years OS: 46% vs 75%, HR 3.04, P = 0.002) [30]. Besides, when retrospectively evaluating application of IFRT(involved-field radiation therapy), R-CHOP and more intensive immunochemotherapy in patients with limited-stage (LS) DHL, similar PFS and OS were observed throughout the treatment groups (IFRT vs no IFRT, and R-CHOP vs more intensive immunochemotherapy) [31].
Several retrospective studies and meta-analyses have compared R-CHOP to more intensive regimens in DHL patients. Petrich et al. analyzed 311 newly diagnosed DHL patients, receiving R-CHOP (31%), R-DA-EPOCH (21%), R-HyperCVAD (21%), and R-CODOX-M/IVAC (14%). They found that intensive regimens were associated with significantly improved PFS, but not OS [32]. Oki et al. found that R-EPOCH was associated with longer EFS (HR = 0.37, P = 0.008) compared to R-CHOP, while R-HyperCVAD did not bring more benefit [33]. A meta-analysis compared the survival outcomes in DHL patients receiving more intensive regimens (R-DA-EPOCH, n = 91, and R-Hyper CVAD or R-CODOX-M/IVAC, n = 123) versus R-CHOP (n = 180) [34]. First-line treatment with R-EPOCH significantly reduced progression risk compared to R-CHOP (relative risk reduction of 34%; P = 0.032), but not OS. In all, these studies supported use of more intensive regimens like R-DA-EPOCH over R-CHOP when treating DHL patients. However, a retrospective multicenter study conducted on 90 cases of DEL patients showed no survival benefit from DA‐EPOCH‐R comparing to R-CHOP [35]. These contradicted results indicate that prospective studies are in need.
Transplantation
Allogeneic/autologous stem cell transplant (allo-SCT/auto-SCT) is a potentially curative option for many hematological malignancies and has been evaluated in the DHL cohort. However, most studies suggest that SCT may have a dismal effect on DHL patients.
In a multicenter study involving 311 DHL patients in North America, the response of patients receiving SCT (allo-SCT or auto-SCT) after induction chemotherapy, which includes R-CHOP, R-HyperCVAD/MA, DA-EPOCH-R, or R-CODOX-M/IVAC, as well as other non-rituximab regimens (5%) were documented. After the first CR, the median OS is between the SCT group(n = 39) and observation group(n = 112) did not have a significant difference [32]. Similar results were obtained by independent studies in 2014(n = 129),2017(two studies, n1 = 159, n2 = 117) and 2021(n = 160). Auto-SCT after a front line or intensive chemotherapy in DHL patients did not affect 3-year PFS or OS after first CR [13, 33, 36, 39]. On the other hand, these studies again emphasize the importance of intensive therapy compared to front-line therapy in improving the 3-year RFS [36]. Nevertheless, according to a 2016 trial, patients with DEL or MYC overexpression may benefit from auto-SCT following CHOP/− + R treatment, presenting a lightly longer PFS and OS [40].
Genetic alterations in DHL
DHL is accompanied by genetic and molecular features that distinguish them from other DLBCL and Burkitt. With several recent studies focusing on the genetic classification of DLBCL [19,20,21,22], we summarized genetic alterations that intensively or exclusively coincides with the translocation of MYC and BCL2 and/or BCL6, and characterized consequent functional disturbance.
Genetic alterations in DHL-BCL2
From previouspublications, DLBCL subgroups C3 [20], EZB(-DHIT+) [21, 22], BCL2 [19], and MYC/BCL2-DH [23] shared similar genetic profiles and unanimously encompassed dual translocations of MYC and BCL2.
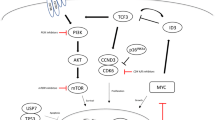
Herein, we summarized genetic similarities of these subgroups and combined them into one group called DHL-BCL2 (Fig. 1). DHL-BCL2 lymphomas were exclusively germinal center lymphomas with aberrant GC formation. DHL-BCL2 lymphomas were exclusively germinal center lymphomas [16, 41], with frequent genetic alterations associated with the formation of GC [42,43,44,45]. The primary genetic lesions are alterations associating epigenetic regulators like KMT2D, EZH2, and CREBBP/EP300. DHL-BCL2 also exhibited an anti-apoptotic and pro-survival profile, with aberrant activation of BCL2 and PI3K-mTOR signaling. Inactivating lesions of the S1PR2-Gα13 axis contributes to tumor cells dissemination, while damaged TNFRSF14 leads to increased CD40 signaling. Other proto-onco mutations also occur in this subtype, including activation of MYC, JAK-STAT, and NF-κB signaling, as well as suppressing p53 pathway. Gene alteration frequency and categorizing foundation were summarized in Table 2.

Genetic and pathways alterations involved in DHL-BCL2
Alterations relating cellular differentiation and transcription factors
In DHL-BCL2, disrupted the transcriptional process of B cell development may contribute to dysregulated GC development, altered apoptosis profile, and eventually lymphomagenesis.
Both interferon regulatory factor8 (IRF8) and myocyte enhancer factor 2B (MEF2B) are vital transcription factors in GC formation and frequently mutated in DHL-BCL2. In normal B cells, IRF8 transactivates BCL6 and AICDA [46] and prevents B cells from p53-induced apoptosis by up-regulating MDM2 [47]. MEF2B modulates the expression of a series of genes from the cell cycle (CCND3), differentiation (BCL6) to apoptosis (TP53 and BCL2) [48]. They are preferentially expressed and frequently altered in B cell lymphomas of GC origin like DLBCL and follicular lymphoma [20,21,22, 49,50,51,52,53]. The mutation of MEF2BD83V contributes to lymphomagenesis by enlarging GC formation, dysregulating level of BCL6 [50] and other gene targets [48], while IRFB mutation and translocation were also believed to promote lymphomagenesis [46, 51].
MYC protein plays a crucial role in cellular processes, including growth, proliferation, metabolism, and biosynthesis [54, 55]. Besides MYC translocation, mutation and amplification are also repeatedly found in DHL-BCL2 [22, 56], leading to dysregulated MYC activity and oncogenic signals. MYC translocation adjacent to IG enhancer [8] and MYC amplification engender elevated MYC expression [57, 58]. MYC mutation, on the other hand, has divergent impacts on MYC expression and clinical outcomes [56]. However, the implications of most mutations have not yet been elucidated.
Forkhead box O1 (FOXO1) fosters B cell differentiation [59], and is expressed and recurrently mutated in DLBCLs [19, 21, 22, 60, 61]. FOXO1 is restricted by PI3K signaling [62], Its absence abrogates toxicity induced by SYK and AKT inhibitor [63]. It has been reported that FOXO1 mutation is associated with inferior prognosis [60], refractory and relapsed GCB-DLBCL [64], as well as correlations with TP53 and BCL2 mutation [64, 65]. Other transcription factors, like octamer-binding protein 2 (OCT2) which is encoded by POU Class 2 Homeobox 2(POU2F2), has been proposed as a therapeutic target due to its role in promoting B cell proliferation in GC [66], inducing the IL-6 expression [67] and regulating SYK expression [68].
Alterations relating apoptosis
The BCL2 family is constituted of three subfamilies: anti-apoptotic proteins (including BCL2, BCL-XL, BCL-W, MCL-1, BFL-1/A1, and BCL-B/BCL2L10), pro-apoptotic BH3-only proteins, and pro-apoptotic effector proteins (BAX and BAK). BH3-only proteins respond to cytotoxic stimuli or extrinsic pathway signals (FAS and TNFR1), expose their BH3 domain and subsequently neutralize anti-apoptotic proteins or directly activate BAX and BAK, to initiate oligomerization, and eventually to trigger caspases-dependent apoptosis [69].
Besides BCL2 translocation juxtaposed to IGH enhancer, BCL2 mutation is also enriched in this subtype [19,20,21,22,23, 70]. Non-synonymous mutations of BCL2 were primarily distributed in three domains: the p53 binding site of flexible loop domain (FLD), the BH4 domain, and the region between BH3 and BH1 [70, 71], thereby promoting its anti-apoptotic effect by disrupting its interaction with p53 [72], inositol 1,4,5-trisphosphate receptor (IP(3)R) [73] and BAX [74], respectively. Moreover, recurrent mutations in the BH3 domain of BCL2 contribute to the resistance of venetoclax [75].
After stimulation by FASL, FAS(CD95) can induce apoptosis and contribute to immune contraction and cell migration [76]. In EZB-DHITsig+, mutation and deletions are frequently observed in the intracellular death domain of FAS, resulting in apoptotic function loss and further inferior survival [77, 78].Additionally, genetic signatures, which were associated with the depletion of immune and stromal cells and the augment of tumor cells, were enriched in the EZB-DHITsig+ cluster with FAS alteration [78].
Alterations relating to epigenetic regulators
Alterations of epigenetic regulating genes are the most prominent in DHL-BCL2 [19,20,21, 23, 79]. Some have been considered promising therapeutical targets.
Histone methylation and demethylation occur at the lysine residue of histone H3 or H4 to different degrees (mono-methylation(me1), di-methylation (me2), or trimethylation (me3)), depending on status of active or inactive chromatin. Upon histone methylation, proteins with methyl-binding domains recognize the methylated lysine and initiate transcriptional activation or repression [80, 81].
KMT2D is a histone lysine methyltransferase that catalyzes the mono- and di-methylation of lysine 4 on histone H3(H3K4). Premature stop codons upstream of the enzymatic region can be introduced by deletion and nonsense mutation, leading to KMT2D inactivation. Missense mutations at C-terminal domains and other possible mechanisms contribute to impaired methyltransferase activity without KMT2D alteration [19,20,21,22,23, 43]. KMT2D lesions cause reduced H3K4 methylation and altered transcriptome, which mainly affects genes regarding cell cycle regulation, anti-apoptosis, and pro-apoptosis. Such process also contributes to increased GC formation, pro-proliferative cellular profile, inhibition of tumor-suppressive pathways, accelerated lymphoma development, and even malignant transformation when cooperate with BCL2 [43, 82].
As for H3K27, hypermethylation may cause lymphomagenesis. EZH2 is a component of polycomb repressive complex 2 (PRC2) which methylates H3K27 and transcriptionally represses genes relating to the cell cycle checkpoint [83]. Missense mutations of EZH2 are enriched at Y641 in the SET domain and have been identified in different patient cohorts and cell lines [19,20,21,22,23, 83,84,85]. Although EZH2 with mutant Y641 showed a reduced ability to directly tri-methylate H3K27 [85], global cellular H3K27me3 level containing EZH2 Y641 is still remarkably promoted owing to the increased substrate preference of EZH2 for H3K27me2 and increased EZH2 stability [84, 86]. EZH2 mutation and aberrantly hypermethylated H3K27 suppress differentiation and promote proliferation of GC B cells. It also alters the expression of EZH2, MYC, and genes relating to B cell regulation decreases MHC expression, and reduces T cell infiltrate in the tumor microenvironment. When cooperating with BCL2, MYC, or BCL6/BCOR complexes, EZH2 mutation promotes lymphomagenesis [87,88,89,90,91]. C10orf12 was also identified to interact with histone methyltransferase PRC2. The interaction enhances activity and substrate preference of PRC2, and thereby upregulate cellular H3K27me3 levels [92].
Normally, histone acetylation dissociates its interaction with DNA, leading to genetic transcription. CREBBP and EP300 are transcriptional coactivators comprising a catalytic histone acetyl-transferase domain [93]. CREBBP and EP300 have high sequence homology, yet their function in GC development turns out to be different. Deletion of both genes results in abrogation of GC formation [94]. In DHL-BCL2, CREBBP alteration (including deletions and mutations) is more frequent than that of EP300 [19,20,21,22,23, 95]. Loss-of-function of CREBBP leads to loss of H3K27ac at putative enhancers, expanded GC compartment, interfered plasma cell differentiation, indirective induction of MYC level, inactive p53 acetylation, proliferative profile, and MHC II deficiency of B cells. It also induces lymphomagenesis in an HDAC3 dependent manner, especially when cooperates with dysregulated BCL2 [42, 96,97,98]. Mutation of EP300 hardly overlaps with that of CREBBP [99]. EP300 function repression due to mutation or BCL6 suppression reduces H3K27ac, induces cell growth, and impairs the effect of BCL6 inhibitors and anti-CD20 antibodies to DLBCL cells [99,100,101]. Additionally, CREBBP and EP300 mutations contribute to the polarization of tumor-associated macrophages by increasing expression of CCL2, CSF1, and IL-10 via the Notch pathway [99].
SWI/SNF chromatin remodeling complexes regulate the transcriptional accessibility of DNA by mobilizing histone octamers to adjacent DNA positions [102, 103]. Point mutations of BCL7A, a subunit of SWI/SNF complexes, is enriched in the first splice donor and results in BCL7A inactivation, damaging BCL7A binding to SWI/SNF complex [19, 21, 22, 104], while wild BCL7A exhibits a tumor-suppressing role [104]. ARID1A is another subunit of SWI/SNF chromatin remodeling complexes. Deletions of ARID1A may affect the regulatory effect on CDKN1A, and promote proliferation and the ubiquitin–proteasome-dependent degradation of ARID1A [105]. The mutation of ARID1A and FOXO1 is also associated with loss of HLA-C [106]. It is worth noting that the ARID1A-deficient cancers are susceptible to the perturbance of homolog ARID1B, indicating that synthetic lethality may be a possible therapeutic option in these specific tumors [107].
Additionally, the P95 mutant of splicing factor SRSF2 identified in DHL-BCL2 alters the binding and global splicing event of RNA, thus affecting the regulation of gene expression and leading to abnormal hematopoiesis [108, 109].
Alterations relating to oncogenic pathways
-
(1)
Alterations relating PI3K-Akt-mTOR and BCR-NF-κB pathway
In GCB-DLBCLs, tonic BCR signaling leads to proliferation via the activation of NF-κB [110]. c-Rel, a protein of the NF-κB family, forms homo- or heterodimer with other NF-κB subunits after upstream stimuli, and translocate to the nucleus to transcriptionally activate genes associated with survival and proliferation [111]. c-Rel is encoded by REL whose amplification has early been identified as a genetic alteration associated with GCB-DLBCL [112] and is recently recognized as an EZB-related event [21, 22]. However, although this amplification arouses increased abundancy of c-Rel mRNA [113], nuclear c-Rel level and NF-κB activity do not correlate with REL amplification [113,114,115].
The activation of co-receptor of BCR, CD19, and SYK kinase can also trigger the signaling of the PI3K-Akt-mTOR pathway, which promotes cell survival [116]. As a negative regulator of Akt, PTEN loss caused by deletion and truncating mutation promotes tonic BCR signaling, Akt activation, and cell proliferation [20, 22, 110, 117].
-
(2)
Alterations relating JAK-STAT pathway
The JAK-STAT signaling cascade consists of three main components: a cell surface receptor, a JAK, and two STAT proteins. Once the cytokine binds to its receptor, the JAK family will be phosphorylated, resulting in the recruitment of phosphorylated and dimerized STAT proteins [118]. Subsequently, STATs are translocated to the nucleus and transcriptionally participate in cell proliferation, survival, angiogenesis, and immunity [119].
Amplification of STAT6 in DHL-BCL2 enhances JAK-STAT signaling [20, 22]. Missense mutations result in repetitive alterations in the DNA-binding domain of STAT6, especially at D419, which is both prevalent in DHL-BCL2 and R/R DLBCL [19, 64]. STAT6-D419 mutations were recognized as the gain of function mutations owing to the increased nuclear localization of STAT6, upregulated STAT6-targeted gene expression, and increased sensitivity to JAK2 inhibitors of STAT6-D419-mutated lymphomas [120,121,122].
-
(3)
Alterations relating p53
P53 is a tumor-suppressing protein encoded by TP53, which transactivates genes regarding apoptosis and cell cycle. It can be degraded by MDM2-mediated ubiquitination and proteasomal degradation and repressed by sumoylation and neddylation [123]. Mutation of TP53 occurs in ~ 20% of all DLBCLs [21, 124], preferentially in DHLs with MYC and BCL2 rearrangements [22, 125]. In DHL-BCL2, most mutations are located in the DNA-binding core domain of TP53, posing a deleterious effect on p53 function [125] since the transcription activity of p53 is requisite for its tumor-suppressing ability [126]. In DHLs, DHITsig positive DLBCLs [127] and the rest of DLBCLs, as well as other lymphoid malignancies mutated TP53 status [123, 124] are correlated with inferior prognosis.
-
(4)
Other alterations relating to oncogenic pathways
Though they have not been fully elucidated, some of the mutations of GNAI2 are orthologous to gain-of-function mutations of GNAI3, indicating an oncogenic effect [128]. Also, activating mutations of GNAI2 at the GTP-binding domain may result in aberrant MAPK/ERK signaling [129]. DDX3X is a tumor-suppressing gene that is recurrently mutated in Burkitt lymphoma and EZH-DHIT + B cell lymphomas, and such mutation results in the truncation or loss of DDX3X [22, 130]. In NK/T cell lymphomas, DDX3X mutant activates NK-κB and MAPK signaling, while losing the effect in inducing apoptosis, inhibiting the activation of ERK and RelB compared to its WT counterpart [131]. HVCN1 is also frequently mutated and truncated, which might induce stronger BCR-dependent signaling, proliferation, and migration [132].
Alterations relating to immune escape
Recurrent TNFRSF14 mutations in DHLs lead to reduced expression of HVEM [19, 20, 22, 23, 78], a TNFR family protein and tumor suppressor which inhibits BCR signaling, abnormal stroma activation, and CD40 signaling through BTLA stimulation [133, 134]. Also, when triggered by LIGHT, it will render tumor B cells higher immunogenicity and sensitivity to FAS-induced apoptosis [135].
Alterations relating to cell migration
S1PR2-Gα13 (encoded by SIPR2 and GNA13, respectively) signaling regulates cell growth, confinement, and localization of B cells to GC through inhibiting Akt phosphorylation under normal physiological conditions [136], while its dysregulation promotes lymphomagenesis [136,137,138,139]. GNA13 mutation, which often co-occurs with BCL2 rearrangement [19, 20, 22, 23, 70, 79], contributes to disrupted GC architecture in lymph nodes and disseminates GC B cells to systematic circulation [137]. Furthermore, loss of function mutation of GNA13 impairs its ability in suppressing BCL2 expression [140].
Genetic alterations in DHL-BCL6
Compared to their counterparts, DHLs with BCL6 translocations are relatively minor described. As reported in multiple centers, DHL-BCL2 are mainly GCB subtypes [19, 20, 22], while GCB, ABC, and undefined lymphoma can all be identified in DHL-BCL6 [141]. Upon previous publications, genetically categorized subtypes C1 [20], BN2 [21], NOTCH-2 [19], and BCL6-DH [23] are all accompanied by the translocation of BCL6, suggesting possible similarities between DHL-BCL6 and these published subtypes.
In this review, we summarized these genetic subtypes as DHL-BCL6. Genetic alterations in DHL-BCL6 closely resemble genetic events in marginal zone lymphomas (Fig. 2) [19,20,21,22, 143]. Genetic lesions in this subtype are associated with the disruption of immune escape, Notch signaling, and ubiquitin–proteasome degradation system. Pro-proliferative signatures, including cell cycle genes mutations, BCR and NF-κB signaling activation, p53 pathways suppression, are also observed in this subtype. Detailed annotations were presented in Table 3.

Genetic and pathways alterations involved in DHL-BCL6. Note: *genes associated with the ubiquitin–proteasome system; #: the consequence of UBE2A mutation is not fully illustrated, might be functionally damaging [142]
Alterations relating Cellular differentiation and transcription factors
DHL-BCL6 is characterized by the dual translocation of MYC and BCL6. BCL6 is a transcriptional repressor, which is vital for the development and maintenance of GC. It regulates complex genetic networks associated with cell differentiation, DNA damage repair, apoptosis, signaling pathways, and T-B cell interactions [144]. Mutations in DHL-BCL6 that coincide with marginal zone lymphomas have been described, and the loss of KLF2 (a transcription factor involved in B cell homeostasis) increases marginal zone B cell [145].
Mutation of other transcription factors may result in the alteration of intercellular signaling. ETS1, a member of the ETS family namely FLI1, sustain proliferation and survival of B cells [146]. In ABC DLBCL, ETS1 is overexpressed, resulting in transactivation of genesrelating to BCR and CD40 signaling, NF-κB/TNFα pathways, immune response, and early differentiation. Thus ETS1 overexpression which promotes B cells growth and proliferation of [147]. ZEB2 is a transcription factor expressed in various immune cells.Loss of ZEB2 leads to impaired differentiation and IL-7 signaling, and aberrant proliferation of myeloid cells [148]. Mutation of POU2F2 is found in both DHL-BCL2 and -BCL6 cohorts [19].
Alterations relating to oncogenic pathways
-
(5)
Alterations relating to BCR and NF-κB signaling
Compared to DHL-BCL2, genes associated with BCR and NF-κB pathways are more prominently mutated in the DHL-BCL6 subtype, leading to up-regulated BCR signaling [19,20,21, 23]. BCL10 and PRKCB encode two components in the BCR and NF-κB signaling transduction [149], which are mutated and amplified in DHL-BCL6 [21, 22]. Inactivating mutations located in TNFAIP3, the negative regulator of NF-κB could cause abnormal activation of NF-κB pathway and proliferation of cancer cells [150]. KLHL21(CRL3, an E3 ubiquitin ligase) negatively regulating NF-κB by degradation of IKKβ [151] and affecting cell migration [152, 153], is also subjected to heterozygous loss and homozygous deletion in DHL-BCL6 [21, 22].
-
(6)
Alterations relating to the Notch pathway
Once Notch ligand binds to the Notch receptor, the latter will be cleaved by disintegrin, metalloproteinase domain-containing protein (ADAM) at S2 site and γ-secretase at S3 site, to generate Notch extracellular domain, Notch transmembrane domain (NTMD), and Notch intracellular domain (NICD). Afterwards, NICD enters the nucleus, combines with cBF1-suppressor of hairless-LAG1 (cSL) and mastermind-like proteins (MAML1, MAML2, or MAML3), and activates the transcription of related genes [154].
The aberrantly activated Notch pathway, which promotes NF-κB signaling and cell proliferation, usually co-occurs with BCL6 translocation [19,20,21,22,23]. Notch targeted genes are up-regulated in this cluster, facilitating their differentiation to marginal zone cells[22, 155]. Mutated or truncated Notch2 receptors, as well as inactive mutation of Notch suppressor SPEN, were often found in this cluster, resulting in Notch2 signaling hyperactivation [156, 157]. DTX1 mutations were also identified, equally in both GCB and non-GCB DLBCLs, and are primarily enriched in the N-terminal protein-interaction domain (WWE1), where the DTX1 binds with Notch and exhibits inhibitory role [158, 159].
Alterations relating cell cycle and p53
Cyclin interacts with CDKs to modulate the cell cycle. Mutations located at the 260–290 amino acids of CCND3 are found both in Burkitt lymphoma and DLBCL, which is likely to increase the stability of cyclin D3, and to further promote cell cycle progression [19, 64, 130, 160]. CDKN2A encodes proteins, including p53 stabilizer ARF and p16INK4a, which halts cell cycle via antagonizing CDK4/6 to [161]. Recurrent mutations found at R80 may result in the loss of CDK4 inhibiting ability [19, 162].
Besides, p53 pathway can also be affected by the mutated TRRAP. TRRAP encodes transformation/transcription domain-associated protein (a cofactor of histone acetyltransferases), which also contributes to p53 accumulation [163]. Heterozygous loss of TRRAP, therefore, may impair p53 signaling [21, 22].
TP63 is a member of TP53 family, which encodes two isoforms of p63, including tumor suppressor TAp63 (with N-terminal transactivation domain), and a truncated form ∆Np63 (without N-terminus), possibly an oncoprotein [164]. Mutation, deletion, and TBL1XR1/TP63 gene fusion may promote oncogenesis via deregulation of p63 and BCL6, BCL2, and MYC [22, 165].
Alterations relating to the ubiquitin–proteasome system
The ubiquitin–proteasome system plays a key role in cellular protein degradation. In the system, polyubiquitination of the target protein is mediated sequentially by a single ubiquitin-activating enzyme 1 (E1), multiple ubiquitin-conjugating enzymes (E2), and ubiquitin-protein ligases (E3). Polyubiquitinated protein is later deubiquitinated and degraded to oligopeptides by proteasome complex [166].
Enzymes related to this process are frequently mutated in DHL-BCL6 [19,20,21,22,23]. The mutation of UBE2A (ubiquitin-conjugating enzyme E2A) might affect histone H2A ubiquitination and in turn, perturb the transcriptome relating to myeloid development [142, 167]. Ubiquitin E3 ligase TRIP12 participates in bioprocesses including cell proliferation, DNA repair, and chromatin remodeling via regulating the stability of proteins including PARP1, CDKN2A, RNF168, USP7, and p53. Mutation and increased expression of ubiquitin E3 ligase TRIP12 are also detected in DHL-BCL6 [168]. As described above, mutation of E3 ubiquitin-protein ligase DTX1 affects the Notch pathway, while KLHL21 mutation results in NF-κB upregulation.
Alterations relating to immune escape
Surface receptors related to immune response, including CD70, B2M, HLA-B, and CD274, are recurrently mutated in DHL-BCL6 [19,20,21]. CD70 interacts with CD27 on T cells, mediating T-cell-dependent cytotoxicity and immune escape [169]. CD70 expression is associated with inferior outcomes, while its mutation and deletion are extensively associated with DHL-BCL6 [21, 22, 170]. Perturbed expression of MHC class I complex on cell membrane, resulting from the mutation of its components Beta-2-microglobulin (encoded by B2M) and HLA-B, leads to decreased expression of MHC I on the cell membrane and immune escape of B cells from T cells [19, 171, 172]. CD274 or PD-L1 is the ligand of the PD-1 immune checkpoint which is expressed on T cells. Ligand binding triggers a series of downstream cascades, eventually leading to the exhaustion of T cells in the form of decreased proliferation and promoted apoptosis. Recurrent gain and amplification of PD-L1 relate to DHL-BCL6, as well as in non-GCB DLBCL [20, 173]. Besides, a higher level of PD-L1 is often associated with poor prognosis [174].
Alterations relating to cell migration
It has been reported that the activation of CXCR4 by its ligand CXCL12 can induce signaling relating to cell migration, survival, and proliferation. In lymphoma, the activation of the CXCL12-CXCR4 axis disseminates B cells into lymph nodes [175] and bone marrow [176] and is correlated with poor survival [177]. It also mediates the immunosuppressive environment in B cell lymphoma through recruitment of regulatory T cells [178]. Besides, the consequence of active CXCR4 signaling in lymphoma also involves oncogenesis [179] and confers resistance to PI3Kδ inhibitor [180], which is disrupted by a missense mutation in DHL-BCL6 [19]. Point mutations at the C-terminal of CXCR4, therefore, may sustain the activation of the CXCL12-CXCR4 axis and contribute to cell dissemination and disease progression [19, 21, 181].
Other alterations
Although mutations relating to epigenetics had been found in DHL-BCL6, its scope and frequency are much less than that in DHL-BCL2 [19,20,21], Lesions affecting histone compartments, as well as epigenetic regulators including KMT2D and ARID1A, had been identified.
Inactivating lesions targeting TMEM30A were identified most frequently in multiple DHL-BCL6 cohorts [19,20,21,22]. The TMEM30A-knockout model suggested a correlation between TMEM30A loss and increased tumor-associated macrophages, up-regulated B cell signaling, as well as better clinical outcomes [182].
Targeted therapy
The discoveries of genetic lesions of two DHL subtypes identified their distinct biological engagements. With this knowledge, the therapeutic sensitivity or resistance of different DHL subtypes might be inferred, facilitating the exploration of corresponding targeted drugs. Below we summarized novel targeted agents and categorized them based on the heterogeneous genetic alterations in different DHL subtypes. For agents not yet investigated in DHL, we discussed their anti-tumor efficacy in DLBCL and other lymphomas for reference.
Targeted therapy for DHL-BCL2
The therapeutic vulnerability of DHL-BCL2 lies in its most perturbed proteins, such as MYC, BCL2, and epigenetic regulators. However, due to the difficulty in MYC inhibition and lack of efficacy in BCL2 inhibitors, significant attention was drawn to the application of epigenetic regulating inhibitors in DHL-BCL2, which exhibited satisfactory effect. Inhibition in signaling pathways, such as PI3K-mTOR and JAK-STAT, was also tested in this subtype. The clinical use of these targeted agents is summarized in Table 4.
Targeting MYC
As described previously, MYC translocations frequently occur in both types of DHL and are often mutated in DHL-BCL2, as well as other hematological malignancies, making it an attractive target for several malignancies. However, this target has been considered undruggable for a long time since the lack of well-defined ligand binding sites and large protein–protein interaction surfaces [211]. Nevertheless, there were still plenty of efforts focusing on discovering effective molecules that directly target MYC by disrupting the MYC/Max dimerization [212, 213], such as MYCi975 [214], 10074-G5 [215], and JY-3-094 [216], or by interfering the formation of c-MYC/Max/DNA complex, like MYRA-A [217] and NSC308848 [218]. Unfortunately, none of them has been promoted to clinic evaluation mainly due to the low efficacy and/or limited in vivo tolerability. Given such a situation, novel medicinal chemistry-based pharmacophore discovery and optimization, or induced-proximity approaches with catalytical mechanisms, such as PROTAC, could be employed to improve the efficacy and broaden the therapeutic window.
Instead of direct strategy, targeting MYC gene transcription or translation is an effective strategy. G-quadruplexes (also known as G4-DNA) are a tertiary structure formed by guanine-rich sequences in nucleic acids. CX-3543 (Quarfloxin) was the first G-quadruplex stabilizer entering clinical trials. It was recently found to down-regulate MYC by damaging the function of G-quadruplex binding nucleolar proteins and MYC transcription [213, 219]. Oligonucleotide and siRNA technology have also been applied to inhibit MYC gene expression [211]. OmoMYC, as a small protein containing the bHLH-zip domain of MYC, is another MYC family inhibitor [211]. OmoMYC inhibits the binding of c-MYC, n-MYC, or l-MYC to Max and prevents MYC-Max heterodimer from interacting with E-box [220, 221]. Recently, a group of natural products (Rocaglates) and their synthetic derivatives have been emerging as promising therapeutical agents for the treatment of MYC-associated lymphoma, especially those Double Hit lymphoma with concurrent MYC and BCL2 dysregulation [222, 223]. These molecules can efficiently inhibit MYC expression and tumor cell viability by stabilizing target mRNA–eIF4A interaction that directly prevents translation. In addition, PLK1 was reported as key factor on sustaining MYC activity through GSK3β-mediated MYC protein stability in DHL. Therefore, inhibition of PLK1 with small molecules, such as volasertib and Ro3280, can also downregulate the protein level of MYC [224].
Although no inhibitor of MYC is effective in DHL, given its specificity, these inhibitors are expected to be the potential therapy for DHL. In addition, MYC function can be affected by BET inhibitors via transcription and destabilization of MYC protein, which leads to the development of BET inhibitors, including JQ1, and will be further discussed in the section of epigenetic regulators [225,226,227].
Targeting apoptosis
The anti-apoptotic nature and up-regulated level of BCL2 in the majority of DHL-BCL2 lymphoma and other malignancies have raised interest in developing inhibitors targeting BCL2 and its family members. BCL2 inhibitors mimic BH3-only proteins which bind to the pro-survival members of the BCL2 family and thus trigger apoptosis.
Venetoclax, a BCL2 inhibitor with good efficiency in leukemia, was observed with limited activity in DLBCL patients with ORR only at 18% [185], and is now being tested as a regimen in several combination therapies including chemotherapy [228], radiotherapy [229] and targeted therapy, such as PI3K, BTK and SYK inhibitors (Fig. 3) [185, 188, 230]. In the CAVALLI phase, 2 studies of venetoclax plus R-CHOP as first-line treatment of DLBCL, the 2-year PFS and OS of venetoclax plus R-CHOP were higher than R-CHOP alone, observed in all DLBCL patients (2-year PFS: 80% vs 67%; 2-year OS: 86% vs 81%), as well as patients with BCL2 + DLBCLs and DELs. AEs were also increased in venetoclax plus the R-CHOP group but were considered manageable [187]. In a preclinical study, the combination of copanlisib (PI3K inhibitor) and venetoclax extended the median survival of the DLBCL mice model bearing BCL2 translocation and DHL-BCL2-like genetic dysregulations [188]. This synergistic effect may contribute to the inhibited PTEN and enhanced Akt pathway function during venetoclax administration [231], which partially explains the similar effect observed in the study of SYK/BTK inhibitor and venetoclax combination [189]. Besides the inhibition of the Akt pathway, inhibitors of EZH2 were also found to sensitize IGH/BCL2 translocated DLBCL cells [190]. In all, the use of BCL2 inhibitor alone may fail to achieve a satisfactory effect in lymphoma, but the combined use of BCL2 inhibitor and other DHL-BCL2 targeted drugs is promising, especially in BCL2 aberrant DLBCLs.

Structures of targeted agents in DHL-BCL2
Other anti-apoptosis proteins in the BCL2 family, like BCL-XL and MCL-1, have also been used in the treatment of other hematological malignancies. Among them, MCL-1 inhibitor has been tested in several clinical trials for the treatment of DLBCLs. Additionally, researches indicate that the upregulation of BCL-XL and MCL-1 may be an underlying mechanism in the BCL2 inhibitors resistance in hematological carcinoma [232, 233]. Also, dual inhibition of BCL2 and MCL-1 on DLBCL exhibits synergic effects both in vitro and in vivo [234, 235]. Therefore, MCL-1 is considered a potential target for DLBCL, and MCL-1 inhibition may be curative for patients with Venetoclax resistance.
AMG 176, an MCL-1 inhibitor that exhibited anti-tumor activity against DLBCL and other hematological malignances in vitro [236], has entered the Phase I study in combination with Venetoclax (NCT03797261). However, the trial is terminated due to cardiac toxicity. Other MCL-1 inhibitors, including PRT1419 (NCT04543305, NCT05107856), MIK665 (as single drug: NCT02992483, terminated, in combination with a BCL2 inhibitor, VOB560: NCT04702425) and AMG 397 (NCT03465540), had also been evaluated in Phase I studies for the treatment of hematological carcinoma including DLBCL. Most results are yet unavailable, while the trial on AMG 397 is currently on voluntary hold.
In addition to the BCL2 family, inhibitors of apoptosis (IAPs) are other sets of anti-apoptotic protein, which are mainly divided into three categories: IAP1 proteins (cIAP1, BIRC2), IAP2 proteins (cIAP2, BIRC3), and XIAP proteins (XIAP, BIRC4). These proteins are essential for activating downstream signaling of the NF-κB pathway, producing pro-survival transcriptional signals [237]. Mitochondrial protein SMAC is an endogenous inhibitor of IAPs which combines and antagonizes IAPs, subsequently activating caspase-3/7/9 and promoting apoptosis [238]. SMAC mimics are used as antagonists of IAPs, and some have entered the clinical trials as a single agent, combination therapies, and immunomodulatory agents [239, 240].
Survivin is a highly conserved member of the apoptotic protein (IAPs) inhibitor family which is overexpressed in up to 60% of DLBCL cases [241]. Therefore, some Survivin inhibitors were tested on DLBCL models. Survivin inhibitor sepantronium Bromide (YM155) entered the clinical study for NHL including DLBCL, yet was terminated in phase II because of insufficient efficacy (NCT00498914) [242]. Combination therapy of YM155 with rituximab therapy was also explored. The combined treatment of YM155 and rituximab enhances the antitumor activity of B-NHL xenografts. Compared with monotherapy, the combined treatment extends the survival time of severely combined immunocompromised mice with WSU-FSCCL and Jeko B-NHL diffuse tumors. Good antitumor activity and low toxic side effects are shown in phase II [243]. When combined with bendamustine and rituximab, stronger tumor lethality and lower toxic side effects were observed, which can decrease FLT-PET signal in lymph nodes and prolong overall survival in mice bearing disseminated DLBCL xenografts, and perhaps relapsed/refractory large B-cell lymphoma as well [244]. Another survivin inhibitor SM1044 is a new water-soluble artemisinin derivative of antimalarial drug, inducing the degradation of survivin through acetylation-dependent interaction with LC3-II to promote the apoptosis of the DLBCL cell line. It also activates the CaMKK2-AMPK-ULK1 axis, which leads to the initiation of autophagy [192].
Inhibitors of other IAP proteins are also under clinical evaluation. Xevinapant is an inhibitor of cIAP protein and is currently in clinical phase I for the treatment of lymphoma. Preclinical evaluations have shown that it has good pharmacokinetic properties in animal models [245] and the dose of less than 400 mg/day combined with daunorubicin and cytarabine can reduce drug resistance during the treatment of acute myeloid leukemia [246]. IAP inhibitor LCL-161 showed therapeutic effects in cell models of rituximab-resistant B-cell lymphoma, suggesting a potential to treat DLBCL patients previously treated with rituximab [191]. LCL161 is currently in clinical trials for a number of hematological malignancies, including B-cell lymphoma [247].
There are also apoptosis inducers, such as PCLX-001. It is an N-myristoyltransferase (NMT) inhibitor, which is currently in Phase I and is mainly used in the treatment of NHL. Treatment of PCLX-001 impacts the global myristoylation of lymphoma cell proteins and inhibits early BCR signaling, which is critical for survival [248]. In addition, PCLX-001 has a high selectivity and significantly promotes the production of numerous non-myristoylated BCR effectors, including c-MYC, NF-κB, and p-ERK. It can be used as a precision drug, with researchers predicting minimal side effects during treatment [249].
Targeting epigenetic regulation
-
(1)
Targeted histone methylation therapy
KMT2D mutation leads to a significant decrease in H3K4 monomethylation in tumor cells, an increase of genomic DNA damage and mutation load, as well as transcriptional instability [250]. As mentioned above, lysine-specific histone methyltransferase KMT2D is one of the most frequently altered genes in DHL-BCL2 [23]. Although few drugs have been reported against KMT2D, novel histone demethylase inhibitors might solve the loss of methylation typically observed in KMT2D-inactivated tumors [251]. In addition, the KMT2D mutation in DHL causes DNA damage and increased transcriptional stress, resulting in the accumulation of a higher mutation load and the generation of more tumor neoantigens [252]. These changes lead to a higher level of immune cell infiltration in the tumor microenvironment, making it more sensitive to immune therapy.
Mutations in EZH2 are also prevalent in DHL patients, suggesting that EZH2 inhibitors may be a potential targeted drug for the treatment of DHL. Tazemetostat is an oral selective potent EZH2 inhibitor. It was firstly approved for clinical trials in 2013. In the phase, I clinical trial, tazemetostat therapy has demonstrated preliminary efficacy in 38% of patients with relapsed/refractory B-cell lymphoma, 14% with CRR, and median progression-free survival (mPFS) of 12.4 months. Grade 3 and above adverse reactions were mainly thrombocytopenia (14%) and neutropenia (14%) [193]. The interim results of phase II clinical trial showed a significant increase in ORR in DLBCL and follicular lymphoma patients with EZH2 mutant compared with wild-type (40% vs. 18%;63% vs. 28%) [194, 195]. In addition, the Phase II clinical trial of tazemetostat combined with R-CHOP (NCT02889523) and PD-L1 antibody atezolizumab (NCT02220842) in the treatment of DLBCL are in progress.
Valemetostat, as a dual inhibitor of EZH 1/2, has shown more potent antitumor efficacy than EZH2 inhibitors [196]. In phase, I a clinical trial with 15 patients with NHL patients treated by Valemetostat showed the ORR of 53% and CBR of 86%. The ORR of patients with peripheral T-cell lymphoma reached 80%, indicating the reliable efficacy of valemetostat in the treatment of NHL. CPI-1205 is a novel oral small molecule EZH2 inhibitor, which achieves demethylation by competitively inhibiting the binding of SAM to the EZH2 catalytic group. Harb et al.[197] found that, among the 28 BCL patients treated with 6 cycles of CPI-1205, one case achieved CR and five cases achieved stable disease (SD). In addition, next-generation EZH2 inhibitors, including CPI169, CPI360, MAK683, PF-06821497, and SHR2554, are currently recruiting lymphoma patients for clinical trials (NCT02900651, NCT03460977, NCT03603951).
-
(2)
Targeted histone acetylation therapy
Loss-of-function mutations in genes encoding proteins with a defined role in histone acetylation (e.g., CREBBP or EP300) are commonly observed in DHL-BCL2 patients (more than 50%). Somatic mutations in CREBBP lead to impaired p53 activation and also promote the oncogenic effects of BCL6 [253]. Loss-of-function mutations in CREBBP also lead to the silencing of some genes involved in MHC-II-mediated antigen-presentation [42, 96], suggesting that it may promote the immune escape of tumor cells. Taken together, these findings indicate that the regulation of histone acetylation is a potential vulnerability of DHL targeted therapy.
Vorinostat is an oral histone deacetylase inhibitor (HDACi), which works by inducing histone acetylation to activate the expression of cell cycle factors and tumor suppressor genes. In 2014, researchers began exploring the use of Vorinostat in B-cell lymphoma (Fig. 3). In phase II clinical trial involving 39 patients with relapsed/refractory follicular lymphoma [198], the ORR was 49% and mPFS was 20 months. The major adverse reactions were thrombocytopenia and neutropenia. These results suggest that Vorinostat can provide sustained clinical benefit for B-cell lymphoma patients with controllable adverse reactions.
Panobinostat was the first HDAC inhibitor for the treatment of multiple myeloma, and in recent years it has also been explored for B-cell lymphoma. The efficacy of panobinostat in combination with rituximab was reported in over 40 patients with relapsed/refractory DLBCL, with an ORR of 28%, CRR of 18%, and MPFS of 14.5 months [199].
Chidamide, a selective HDAC inhibitor, has been shown to have a synergistic effect with rituximab in the treatment of DLBCL in vitro and in vivo [200]. The loss of CD20 on the cell surface is the main difficulty in the treatment of relapsed/refractory DLBCL with rituximab. Chidamide significantly enhanced the expression of CD20 on the surface of DLBCL cells, thus synergizing with rituximab to exert anti-tumor effects [254]. In DLBCL xenograft mice, chidamide and rituximab synergically induce cell death and inhibited tumor growth [200].
Batlevi et al. completed a phase II clinical trial to evaluate the effect of mocetinostat in FL and DLBCL, and the ORR and clinical benefit rate (CBR) in DLBCL patients were 18.9% and 54.1%, respectively [202]. Romidepsin, an approved macrocyclic pan-HDAC inhibitor, was shown to exert synergistic antitumor effects with GSK126, an EZH2 inhibitor, in the SU-DHL-10 xenograft model [201]. Notably, the novel dual HDAC and PI3K inhibitor CUDC-907 demonstrated excellent activity (ORR 55%) and tolerability (43% of grade ≥ 3 adverse events) in a phase I trial in patients with DLBCL, FL, and HL [203]. CUDC-907 is currently in phase II clinical trial in patients with DLBCL (NCT02674750).
-
(3)
Targeted bromodomain and extra-terminal domain family therapy
The bromodomain and extra-terminal domain (BET) family consists of four proteins: BRD2, BRD3, BRD4, and BRDT, while its N-terminal contains two BRD modules involved in acetyllysine recognition. BRD4 is presented in several transcriptional complexes, including the cofactor regulator and p-TEFB extension factor [255]. The C-terminal domain also regulates the interactions between BRD4 and many well-known transcriptional regulators, notably p-TEFB, MYC, NF-κB, and p53 [255,256,257]. Although BET mutations or translocations are rare, BET may be overexpressed [258]. Therefore, BET inhibition has been effective in preclinical studies of multiple cancer types, especially for many hematopoietic system cancers that rely on constant BRD4 activity to express MYC [259,260,261].
OTX015 is the first BET inhibitor (BETi) to reach clinical trials. Amorim et al. [204] conducted a phase I clinical trial in 17 DLBCL patients treated with OTX015, which showed a CBR of 47%, suggesting that OTX015 may have potential antitumor activity against lymphoma. The most common grade 3/4 ADRs included thrombocytopenia (58%), anemia (27%), and neutropenia (22%). Further studies showed that OTX015 can inhibit TLR/NF-κB and JAK/STAT signaling pathways by down-regulating MYC expression and play a targeted therapeutic role [259]. In DHL/THL model, OTX015 (Birabresib) combined with BCL2 antagonist (such as Venetoclax) synergically inhibited the malignant proliferation of DHL/THL cells [262], supporting the combining use of BETi and BCL2 inhibitors against MYC-driven lymphoma. A recent study showed that nonbenzodiazepine BETi, PLX-2853, synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL2 expression [206]. Similarly, in DHL patients, resistance to venetoclax was overcome by the use of the BET inhibitor CPI-203, possibly due to the down-regulation of BCL2-like protein (BFL1) [205]. In phase I clinical trial with 44 patients with relapsed/refractory lymphoma (DLBCL, FL, and HL) enrolled, 12 patients achieved a moderate response, and 2 patients with DLBCL achieved CR after 5 and 6 cycles of CPI-0610 (a highly selective BETi) treatment, respectively [207]. The treatment of JQ1 (a classic inhibitor of BRD4) extended survival in a mouse xenograft model of MYC-driven lymphoma [208], suggesting that BETi has broad therapeutic potential in DHL that is highly associated with MYC.
-
(4)
Targeted DNA methylation therapy
DNA methylation is one of the most widely studied epigenetic modifications [251]. DNA methylation refers to the formation of 5-methylcytosine by adding methyl at the 5' end of cytosine nucleotide of C-phosphodiester-G (CpG) under the catalysis of DNA methyltransferase (DNMT). A conformational change in the methylated CpG sequence prevents transcription factors from binding, thus silencing the expression of methylated genes [263]. In normal cells, CpG islands are usually in a non-methylated or hypomethylated state, and hypermethylation will inhibit the expression of genes. Some studies suggested that high expression of DNMT3b may be associated with a poor prognosis of DLBCL. The FDA-approved DNMT inhibitors, 5-azacitidine, and decitabine, effectively demethylate DNA and induce the expression of related tumor suppressor genes [251]. In phase I clinical trial [209] involving 33 patients with DLBCL and high-grade follicular lymphoma, 5-azacitidine combined with R-CHOP regimen showed a higher objective response rate (ORR), complete response rate (CRR), and lower incidence of ADRs than R-CHOP.
Targeting oncogenic pathways
-
(1)
Targeting JAK-STAT Signaling
The JAK/STAT pathway is often abnormally activated in patients with lymphoma, especially in DHL-BCL2, making it a promising target [119]. Disrupted or dysregulated JAK-STAT functionality could result in an altered microenvironment to allow for immune evasion of tumor cells [264]. STAT3 expression and activation are significantly higher in ABC DLBCL cell lines and these cell lines demonstrate higher NF-κB activity than those with low STAT3 activity [265]. Knockdown of STAT3 in mouse xenografts models suppressed the growth of ABC DLBCL tumors, validating STAT3 as a therapeutic target in this subtype of DLBCL. Due to the difficulties in developing inhibitory molecules for STAT3, plenty of effort was put into the downregulation of STAT3.
SD-36, a novel selective STAT3 degrader, can significantly and selectively degrade STAT3 in different cell lines [266]. According to preclinical data, SD-36 had a significant inhibitory effect on the growth of five DLBCL cell lines. What’s more, SD-36 effectively inhibited tumor growth at 100 mg/kg in the SU-DHL-1 xenograft model and had a complete and long-lasting PD effect in inducing STAT3 degradation in vivo [267].
Danvatirsen (AZD9150) is an antisense oligonucleotide designed to reduce the production of STAT3. Depletion of STAT3 with AXD9150 in KARPAS-299 and SUP-M2 cells and xenograft models rapidly induced apoptosis and reduced the expression of STAT3 pro-survival targets, including MCL-1, c-MYC, BCL6, CYCLIN D1, BIRC5 (SURVIVIN), and IL2Rα [268]. These findings led to a phase I dose-escalation study of AZD9150 in patients with advanced cancer, including 12 lymphoma patients, 7 of them with DLBCL. Anti-tumor activity was shown in these heavily pretreated patients [269, 270]. It is currently used as the treatment of solid tumors and lymphomas (including DLBCL) that have relapsed or are ineffective against multiple chemotherapy regimens.
The targeting inhibition of STAT3 also can be achieved by inhibition of its upstream kinase including JAK1 and JAK2. Ruxolitinib, a non-selective JAK1 and 2 inhibitor that blocks phosphorylation of STAT1 and STAT3, has been approved for primary myelofibrosis [271]. Phase I/II studies evaluating this agent either as monotherapy or in combination with bortezomib in relapsed/refractory DLBCL are ongoing [272]. Pacritinib (formerly SB1518) is an oral small molecule that selectively and potently inhibits JAK2, showing promising in vitro activity in JAK2-dependent DLBCL cell lines independent of JAK2 mutational status [273]. In a phase I study in 34 patients with relapsed or refractory lymphomas, including DLBCL, pacritinib demonstrated a favorable toxicity profile [274].
-
(2)
Targeting PI3K-AKT-mTOR signaling pathway
As previously mentioned, the PI3K/AKT/mTOR pathway was proved to play an essential role in the development and progression of many hematological malignancies, including DLBCL [275]. Inhibitors of this pathway are mainly composed of PI3K, Akt, and mTOR inhibitors.
There are many PI3K inhibitors currently in the market or clinical trials stage for the treatment of hematological tumors. Idelalisib has demonstrated antitumor activity in indolent B-NHL (iB-NHL) [276] but it is less effective in the treatment of DLBCL as a single agent regimen (Fig. 3). When combined with anti-CD20 monoclonal antibodies or BTK inhibitors, anti-tumor efficacy could be. Through complementary mechanisms, combined use with BTK inhibitors can enhance the sensitivity of DLBCL cells, overcome drug resistance, and enhance tumor lethality [277]. So far, some combination regimens have reached clinical trials [278].
Copanlisib was approved for the treatment of follicular center lymphoma [210]. Preclinical experiments have shown that Copanlisib exhibits cell-type-specific cytotoxicity at nanomolar concentrations against DLBCL cell lines. A phase II clinical trial showed that the ORR of all types of DLBCL is 25%, and the ORR for ABC-DLBCL is 38%[279].
Enzastaurin is currently in phase III. It can delay the deterioration of DLBCL despite its poor treatment effect, and have low toxicity and side effects [280]. The first result about enzastaurin for relapsed/refractory DLBCL patients in Phase II was published in 2007 [281]. it showed that none of the disease-related parameters were correlated with response to enzastaurin. Despite the low response rate, the long-term response and the good safety in Phase II warrant further evaluation of enzastaurin together with R-CHOP therapy. Besides, a synergistic effect of enzastaurin with bortezomib and gemcitabine was found in a xenograft model of GCB-DLBCL [282].
The AKT inhibitor MK-2206 is in phase II and is mainly used for the treatment of non-small cell lung cancer, colorectal cancer, while also exhibiting a therapeutic effect on DLBCL. The combination of MK-2206 and mTOR inhibitor nelfinavir can overcome the drug resistance of mTOR inhibitors in DLBCL, reduce the viability of DLBCL cells and halt the cell cycle, promoting cell apoptosis [283]. MK-2206 monotherapy kills lymphoma cells by reducing the level of p-AKT, inhibiting the downstream targets of AKT signaling, and inducing Rictor and phosphatidylinositol 3-kinase expression. The sensitivity of MK-2206 is related to the activation state of AKT in DLBCL cells. Although monomer therapy is also effective, AKT inhibitors are combined with other targeted drugs for better clinical efficacy [284].
Everolimus is an mTOR inhibitor that was approved in 2003 to treat breast cancer and kidney disease. Its research for DLBCL is in phase III studies which showed that oral Everolimus can induce BLDCL cell cycle arrest. According to the result of the Phase I study, the best dose of Everolimus is 10 mg/d [285]. Results from the phase II study showed the ORR of Everolimus DLBCL is 30%. Meanwhile, Everolimus combined with rituximab hassled to stronger cell lethality. The objective response rate was 38%, the complete response rate was 13%, and there was no increase in toxicity [286].
Temsirolimus is also an mTOR inhibitor approved for the treatment of NHL in 2007 [287]. It is in phase II for DLBCL. It can inhibit the growth of GCB-DLBCL and ABC-BLDCL cells (GCB = 30–66%, ABC = 45–57%). Combination therapy enhances its therapeutic effect, and combined with different drugs can be used to treat different types of DLBCL. For example, its combination with idelalisib enhanced the lethality of GCB-DLBCL and ABC-BLDCL cells, while combining with ibrutinib or bortezomib, a better therapeutic effect on ABC-DLBCL or GCB-DLBCL was observed, respectively [288].
Other therapies
Since DHL-BCL2 is accompanied by a high frequency of TNFRSF14 and ARID1A mutation, certain unique vulnerabilities were found in these subsets of tumors.
As mentioned above, TNFRSF14 mutation brings about reduced HVEM levels, resulting in BTLA-mediated proliferation and CD40 signaling. Stimulation of HVEM by human LIGHT, its ligand, renders NHL more immunogenetic and sensitive to Fas-induced apoptosis, without inducing proliferation [135]. Also, restoration of HEVM ectodomain by soluble HVEM protein or HVEM-producing and CD19-targeted CAR-T cells were tumor-suppressive in MYC + /BCL2 + DLBCL cell lines and BCL2 overexpressing lymphoma xenograft model, respectively [134]. These results indicated the breakthrough point for the intervention of immunotherapy in DHL-BCL2.
ARID1A and its homolog ARID1B have a synthetic lethal relationship. In ARID1A mutated cancers, the co-mutation of ARID1B destabilizes the SWI/SNF complex and damages cell proliferation, suggesting a potential target for malignancies harboring ARID1A mutations [107]. Also, researchers found that tumors with ARID1A deficiency were sensitive to immune checkpoint blockade (targeting PD-1–PD-L1), bringing a novel perspective for treating DHL-BCL2 [289].
Targeted therapy for DHL-MYC/BCL6
Compared to DHL-BCL2, targeted agents for DHL-BCL6 were less subtype-specific due to insufficient understanding of its biology and diverse cell-of-origin. Here, we selected drugs targeting genetically disturbed proteins or pathways found in DHL-BCL6, hopefully providing insight into the treatment of DHL-BCL6. Clinical tests of these inhibitors are summarized in Table 5.
Targeting oncogenic pathways
-
(1)
Targeting Notch pathway
Despite the frequent activation of the Notch pathway in DHL-BCL6, drugs targeting this cascade were rarely tested in DHLs, even DLBCLs. Inhibition of the γ-secretase is a common strategy in targeting the Notch pathway, which is mainly used in solid tumors [308], with a few were tested in hematology malignancies in pre-clinical studies. Z-IL-CHO (GSI-XII) can reduce the mRNA content of HES1 in TMD8 cell lines established from DLBCL patients, proving its potential therapeutic effect in DLBCL [291]. A combination of Z-IL-CHO and bortezomib can enhance the cytotoxicity of bortezomib in multiple myeloma. This process is mainly caused by the synergistic inhibition of chymotrypsin-like proteasome by Z-IL-CHO and bortezomib, rather than the inhibition of the Notch pathway, which provides a choice for the combined treatment of multiple myeloma [309]. Z-LLNle-CHO (GSI-I), crenigacestat, and MK-0752 all exhibited inhibitory activity against γ-secretase and were also tested in different hematology malignancies, yet no studies in DLBCL were launched [290, 310,311,312].
Another molecule, CB-103, acts as a disruptor of NICD protein–protein interaction and inhibitor of the active forms of Notch receptors. It targets various cancers with Notch overexpression resistant to Notch inhibition by GSIs and monoclonal antibodies while eliminating the gastrointestinal toxicity and expanding the therapeutic window of the first—and second-generation Notch inhibitors [313, 314]. Likely, evaluation of these inhibitors may yield unexpected results in the DHL-BCL6 subtype.
-
(2)
Targeting BCR Signaling
The BCR complex and associated protein tyrosine kinases are essential for normal B-cell function and antibody production. BCR cross-linking activates 3 main pathways: BTK, PLC-γ2, and PI3K [315]. Constitutively activated BCR signaling is linked to the initiation and maintenance of B-cell malignancies, especially in the ABC-DLBCL. Drugs targeting these proteins either as adjuncts to R-CHOP in the frontline setting or as monotherapy/combination therapy in the relapsed/refractory setting are under evaluation [316].
Ibrutinib, a first-in-class small molecule that selectively covalently binds to the cysteine residue (Cys-481) of the active site of BTK, irreversibly inhibits its activity (Fig. 4) [317]. For patients with ABC DLBCL, the effective rate of single ibrutinib in relapsed/refractory patients can be as high as 37%, but for patients with GCB DLBCL, the effective rate is only 15%. In terms of combination therapy, the combination of ibrutinib and R-CHOP can significantly improve the efficacy of R/R DLBCL, and the effectiveness of the GCB subtype is still inferior to that of the ABC subtype [318].

Structures of targeted agents in DHL-BCL6
Fostamatinib Disodium (R788) is a prodrug of the active metabolite R406, which is an oral SYK inhibitor with IC50 of 41 nM. It strongly inhibits SYK but does not inhibit Lyn, while the effect on Flt3 is 5 times weaker [319]. However, the results of two pivotal phases III clinical trials OSKIRA-2 and OSKIRA-3 showed that compared with the placebo group, fostamatinib did not show significant clinical benefit in ABC DLBCL [320]. At the same time, phase II clinical studies of hematological malignancies did not show positive results.
Cerdulatinib (PRT-062070) is an orally active, multi-targeted tyrosine kinase inhibitor with IC50 of 12 nM/6 nM/8 nM/0.5 nM and 32 nM for JAK1/JAK2/JAK3/TYK2 and SYK, respectively [321]. It has broad anti-tumor activity in both ABC and GC cell lines of diffuse large B cells at least in part by inhibiting SYK and JAK pathways [322].
Enzastaurin, an oral PKC-β agent, showed limited cytotoxicity in clinical phase I trials and achieved encouraging clinical effects in the phase II trial of DLBCL as induction therapy [323]. However, the PRELUDE randomized phase III trial of R-CHOP ± enzastaurin did not improve PFS as a primary endpoint.
-
(3)
Targeting NF-κB Signaling
The excessive activation of NF-κB dependent gene expression is an important characteristic of the more aggressive ABC DLBCL subtype [324]. The formation of the CARD11/ BCL10/MALTI complex activates IKK, causing phosphorylation and breakdown of IκB, and ultimately leading to the activation of NF-κB [325]. In addition, the adaptor protein MYD88 acts as a mediator, which can accept Toll-like receptor signals to activate NF-κB, and MYD88 is highly mutated in ABC DLBCL [326]. The NF-κB pathway as a therapeutic target was first demonstrated by the IκB kinase (IKK) complex inhibitors in selective inhibition of ABC DLBCL cell lines. Moreover, proteasome inhibition has been shown particular efficacy in patients with ABC DLBCL.
KT-474, a novel heterobifunctional IRAK4 degrader that targets both IRAK4 and IMiD biology leads to potent cell kill in ABC DLBCL lines harboring MYD88 L265P through down modulating survival signals, including NF-κB and autocrine IL-6/IL-10 engagement of the JAK-STAT3 pathway. KT-474 induced superior cellular toxicity compared to IRAK4 kinase inhibition as determined by lower IC50 and induction of apoptosis. MYD88-mutated ABC-DLBCL cell lines are more sensitive to KT-474 exposure as compared to wild-type. A combination of KT-474 in conjunction with ibrutinib, venetoclax and umbralisib shows a synergistic effect in the OCI-LY10 cell line.
Targeting ubiquitin–proteasome system
As many pathways are interfered with by genetic alteration, remodeling intercellular signaling via the inhibition of ubiquitin–proteasome-dependent degradation has been considered as a possible strategy, since several important proteins, including IκB, the inhibitory switch of NF-κB, p53, a tumor suppressor, and other ubiquitin-conjugating enzymes found to be mutated in the DHL-BCL6 are associated with ubiquitin–proteasome system. Although no cogent evidence supports the specialty of ubiquitin–proteasome regulator in this subtype, inhibitors regulating the NF-κB pathway exhibited cytotoxicity selectively towards ABC-DLBCL [327], which is more prominent in the DHL-BCL6.
Lenalidomide targets the E3 ubiquitin ligase component cereblon that modulates the interferon regulatory factor 4 and down-regulates the NF-κB pathway in ABC-DLBCL (Fig. 4) [328]. In clinical studies, lenalidomide exhibited promising efficacy in DLBCLs, especially in non-GCB subtype, with significantly prolonged PFS and OS, either as a single agent [293] or in combination with chemotherapies (R-CHOP and similar therapies) [293, 294] or other regiments(rituximab or ibrutinib) [295, 296], as well as being maintenance strategy [297]. For DHL, the response to lenalidomide plus DA-EPOCH-R is less satisfied compared to DELs in terms of disease remission, PFS, and CNS involvement [292]. The difference may be attributed to the small DHL patient group or the higher correlation between DEL and non-GCB lymphoma, which respond better to lenalidomide-based therapies [292, 329]. Durable remission was observed in a DHL-BCL6 case when treated with lenalidomide following salvage chemotherapy [330]. Hence, more and larger clinical trials may be beneficial.
Bortezomib (PS-341) is the first generation of protease inhibitor with Ki of 0.6 nM, which shows good selectivity for tumor cells (Fig. 4) [331]. Bortezomib can prevent the degradation of IkB, keeping NF‑κB in its inactive form [332]. In the R/R DLBCL population, bortezomib alone did not bring great efficacy, but when combined with chemotherapy (DAEPOCH-R), the combination achieved better efficacy (response rate to 83% vs 13%; P < 0.001) and overall survival (10.8 vs 3.4 months; P = 0.003) in ABC vs GC DLBCL [333]. More recently, results of the randomized phase II PYRAMID study revealed no benefit to the use of bortezomib as an adjunct to R-CHOP in the frontline setting for non-GC DLBCL patients [334]. In a word, its clinical application is limited due to short drug effect time and severe dose-dependent neurological side effects [335].
Compared with the first generation of proteasome inhibitors, the second generation has oral biological activity, improved pharmacokinetics, and reduced neuropathy. Ixazomib is the first proteasome inhibitor that can be administered orally [336]. At present, preclinical experiments have proved that ixazomib has not only anti-tumor activity in DLBCL cell lines, but also inhibitory activity in DLBCL cell lines that have translocation of MYC and BCL2, and its IC50 value is 40-200 nm. A DHL xenograft mouse model showed anti-tumor activity at a clinically achievable drug concentration. At a dose of 7 mg/kg, the maximum tumor inhibition rate (TGImax) is 64%. Therefore, ixazomib is a potential drug for the treatment of DHL-BCL2, and its combination with CHK2 inhibitors provides a potential treatment option for drug-resistant DLBCL and DHL-BCL2 [337].
B-AP15 is a specific inhibitor of the deubiquitinating enzymes, and subsequently, inhibits the migration of DLBCL tumor cells and induces apoptosis. Preclinical experiments have shown that B-AP15 promotes apoptosis by inhibiting the activity of proteasome DUB (USP14 and UCHL5) in GCB and ABC-DLBCL cell lines [338], and inhibits WNT/β-catenin and TGFβ/Smad pathways to prevent DLBCL tumor cells migration. B-AP15 directly inhibits c-MYC protein, rather than inhibiting BCL2, but it can cause a decrease in BCL2 mRNA levels. Therefore, b-AP15 is not only a potential treatment for DLBCL but also DHL-BCL2 [298].
Targeting p53 signaling
Although 50% of tumors carry TP53 mutations, the development of p53-targeted therapies is challenging [339]. Some TP53 mutants do not encode a complete protein, and p53 itself, as a transcriptional factor, does not have an "active pocket" for small molecules to bind [340]. Current strategies targeting p53 mainly include targeting mutant p53 [341], synthetic lethality, and targeting MDM2-dependent p53 degradation [342]. However, these agents are only tested in leukemia, while studies in lymphoma remain unperformed [343,344,345].
XPO1 is a nuclear export receptor, which is involved in the nuclear-cytoplasmic transport of proteins bearing nuclear export signal (NES), and multiple RNA species [346]. XPO1 is usually overexpressed and/or mutated in the DLBCL cell lines, which causes poor nuclear retention of several proteins including several tumor suppressors like p53, BRCA1/2, and p27 [346, 347]. In addition, clinical research shows that high XPO1 expression has a significant adverse prognostic impact in DHL patients, especially in those with BCL2 overexpression [348]. Inhibition of XPO1 can down-regulate the expression of MYC in a variety of DHL cell lines [349].
Selinexor (KPT-330) is a first-in-class oral, biologically effective, and selective XOP1 inhibitor, which has been approved by the FDA for the treatment of heavily pre-treated R/R DLBCL and R/R multiple myeloma (Fig. 4) [299]. Selinexor can effectively down-regulate MYC protein expression and thus reprogram the gene expression of MYC downstream. Combined use of XPO1 and BCL2 inhibitors can synergistically induce the apoptosis of DHL tumor cells in vitro, and most importantly, block tumor progression and spread in vivo [349]. The effect of Selinexor has been confirmed in high-risk molecular DLBCL (both GCB and non-GCB subtypes), de novo, and transformed DLBCL and double-hit lymphomas [350].
Targeting cell cycle
CDKN2A is a tumor suppressor gene located on chromosome 9, encoding p16CDKN2A protein, which can inhibit the activity of CDK and regulate the G1 cell cycle [351]. Inactivation of CDKN2A may result in uncontrolled cell growth and proliferation. CDKN2A is frequently deleted, mutated, or hypermethylated in many tumors, including T-cell lymphoma, and is believed to be the second most commonly inactivated gene in cancer after p53 [351]. It is generally thought that CDKN2A gene loss-of-function mutations may lead to abnormal activation of CDK2/4/6, so CDK inhibitors may be a potential treatment for patients with CDKN2A mutations [352]. In addition, the same highly frequent alteration of the CCND3 gene was observed in MYC-BCL6 DHL patients, and the high expression of CCND3 can lead to downstream CDK4/6 activation, resulting in abnormal cell proliferation [353]. Therefore, CDK inhibitors may serve as potential therapeutic agents against MYC-BCL6 DHL.
Both palbociclib and abemaciclib are orally selective small-molecule CDK4/6 inhibitors that inhibit the progression of the cell cycle from G1 to S phase and block DNA synthesis [354]. A recent study found that both palbociclib and abemaciclib play a role in high-grade B-cell lymphoma with abnormal expression of CCND3 [355]. Pan-CDK inhibitor dinaciclib is proved to play synergistic induction of apoptosis in high-risk DLBCL when in combination with BCL2 inhibitor venetoclax [234]. In addition, CDK inhibitors such as flavopiridol and seliciclib were also found to be effective in BCL [356, 357], suggesting that targeted inhibition of CDK activity may indeed be a feasible treatment for DHL-BCL6. It is worth mentioning that a series of PROTACs studies targeting CDK has been progressively reported recently [358, 359], which may provide more treatment options for DHL.
Targeting CXCR4
As mentioned in the section of “3.1.6 Alterations relating cell migration”, the activation of the CXCR4 axis leads to cell dissemination to extranodal location. Two major strategies to target CXCR4 are direct inhibition and combination agents, aiming to induce cell death in CXCR4 + tumor cells.
Direct CXCR4 antagonists, including AMD3100(Plerixafor), AMD070, and WKI, demonstrated a pro-apoptotic effect in CXCR4 + DLBCL cells via modulation of JNK, ERK, NF-κB/ BCR, and BCL2 targets [176]. PF-06747143, a CXCR4 inhibiting IgG1 antibody, inhibits the signaling pathway and cell migration of lymphoma cells, reducing the bone marrow infiltration of Burkitt’s lymphoma in vivo [301]. In light of this, the safety and effect of CXCR4 targeted agents have been tested in human subjects in clinical trials. However, instead of cytotoxic agents, CXCR4 targeted drugs were mostly used as conditioning regimens. AMD3100, an extensively studied CXCR4 antagonist, was approved to mobilize hematopoietic stem cells for ASCT in NHL patients combined with G-CSF.
For conjugated agents, the liganded drug achieved higher tumor delivery and uptake than the free drug [360, 361]. T22-AUR is an auristatin E nanoconjugate that selectively targets CXCR4-overexpressing DLBCL cells. The cytotoxic effect of T22-AUR to CXCR4 + cells is significantly higher than free auristatin E since the CXCR4-dependent internalization of the conjugates facilitates the binding of auristatin E to tubulin, where auristatin E induces cell death. The use of T22-AUR in DLBCL xenograft reduced the dissemination of tumor cells into bone marrow and the central nervous system [302].
One clinical study was initiated recently to investigate the safety of CXCR4 modified CD19 CAR-T cells in refractory B-cell NHL (NCT04684472). Another preclinical study recruited six patients with heavily pretreated DLBCL. They were administered with CXCR4-directed radioligand therapy in combination with conditioning chemotherapy and allogeneic stem cell transplantation. The regimen was well tolerated, resulting in CR of all-CXCR4 + lymphoma but the appearance of CXCR- lesions, supporting the use of CXCR4-directed radioligand therapy as a conditioning regimen [303].
In all, CXCR4 antagonists can reduce tumor burden and the dissemination of lymphoma to organs. However, the use of these regimens is limited to CXCR4 + lymphomas like CXCR4 + disseminated refractory or relapsed DLBCL patients. For a broader spectrum of patients, the combination of CXCR4 antagonist and other therapy (chemotherapy, targeted therapy, or radiotherapy) is needed.
Targeting immune escape
Recurrent mutations associated with immune escape genes in the DHL-BCL6 may suggest that this subtype is a better candidate for immunotherapy than DHL-BCL2. A number of ongoing trials are investigating the monotherapy and combination therapy of anti-PD-1 antibodies in DLBCL. The results of several clinical trials have been published.
In a phase Ib dose-escalation study of nivolumab, the ORR was 36% in R/R DLBCL with most adverse events (80%) under grade 3 [304]. However, in another phase II study, the ORR was lower, 10% and 3% in relapsed patients after or were ineligible for autologous hematopoietic cell transplantation, respectively. It is worth noting that the membranous PD-L1 level of DLBCLs in this study is only 9%, which may contribute to the differences in the two studies [305]. A study of 105 DLBCL patients showed that when the PD-L1 gene is altered, increased PD-L1 protein downregulated HLA expression, activated NF-κB, and higher response to PD-1 blockade (anti-PD-1 antibody pembrolizumab) were observed (NCT01953692) [306]. Another study observed a similar result when treatment naïve DLBCL patients were treated with pembrolizumab in combination with R-CHOP. The toxicity was comparable to R-CHOP alone. The ORR was 90% and the CR rate was 77%. Similar to the study above, higher PD-L1 expression was associated with non-GCB subtype and improved PFS and survival [307]. Based on the study above, together with results in primary CNS lymphoma and primary testicular lymphoma [362], B cell lymphomas with PD-L1 amplification, gain or aberration are likely to have a promising response to PD-1 blockade therapy.
Combined regimens for DHL
Although the targeted agents showed good efficacy or potential for the treatment of DHL with monotherapy, combined regimens had been evaluated on DHL patients, or specific clinical trials on DHL patients are ongoing, such as NCT04479267. We summarized therapeutic targets that had been under clinical evaluation with combined regimens, including the status of clinical trials, clinical response rate, adverse events and so on in Table 6. All these clinical trials covered DHL in the inclusion criteria or reported DHL cases in the relevant publications.
Conclusions and future perspectives
Due to lack of effective treatment and late diagnosis, DHL patients are subjected to intensive chemotherapy and poor outcome. To avoid the late diagnosis, the study result supports the performance of the routine FISH test in all DLBCL patients, which considerably improves the outcome of DHL (ST vs RT: 2-year RFS, 38% v 70% and 2-year overall survival, 38% v 74%). At the same time, routine testing identified a cohort of low-risk DHL patients and the lack of benefit from intensive immunochemotherapy [28]. Independent research further stratifies DHL into high-risk and low-risk (DHIT + SIG) subgroups, suggesting tremendous heterogenicity lies in the DHL cohort [26]. However, despite the satisfiable efficacy of standard R-CHOP in low-risk DHL, the poor outcome of high-risk DHL remains a challenge, while dose-adjusted intensive regimens become appropriate induction options for most patients with double-hit lymphoma in clinical practice.
To facilitate risk stratification and treatment selection, approaches have been applied to classify DLBCL into different subgroups. Based on cell-of-origin, DLBCLs can be categorized into GCB and non-GCB types, which further include ABC DLBCL and unclassifiable DLBCL. These subgroups differ in biological features, clinical outcomes, and treatment response. For instance, GCB-DLBCL tends to present genetic alterations in the PI3K pathway, which are related to “tonic” BCR signaling, while ABC-DLBCL was observed to harbor mutations characterized as chronic active BCR signaling [143, 371]. Hence, this fundamental divergence contributes to the different responses of DLBCL subtypes to NF-κB pathway inhibition [327]. Similarly, high sensitivity to EZH2 inhibitors was also observed in DLBCLs with hyper-H3K27me3 status, which is closely related to the GCB cohort [372].
Considering heterogenicity in the DLBCL group, we aim to link distinct genetic events to different DLBCL subgroups, i.e. DHL-BCL2 and DHL-BCL6. After extensive literature, we summarized unique genetic events encompassed in the DHL subtypes. We further collected potential agents targeting these abnormalities, hopefully providing insights into the treatment of DHL. However, this review has certain limitations. Owing to the dominant proportion in DHL and close coincidence with GCB DLBCL, mutations and treatment strategies concerning the DHL-BCL2 cohort are far more elaborate than the DHL-BCL6, which was the minority of DHL with a mixed composition of GCB, ABC, and unclassifiable DLBCLs. More studies and investigations should be done to this subtype, as the incidence of DHL-BCL6 in east Asia is considerable (comparison of two centers: South China: DHL-BCL2 (37%), DHL-BCL6 (63%) [141] vs North American: DHL-BCL2 (87%), DHL-BCL6 (23%) [32].
Although alteration of some pathways was repeated spotted in DHL, agents targeting corresponding proteins or pathways were rarely tested in DLBCLs or yielded unsatisfactory results. As more studies highlight the unique signaling in DHL, novel agents or therapeutic strategies, like CAR-T and CAR-NK, can be tested to accelerate treatment development for these patients.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Abbreviations
- ARID1A:
-
AT-rich interaction domain 1A
- AICDA:
-
Activation-induced cytidine deaminase
- ASCT:
-
Autologous stem cell transplant
- BAK:
-
BCL2 homologous antagonist killer
- BAX:
-
BCL2-associated X protein
- BCL:
-
B-cell lymphoma
- BCR:
-
Breakpoint cluster region protein
- BET:
-
Bromodomain and extraterminal protein
- BFL:
-
BCL2-related protein A1
- BIRC5:
-
Baculoviral IAP repeat containing 5
- BRD4:
-
Bromodomain containing 4 protein
- BTK:
-
Bruton's tyrosine kinase
- CAR-T:
-
Chimeric antigen receptor T-cell
- CDK4/6:
-
Cyclin dependent kinase 4 and 6
- CHK2:
-
Checkpoint kinase 2
- CR:
-
Complete response
- CRBN:
-
Cereblon
- CREBBP:
-
CAMP response element-binding protein
- CXCR4:
-
C-X-C motif chemokine receptor 4
- DEL:
-
Double-expressor lymphoma
- DHL:
-
Double-hit lymphoma
- DLBCL:
-
Diffuse large B-cell lymphoma
- DSS:
-
Disease-specific survival
- EBF1:
-
Early B cell factor 1
- EFS:
-
Event-free survival
- ERK:
-
Extracellular signal-regulated kinase
- EZH2:
-
Enhancer of zeste homolog 2
- FAS:
-
Fas cell surface death receptor
- FISH:
-
Fluorescence in situ hybridization
- FOXO1:
-
Forkhead box O1
- FLT3:
-
Fms related receptor tyrosine kinase 3
- GCB:
-
Germinal center B-cell like
- GEP:
-
Gene expression profile
- GNA13:
-
G-Protein subunit alpha 13
- HES1:
-
Hes family BHLH transcription factor 1
- HDAC:
-
Histone deacetylase
- HLA-DMB:
-
Major histocompatibility complex, class II, DM beta
- HVCN1:
-
Hydrogen voltage gated channel 1
- IAP2:
-
Inhibition of apoptosis protein 2
- IFRT:
-
Involved-field radiation therapy
- IRF:
-
Interferon regulatory factor
- IKK:
-
IκB kinase
- IMiD:
-
Immunomodulatory imide drug
- JAK:
-
Janus kinase
- KMT2D:
-
Lysine methyltransferase 2D
- MCL1:
-
Induced myeloid leukemia cell differentiation protein
- MEF2B:
-
Myocyte enhancer factor 2B
- mTOR:
-
Mammalian target of rapamycin
- MYC:
-
MYC proto-oncogene
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NHL:
-
Non-Hodgkin's lymphoma
- NMT:
-
N-myristoyltransferase
- OCT2:
-
Octamer-binding protein 2
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphoinositide 3-kinase
- PIK3R1:
-
Phosphoinositide-3-kinase regulatory subunit 1
- PKC:
-
Protein kinase C
- POU2F2:
-
POU class 2 homeobox 2
- PTEN:
-
Phosphatase and tensin homolog
- PLK1:
-
Polo like kinase 1
- REL:
-
Proto-oncogene c-Rel
- RFS:
-
Relapse-free survival
- R-CHOP:
-
Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone
- R-DA-EPOCH:
-
Rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin
- R-Hyper-CVAD:
-
Rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with cytarabine plus methotrexate
- R-CODOX-M/IVAC:
-
Rituximab, cyclophosphamide, vincristine, doxorubicin, methotrexate/ ifosfamide, etoposide, cytarabine
- STAT:
-
Signal transducer and activator of transcription
- SMAC:
-
Second mitochondria-derived activator of caspase
- SRSF2:
-
Serine/arginine-rich splicing factor 2
- SIPR2:
-
Sphingosine-1-phosphate receptor 2
- SYK:
-
Spleen associated tyrosine kinase
- TEFB:
-
Transcription elongation factor b
- TGFβ:
-
Transforming growth factor beta
- THL:
-
Triple-hit lymphoma
- TNFRSF14:
-
TNF receptor superfamily member 14
- TNFR1:
-
Tumor necrosis factor receptor 1
- TYK2:
-
Tyrosine kinase 2
- XPO1:
-
Exportin 1
References
Siegel RL, Miller KD, Jemal A. Cancer statistics 2020. CA Cancer J Clin. 2020;70(1):7–30.
Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(28):3452–9.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90.
Swerdlow SH. Diagnosis of “double hit” diffuse large B-cell lymphoma and B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma: when and how, FISH versus IHC. Hematol Am Soc Hematol Educ Program. 2014;2014(1):90–9.
Sesques P, Johnson NA. Approach to the diagnosis and treatment of high-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood. 2017;129(3):280–8.
Ma Z, Niu J, Cao Y, Pang X, Cui W, Zhang W, Li X. Clinical significance of ‘double-hit’ and ‘double-expression’ lymphomas. J Clin Pathol. 2020;73(3):126–38.
Merron B, Davies A. Double hit lymphoma: How do we define it and how do we treat it? Best Pract Res Clin Haematol. 2018;31(3):233–40.
Copie-Bergman C, Cuilliere-Dartigues P, Baia M, Briere J, Delarue R, Canioni D, Salles G, Parrens M, Belhadj K, Fabiani B, et al. MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: a GELA/LYSA study. Blood. 2015;126(22):2466–74.
McPhail ED, Maurer MJ, Macon WR, Feldman AL, Kurtin PJ, Ketterling RP, Vaidya R, Cerhan JR, Ansell SM, Porrata LF, et al. Inferior survival in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements is not associated with MYC/IG gene rearrangements. Haematologica. 2018;103(11):1899–907.
Pedersen MO, Gang AO, Poulsen TS, Knudsen H, Lauritzen AF, Nielsen SL, Klausen TW, Norgaard P. MYC translocation partner gene determines survival of patients with large B-cell lymphoma with MYC- or double-hit MYC/BCL2 translocations. Eur J Haematol. 2014;92(1):42–8.
Aukema SM, Kreuz M, Kohler CW, Rosolowski M, Hasenclever D, Hummel M, Kuppers R, Lenze D, Ott G, Pott C, et al. Biological characterization of adult MYC-translocation-positive mature B-cell lymphomas other than molecular Burkitt lymphoma. Haematologica. 2014;99(4):726–35.
Rosenwald A, Bens S, Advani R, Barrans S, Copie-Bergman C, Elsensohn MH, Natkunam Y, Calaminici M, Sander B, Baia M, et al. Prognostic significance of MYC rearrangement and translocation partner in diffuse large B-cell lymphoma: a study by the lunenburg lymphoma biomarker consortium. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37(35):3359–68.
Herrera AF, Mei M, Low L, Kim HT, Griffin GK, Song JY, Merryman RW, Bedell V, Pak C, Sun H, et al. Relapsed or Refractory double-expressor and double-hit lymphomas have inferior progression-free survival after autologous stem-cell transplantation. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35(1):24–31.
Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, Liu WM, Visco C, Li Y, Miranda RN, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from the International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;121(20):4021–31 (quiz 4250).
Sha C, Barrans S, Cucco F, Bentley MA, Care MA, Cummin T, Kennedy H, Thompson JS, Uddin R, Worrillow L, et al. Molecular high-grade B-cell lymphoma: defining a poor-risk group that requires different approaches to therapy. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37(3):202–12.
Scott DW, King RL, Staiger AM, Ben-Neriah S, Jiang A, Horn H, Mottok A, Farinha P, Slack GW, Ennishi D, et al. High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood. 2018;131(18):2060–4.
Huang W, Medeiros LJ, Lin P, Wang W, Tang G, Khoury J, Konoplev S, Yin CC, Xu J, Oki Y, et al. MYC/BCL2/BCL6 triple hit lymphoma: a study of 40 patients with a comparison to MYC/BCL2 and MYC/BCL6 double hit lymphomas. Mod Pathol Off J US Can Acad Pathol. 2018;31(9):1470–8.
Pillai RK, Sathanoori M, Van Oss SB, Swerdlow SH. Double-hit B-cell lymphomas with BCL6 and MYC translocations are aggressive, frequently extranodal lymphomas distinct from BCL2 double-hit B-cell lymphomas. Am J Surg Pathol. 2013;37(3):323–32.
Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, Cooke SL, Ruiz C, Glover P, Van Hoppe SJL, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood. 2020;135(20):1759–71.
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679–90.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396–407.
Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer cell. 2020;37(4):551–68.
Cucco F, Barrans S, Sha C, Clipson A, Crouch S, Dobson R, Chen Z, Thompson JS, Care MA, Cummin T, et al. Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia. 2020;34(5):1329–41.
Li S, Desai P, Lin P, Yin CC, Tang G, Wang XJ, Konoplev SN, Khoury JD, Bueso-Ramos CE, Medeiros LJ. MYC/BCL6 double-hit lymphoma (DHL): a tumour associated with an aggressive clinical course and poor prognosis. Histopathology. 2016;68(7):1090–8.
Ye Q, Xu-Monette ZY, Tzankov A, Deng L, Wang X, Manyam GC, Visco C, Montes-Moreno S, Zhang L, Dybkær K, et al. Prognostic impact of concurrent MYC and BCL6 rearrangements and expression in de novo diffuse large B-cell lymphoma. Oncotarget. 2016;7(3):2401–16.
Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, Rushton C, Tang J, Thomas N, Slack GW, et al. Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37(3):190–201.
Frosch ZAK, Landsburg DJ. Molecular risk stratification in aggressive B-cell lymphomas. J Clin Oncol Off J Am Soc Clin Oncol. 2020;38(18):2014–7.
Frosch ZAK, Dwivedy Nasta S, Schuster SJ, Svoboda J, Barta SK, Gerson JN, Chong EA, Ayers EC, Rhodes JM, Koike A, et al. Outcomes for double hit lymphoma patients identified via routine vs selective testing for MYC rearrangement. Blood. 2019;134(Supp 1):1607–1607.
B-Cell Lymphomas [https://www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf]
Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, Nielsen O, Gadeberg OV, Mourits-Andersen T, Frederiksen M, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(28):3460–7.
Torka P, Kothari SK, Sundaram S, Li S, Medeiros LJ, Ayers EC, Landsburg DJ, Bond DA, Maddocks KJ, Giri A, et al. Outcomes of patients with limited-stage aggressive large B-cell lymphoma with high-risk cytogenetics. Blood Adv. 2020;4(2):253–62.
Petrich AM, Gandhi M, Jovanovic B, Castillo JJ, Rajguru S, Yang DT, Shah KA, Whyman JD, Lansigan F, Hernandez-Ilizaliturri FJ, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood. 2014;124(15):2354–61.
Oki Y, Noorani M, Lin P, Davis RE, Neelapu SS, Ma L, Ahmed M, Rodriguez MA, Hagemeister FB, Fowler N, et al. Double hit lymphoma: the MD Anderson cancer center clinical experience. Br J Haematol. 2014;166(6):891–901.
Howlett C, Snedecor SJ, Landsburg DJ, Svoboda J, Chong EA, Schuster SJ, Nasta SD, Feldman T, Rago A, Walsh KM, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol. 2015;170(4):504–14.
D’Angelo CR, Hanel W, Chen Y, Yu M, Yang D, Guo L, Karmali R, Burkart M, Ciccosanti C, David K, et al. Impact of initial chemotherapy regimen on outcomes for patients with double-expressor lymphoma: a multi-center analysis. Hematol Oncol. 2021;39(4):473–82.
Landsburg DJ, Falkiewicz MK, Maly J, Blum KA, Howlett C, Feldman T, Mato AR, Hill BT, Li S, Medeiros LJ, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35(20):2260–7.
Sun H, Savage KJ, Karsan A, Slack GW, Gascoyne RD, Toze CL, Sehn LH, Abou Mourad Y, Barnett MJ, Broady RC, et al. Outcome of patients with non-hodgkin lymphomas with concurrent MYC and BCL2 rearrangements treated with CODOX-M/IVAC with rituximab followed by hematopoietic stem cell transplantation. Clin Lymphoma Myeloma Leuk. 2015;15(6):341–8.
Li S, Lin P, Fayad LE, Lennon PA, Miranda RN, Yin CC, Lin E, Medeiros LJ. B-cell lymphomas with MYC/8q24 rearrangements and IGH@BCL2/t(14;18)(q32;q21): an aggressive disease with heterogeneous histology, germinal center B-cell immunophenotype and poor outcome. Mod Pathol Off J US Can Acad Pathol. 2012;25(1):145–56.
Laude MC, Lebras L, Sesques P, Ghesquieres H, Favre S, Bouabdallah K, Croizier C, Guieze R, Drieu La Rochelle L, Gyan E, et al. First-line treatment of double-hit and triple-hit lymphomas: Survival and tolerance data from a retrospective multicenter French study. Am J Hematol. 2021;96(3):302–11.
Puvvada SD, Stiff PJ, Leblanc M, Cook JR, Couban S, Leonard JP, Kahl B, Marcellus D, Shea TC, Winter JN, et al. Outcomes of MYC-associated lymphomas after R-CHOP with and without consolidative autologous stem cell transplant: subset analysis of randomized trial intergroup SWOG S9704. Br J Haematol. 2016;174(5):686–91.
Huang W, Medeiros LJ, Lin P, Wang W, Tang G, Khoury J, Konoplev S, Yin CC, Xu J, Oki Y, et al. MYC/BCL2/BCL6 triple hit lymphoma: a study of 40 patients with a comparison to MYC/BCL2 and MYC/BCL6 double hit lymphomas. Mod Pathol. 2018;31(9):1470–8.
Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, Meyer SN, Mo T, Basso K, Brindle PK, et al. The CREBBP acetyltransferase is a haploinsufficient tumor suppressor in B-cell lymphoma. Cancer Discov. 2017;7(3):322–37.
Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21(10):1190–8.
Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, Basso K, Dalla-Favera R. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell. 2018;34(3):453-465.e459.
Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92.
Lee CH, Melchers M, Wang H, Torrey TA, Slota R, Qi CF, Kim JY, Lugar P, Kong HJ, Farrington L, et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med. 2006;203(1):63–72.
Zhou JX, Lee CH, Qi CF, Wang H, Naghashfar Z, Abbasi S, Morse HC 3rd. IFN regulatory factor 8 regulates MDM2 in germinal center B cells. J Immunol. 2009;183(5):3188–94.
Brescia P, Schneider C, Holmes AB, Shen Q, Hussein S, Pasqualucci L, Basso K, Dalla-Favera R. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer cell. 2018;34(3):453–65.
Martinez A, Pittaluga S, Rudelius M, Davies-Hill T, Sebasigari D, Fountaine TJ, Hewitt S, Jaffe ES, Raffeld M. Expression of the interferon regulatory factor 8/ICSBP-1 in human reactive lymphoid tissues and B-cell lymphomas: a novel germinal center marker. Am J Surg Pathol. 2008;32(8):1190–200.
Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, Califano A, Pasqualucci L, Basso K, Dalla-Favera R. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013;14(10):1084–92.
Bouamar H, Abbas S, Lin AP, Wang L, Jiang D, Holder KN, Kinney MC, Hunicke-Smith S, Aguiar RC. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood. 2013;122(5):726–33.
Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, Fox H, Jacobi K, Saiya-Cork K, Bixby D, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123(10):1487–98.
Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1398–403.
Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5.
Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511(7510):488–92.
Xu-Monette ZY, Deng Q, Manyam GC, Tzankov A, Li L, Xia Y, Wang XX, Zou D, Visco C, Dybkaer K, et al. Clinical and biologic significance of MYC genetic mutations in De Novo diffuse large B-cell lymphoma. Clin Cancer Res. 2016;22(14):3593–605.
Valera A, Lopez-Guillermo A, Cardesa-Salzmann T, Climent F, Gonzalez-Barca E, Mercadal S, Espinosa I, Novelli S, Briones J, Mate JL, et al. MYC protein expression and genetic alterations have prognostic impact in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Haematologica. 2013;98(10):1554–62.
Stasik CJ, Nitta H, Zhang W, Mosher CH, Cook JR, Tubbs RR, Unger JM, Brooks TA, Persky DO, Wilkinson ST, et al. Increased MYC gene copy number correlates with increased mRNA levels in diffuse large B-cell lymphoma. Haematologica. 2010;95(4):597–603.
Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol. 2008;9(6):613–22.
Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJ, Mungall AJ, Zhao Y, Schein J, Steidl C, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013;121(18):3666–74.
Xie L, Ushmorov A, Leithauser F, Guan H, Steidl C, Farbinger J, Pelzer C, Vogel MJ, Maier HJ, Gascoyne RD, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119(15):3503–11.
Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, Li S, Imami K, Selbach M, Di Virgilio M, et al. PI3 kinase and FOXO1 transcription factor activity differentially control b cells in the germinal center light and dark zones. Immunity. 2015;43(6):1075–86.
Szydlowski M, Kiliszek P, Sewastianik T, Jablonska E, Bialopiotrowicz E, Gorniak P, Polak A, Markowicz S, Nowak E, Grygorowicz MA, et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B-cell lymphomas. Blood. 2016;127(6):739–48.
Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, Grewal J, Yu S, Fornika D, Bushell K, et al. Genetic Landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22(9):2290–300.
Ushmorov A, Wirth T. FOXO in B-cell lymphopoiesis and B cell neoplasia. Semin Cancer Biol. 2018;50:132–41.
Hodson DJ, Shaffer AL, Xiao W, Wright GW, Schmitz R, Phelan JD, Yang Y, Webster DE, Rui L, Kohlhammer H, et al. Regulation of normal B-cell differentiation and malignant B-cell survival by OCT2. Proc Natl Acad Sci USA. 2016;113(14):E2039-2046.
Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D’Costa K, Tarlinton DM, Kallies A, Corcoran LM. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med. 2012;209(11):2049–64.
Corcoran L, Emslie D, Kratina T, Shi W, Hirsch S, Taubenheim N, Chevrier S. Oct2 and Obf1 as facilitators of B: T cell collaboration during a humoral immune response. Front Immunol. 2014;5:108.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63.
Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, Boyle M, Slack GW, Marra MA, Connors JM, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26(6):1383–90.
Ennishi D, Mottok A, Ben-Neriah S, Shulha HP, Farinha P, Chan FC, Meissner B, Boyle M, Hother C, Kridel R, et al. Genetic profiling of MYC and BCL2 in diffuse large B-cell lymphoma determines cell-of-origin-specific clinical impact. Blood. 2017;129(20):2760–70.
Deng X, Gao F, Flagg T, Anderson J, May WS. Bcl2’s flexible loop domain regulates p53 binding and survival. Mol Cell Biol. 2006;26(12):4421–34.
Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of BCL2 versus Bcl-Xl. Cell Death Differ. 2012;19(2):295–309.
Ding J, Zhang Z, Roberts GJ, Falcone M, Miao Y, Shao Y, Zhang XC, Andrews DW, Lin J. BCL2 and Bax interact via the BH1-3 groove-BH3 motif interface and a novel interface involving the BH4 motif. J Biol Chem. 2010;285(37):28749–63.
Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123(26):4111–9.
Guegan JP, Ginestier C, Charafe-Jauffret E, Ducret T, Quignard JF, Vacher P, Legembre P. CD95/Fas and metastatic disease: What does not kill you makes you stronger. Semin Cancer Biol. 2020;60:121–31.
Takahashi H, Feuerhake F, Kutok JL, Monti S, Dal Cin P, Neuberg D, Aster JC, Shipp MA. FAS death domain deletions and cellular FADD-like interleukin 1beta converting enzyme inhibitory protein (long) overexpression: alternative mechanisms for deregulating the extrinsic apoptotic pathway in diffuse large B-cell lymphoma subtypes. Clin Cancer Res. 2006;12(11 Pt 1):3265–71.
Razzaghi R, Agarwal S, Kotlov N, Plotnikova O, Nomie K, Huang DW, Wright GW, Smith GA, Li M, Takata K et al: Compromised counterselection by FAS creates an aggressive subtype of germinal center lymphoma. J Exp Med 2021, 218(3).
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303.
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48(4):491–507.
Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–57.
Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–208.
Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, Elemento O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116(24):5247–55.
Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–9.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KS. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene. 2015;34(4):445–54.
Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92.
Deng Y, Chen X, Huang C, Chen G, Chen F, Lu J, Shi X, He C, Zeng Z, Qiu Y, et al. EZH2/BCL2 coexpression predicts worse survival in diffuse large B-cell lymphomas and demonstrates poor efficacy to rituximab in localized lesions. J Cancer. 2019;10(9):2006–17.
Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, Bashashati A, Saberi S, Boyle M, Meissner B, et al. Molecular and genetic characterization of mhc deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov. 2019;9(4):546–63.
Dersh D, Phelan JD, Gumina ME, Wang B, Arbuckle JH, Holly J, Kishton RJ, Markowitz TE, Seedhom MO, Fridlyand N, et al. Genome-wide screens identify lineage- and tumor-specific genes modulating MHC-I- and MHC-II-restricted immunosurveillance of human lymphomas. Immunity. 2021;54(1):116–31.
Berg T, Thoene S, Yap D, Wee T, Schoeler N, Rosten P, Lim E, Bilenky M, Mungall AJ, Oellerich T, et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood. 2014;123(25):3914–24.
Shi Y, Ma HL, Zhuang YW, Wang XX, Jiang Y, Xu HE. C10ORF12 modulates PRC2 histone methyltransferase activity and H3K27me3 levels. Acta Pharmacol Sin. 2019;40(11):1457–65.
Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115(6):2419–52.
Meyer SN, Scuoppo C, Vlasevska S, Bal E, Holmes AB, Holloman M, Garcia-Ibanez L, Nataraj S, Duval R, Vantrimpont T, et al. Unique and shared epigenetic programs of the CREBBP and EP300 acetyltransferases in germinal center B cells reveal targetable dependencies in lymphoma. Immunity. 2019;51(3):535–47.
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–95.
Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, McNally D, Wang L, Doane AS, Agirre X, et al. CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discov. 2017;7(1):38–53.
Garcia-Ramirez I, Tadros S, Gonzalez-Herrero I, Martin-Lorenzo A, Rodriguez-Hernandez G, Moore D, Ruiz-Roca L, Blanco O, Alonso-Lopez D, Rivas JL, et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood. 2017;129(19):2645–56.
Hashwah H, Schmid CA, Kasser S, Bertram K, Stelling A, Manz MG, Muller A. Inactivation of CREBBP expands the germinal center B cell compartment, down-regulates MHCII expression and promotes DLBCL growth. Proc Natl Acad Sci U S A. 2017;114(36):9701–6.
Huang YH, Cai K, Xu PP, Wang L, Huang CX, Fang Y, Cheng S, Sun XJ, Liu F, Huang JY, et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-NOTCH-CCL2/CSF1 axis. Signal Transduct Target Ther. 2021;6(1):10.
Cerchietti LC, Hatzi K, Caldas-Lopes E, Yang SN, Figueroa ME, Morin RD, Hirst M, Mendez L, Shaknovich R, Cole PA, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J Clin Invest. 2010;120(12):4569–82.
Scialdone A, Khazaei S, Hasni MS, Lennartsson A, Gullberg U, Drott K. Depletion of the transcriptional coactivators CREB-binding protein or EP300 downregulates CD20 in diffuse large B-cell lymphoma cells and impairs the cytotoxic effects of anti-CD20 antibodies. Exp Hematol. 2019;79:35–46.
Roberts CW, Orkin SH. The SWI/SNF complex–chromatin and cancer. Nat Rev Cancer. 2004;4(2):133–42.
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601.
Balinas-Gavira C, Rodriguez MI, Andrades A, Cuadros M, Alvarez-Perez JC, Alvarez-Prado AF, de Yebenes VG, Sanchez-Hernandez S, Fernandez-Vigo E, Munoz J, et al. Frequent mutations in the amino-terminal domain of BCL7A impair its tumor suppressor role in DLBCL. Leukemia. 2020;34(10):2722–35.
Guan B, Gao M, Wu CH, Wang TL, Shih Ie M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia. 2012;14(10):986–93.
Shen R, Xu PP, Wang N, Yi HM, Dong L, Fu D, Huang JY, Huang HY, Janin A, Cheng S, et al. Influence of oncogenic mutations and tumor microenvironment alterations on extranodal invasion in diffuse large B-cell lymphoma. Clin Transl Med. 2020;10(7):e221.
Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20(3):251–4.
Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, Chen L, Monterroza DD, Li H, DeKelver RC, et al. SRSF2 is essential for hematopoiesis, and its myelodysplastic syndrome-related mutations dysregulate alternative Pre-mRNA splicing. Mol Cell Biol. 2015;35(17):3071–82.
Kon A, Yamazaki S, Nannya Y, Kataoka K, Ota Y, Nakagawa MM, Yoshida K, Shiozawa Y, Morita M, Yoshizato T, et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood. 2018;131(6):621–35.
Havranek O, Xu J, Kohrer S, Wang Z, Becker L, Comer JM, Henderson J, Ma W, Man Chun Ma J, Westin JR, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood. 2017;130(8):995–1006.
Kober-Hasslacher M, Schmidt-Supprian M. The unsolved puzzle of c-Rel in B cell lymphoma. Cancers (Basel) 2019, 11(7).
Houldsworth J, Mathew S, Rao PH, Dyomina K, Louie DC, Parsa N, Offit K, Chaganti RS. REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood. 1996;87(1):25–9.
Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, Kurtin P, Pinkus GS, de Leval L, Harris NL, et al. NFkappaB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood. 2005;106(4):1392–9.
Houldsworth J, Olshen AB, Cattoretti G, Donnelly GB, Teruya-Feldstein J, Qin J, Palanisamy N, Shen Y, Dyomina K, Petlakh M, et al. Relationship between REL amplification, REL function, and clinical and biologic features in diffuse large B-cell lymphomas. Blood. 2004;103(5):1862–8.
Li L, Xu-Monette ZY, Ok CY, Tzankov A, Manyam GC, Sun R, Visco C, Zhang M, Montes-Moreno S, Dybkaer K, et al. Prognostic impact of c-Rel nuclear expression and REL amplification and crosstalk between c-Rel and the p53 pathway in diffuse large B-cell lymphoma. Oncotarget. 2015;6(27):23157–80.
Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–43.
Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, Dietze K, Nogai H, Storek B, Madle H, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110(30):12420–5.
Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984–2009.
Aittomäki S, Pesu M. Therapeutic targeting of the Jak_STAT pathway. Basic Clin Pharmacol Toxicol. 2014;114:18–23.
Ritz O, Guiter C, Dorsch K, Dusanter-Fourt I, Wegener S, Jouault H, Gaulard P, Castellano F, Moller P, Leroy K. STAT6 activity is regulated by SOCS-1 and modulates BCL-XL expression in primary mediastinal B-cell lymphoma. Leukemia. 2008;22(11):2106–10.
Ritz O, Rommel K, Dorsch K, Kelsch E, Melzner J, Buck M, Leroy K, Papadopoulou V, Wagner S, Marienfeld R, et al. STAT6-mediated BCL6 repression in primary mediastinal B-cell lymphoma (PMBL). Oncotarget. 2013;4(7):1093–102.
Yildiz M, Li H, Bernard D, Amin NA, Ouillette P, Jones S, Saiya-Cork K, Parkin B, Jacobi K, Shedden K, et al. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125(4):668–79.
Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, Greiner TC, McDonnell TJ, Young KH. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood. 2012;119(16):3668–83.
Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL rituximab-CHOP Consortium Program Study. Blood. 2012;120(19):3986–96.
Gebauer N, Bernard V, Gebauer W, Thorns C, Feller AC, Merz H. TP53 mutations are frequent events in double-hit B-cell lymphomas with MYC and BCL2 but not MYC and BCL6 translocations. Leuk Lymphoma. 2015;56(1):179–85.
Gaidarenko O, Xu Y. Transcription activity is required for p53-dependent tumor suppression. Oncogene. 2009;28(49):4397–401.
Song JY, Perry AM, Herrera AF, Chen L, Skrabek P, Nasr MR, Ottesen RA, Nikowitz J, Bedell V, Murata-Collins J, et al. Double-hit signature with TP53 abnormalities predicts poor survival in patients with germinal center type diffuse large B-cell lymphoma treated with R-CHOP. Clin Cancer Res. 2021;27(6):1671–80.
Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7):1256–65.
Nairismagi ML, Tan J, Lim JQ, Nagarajan S, Ng CC, Rajasegaran V, Huang D, Lim WK, Laurensia Y, Wijaya GC, et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia. 2016;30(6):1311–9.
Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490(7418):116–20.
Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, Pan CM, Hu Y, Cai CP, Dong Y, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–6.
Hondares E, Brown MA, Musset B, Morgan D, Cherny VV, Taubert C, Bhamrah MK, Coe D, Marelli-Berg F, Gribben JG, et al. Enhanced activation of an amino-terminally truncated isoform of the voltage-gated proton channel HVCN1 enriched in malignant B cells. Proc Natl Acad Sci USA. 2014;111(50):18078–83.
Mintz MA, Felce JH, Chou MY, Mayya V, Xu Y, Shui JW, An J, Li Z, Marson A, Okada T, et al. The HVEM-BTLA Axis Restrains T Cell Help to Germinal Center B Cells and Functions as a Cell-Extrinsic Suppressor in Lymphomagenesis. Immunity. 2019;51(2):310–23.
Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell. 2016;167(2):405–18.
Costello RT, Mallet F, Barbarat B, De Colella JMS, Sainty D, Sweet RW, Truneh A, Olive D. Stimulation of non-Hodgkin’s lymphoma via HVEM: an alternate and safe way to increase Fas-induced apoptosis and improve tumor immunogenicity. Leukemia. 2003;17(12):2500–7.
Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CD, Schmidt TH, Xu Y, Proia RL, Coughlin SR, et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12(7):672–80.
Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, An J, Xu Y, Rosenwald A, Ott G, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516(7530):254–8.
Cattoretti G, Mandelbaum J, Lee N, Chaves AH, Mahler AM, Chadburn A, Dalla-Favera R, Pasqualucci L, MacLennan AJ. Targeted disruption of the S1P2 sphingosine 1-phosphate receptor gene leads to diffuse large B-cell lymphoma formation. Cancer Res. 2009;69(22):8686–92.
Stelling A, Hashwah H, Bertram K, Manz MG, Tzankov A, Muller A. The tumor suppressive TGF-beta/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood. 2018;131(20):2235–46.
Xia Z, Zhang X, Liu P, Zhang R, Huang Z, Li D, Xiao X, Wu M, Ning N, Zhang Q, et al. GNA13 regulates BCL2 expression and the sensitivity of GCB-DLBCL cells to BCL2 inhibitors in a palmitoylation-dependent manner. Cell Death Dis. 2021;12(1):54.
Zhang J, Weng Z, Huang Y, Li M, Wang F, Wang Y, Rao H. High-grade B-Cell Lymphoma With MYC, BCL2, and/or BCL6 translocations/rearrangements: clinicopathologic features of 51 cases in a single Institution of South China. Am J Surg Pathol. 2020;44(12):1602–11.
Magistroni V, Mauri M, D’Aliberti D, Mezzatesta C, Crespiatico I, Nava M, Fontana D, Sharma N, Parker W, Schreiber A, et al. De novo UBE2A mutations are recurrently acquired during chronic myeloid leukemia progression and interfere with myeloid differentiation pathways. Haematologica. 2019;104(9):1789–97.
Mlynarczyk C, Fontan L, Melnick A. Germinal center-derived lymphomas: the darkest side of humoral immunity. Immunol Rev. 2019;288(1):214–39.
Laidlaw BJ, Cyster JG. Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol. 2021;21(4):209–20.
Hart GT, Wang X, Hogquist KA, Jameson SC. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci U S A. 2011;108(2):716–21.
Bonetti P, Testoni M, Scandurra M, Ponzoni M, Piva R, Mensah AA, Rinaldi A, Kwee I, Tibiletti MG, Iqbal J, et al. Deregulation of ETS1 and FLI1 contributes to the pathogenesis of diffuse large B-cell lymphoma. Blood. 2013;122(13):2233–41.
Priebe V, Sartori G, Napoli S, Chung EYL, Cascione L, Kwee I, Arribas AJ, Mensah AA, Rinaldi A, Ponzoni M et al. Role of ETS1 in the transcriptional network of diffuse large B Cell lymphoma of the activated B cell-like type. Cancers (Basel) 2020, 12(7).
Li J, Riedt T, Goossens S, Carrillo Garcia C, Szczepanski S, Brandes M, Pieters T, Dobrosch L, Gutgemann I, Farla N, et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood. 2017;129(4):460–72.
Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, Shaffer AL 3rd, Ceribelli M, Wang JQ, Schmitz R, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560(7718):387–91.
Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci USA. 2012;109(20):7865–70.
Mei ZZ, Chen XY, Hu SW, Wang N, Ou XL, Wang J, Luo HH, Liu J, Jiang Y. Kelch-like protein 21 (KLHL21) targets IkappaB kinase-beta to regulate nuclear factor kappa-light chain enhancer of activated B cells (NF-kappaB) signaling negatively. J Biol Chem. 2016;291(35):18176–89.
Courtheoux T, Enchev RI, Lampert F, Gerez J, Beck J, Picotti P, Sumara I, Peter M. Cortical dynamics during cell motility are regulated by CRL3(KLHL21) E3 ubiquitin ligase. Nat Commun. 2016;7:12810.
Li L, Zhang W, Liu Y, Liu X, Cai L, Kang J, Zhang Y, Chen W, Dong C, Zhang Y, et al. The CRL3(BTBD9) E3 ubiquitin ligase complex targets TNFAIP1 for degradation to suppress cancer cell migration. Signal Transduct Target Ther. 2020;5(1):42.
Shanmugam V, Craig JW, Hilton LK, Nguyen MH, Rushton CK, Fahimdanesh K, Lovitch S, Ferland B, Scott DW, Aster JC. Notch activation is pervasive in SMZL and uncommon in DLBCL: implications for Notch signaling in B-cell tumors. Blood Adv. 2021;5(1):71–83.
Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K, et al. Notch2 Is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18(5):675–85.
Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, Nannya Y, Suzuki R, Ota S, Ota Y, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009;100(5):920–6.
Zhang X, Shi Y, Weng Y, Lai Q, Luo T, Zhao J, Ren G, Li W, Pan H, Ke Y, et al. The truncate mutation of Notch2 enhances cell proliferation through activating the NF-kappaB signal pathway in the diffuse large B-cell lymphomas. PLoS ONE. 2014;9(10):e108747.
de Miranda NF, Georgiou K, Chen L, Wu C, Gao Z, Zaravinos A, Lisboa S, Enblad G, Teixeira MR, Zeng Y, et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 2014;124(16):2544–53.
Meriranta L, Pasanen A, Louhimo R, Cervera A, Alkodsi A, Autio M, Taskinen M, Rantanen V, Karjalainen-Lindsberg ML, Holte H, et al. Deltex-1 mutations predict poor survival in diffuse large B-cell lymphoma. Haematologica. 2017;102(5):e195–8.
Filipits M, Jaeger U, Pohl G, Stranzl T, Simonitsch I, Kaider A, Skrabs C, Pirker R. Cyclin D3 is a predictive and prognostic factor in diffuse large B-cell lymphoma. Clin Cancer Res. 2002;8(3):729–33.
Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127(2):265–75.
Kannengiesser C, Brookes S, del Arroyo AG, Pham D, Bombled J, Barrois M, Mauffret O, Avril MF, Chompret A, Lenoir GM, et al. Functional, structural, and genetic evaluation of 20 CDKN2A germ line mutations identified in melanoma-prone families or patients. Hum Mutat. 2009;30(4):564–74.
Jethwa A, Slabicki M, Hullein J, Jentzsch M, Dalal V, Rabe S, Wagner L, Walther T, Klapper W, Project MN et al. TRRAP is essential for regulating the accumulation of mutant and wild-type p53 in lymphoma. Blood 2018; 131(25):2789–2802.
Xu-Monette ZY, Zhang S, Li X, Manyam GC, Wang XX, Xia Y, Visco C, Tzankov A, Zhang L, Montes-Moreno S, et al. p63 expression confers significantly better survival outcomes in high-risk diffuse large B-cell lymphoma and demonstrates p53-like and p53-independent tumor suppressor function. Aging. 2016;8(2):345–65.
Scott DW, Mungall KL, Ben-Neriah S, Rogic S, Morin RD, Slack GW, Tan KL, Chan FC, Lim RS, Connors JM, et al. TBL1XR1/TP63: a novel recurrent gene fusion in B-cell non-Hodgkin lymphoma. Blood. 2012;119(21):4949–52.
Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14(7):417–33.
de Oliveira JF, do Prado PFV, da Costa SS, Sforca ML, Canateli C, Ranzani AT, Maschietto M, de Oliveira PSL, Otto PA, Klevit RE, et al. Mechanistic insights revealed by a UBE2A mutation linked to intellectual disability. Nat Chem Biol. 2019;15(1):62–70.
Brunet M, Vargas C, Larrieu D, Torrisani J, Dufresne M: E3 ubiquitin ligase TRIP12: regulation, structure, and physiopathological functions. Int J Mol Sci 2020, 21(22).
Jacobs J, Deschoolmeester V, Zwaenepoel K, Rolfo C, Silence K, Rottey S, Lardon F, Smits E, Pauwels P. CD70: an emerging target in cancer immunotherapy. Pharmacol Ther. 2015;155:1–10.
Bertrand P, Maingonnat C, Penther D, Guney S, Ruminy P, Picquenot JM, Mareschal S, Alcantara M, Bouzelfen A, Dubois S, et al. The costimulatory molecule CD70 is regulated by distinct molecular mechanisms and is associated with overall survival in diffuse large B-cell lymphoma. Genes Chromosom Cancer. 2013;52(8):764–74.
Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, Dalla-Favera R. Combined genetic inactivation of beta2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20(6):728–40.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29.
Georgiou K, Chen L, Berglund M, Ren W, de Miranda NF, Lisboa S, Fangazio M, Zhu S, Hou Y, Wu K, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127(24):3026–34.
Song MK, Park BB, Uhm J. Understanding immune evasion and therapeutic targeting associated with PD-1/PD-L1 pathway in diffuse large B-cell lymphoma. Int J Mol Sci 2019, 20(6).
Zabel BA, Lewen S, Berahovich RD, Jaen JC, Schall TJ. The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol Cancer. 2011;10:73.
Pansy K, Feichtinger J, Ehall B, Uhl B, Sedej M, Roula D, Pursche B, Wolf A, Zoidl M, Steinbauer E et al. The CXCR4-CXCL12-axis is of prognostic relevance in DLBCL and Its antagonists exert pro-apoptotic effects in vitro. Int J Mol Sci 2019, 20(19).
Moreno MJ, Bosch R, Dieguez-Gonzalez R, Novelli S, Mozos A, Gallardo A, Pavon MA, Cespedes MV, Granena A, Alcoceba M, et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J Pathol. 2015;235(3):445–55.
Durr C, Pfeifer D, Claus R, Schmitt-Graeff A, Gerlach UV, Graeser R, Kruger S, Gerbitz A, Negrin RS, Finke J, et al. CXCL12 mediates immunosuppression in the lymphoma microenvironment after allogeneic transplantation of hematopoietic cells. Cancer Res. 2010;70(24):10170–81.
Peled A, Klein S, Beider K, Burger JA, Abraham M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine. 2018;109:11–6.
Kim JH, Kim WS, Ryu KJ, Kim SJ, Park C. CXCR4 can induce PI3Kdelta inhibitor resistance in ABC DLBCL. Blood Cancer J. 2018;8(2):23.
Treon SP, Xu L, Guerrera ML, Jimenez C, Hunter ZR, Liu X, Demos M, Gustine J, Chan G, Munshi M, et al. Genomic landscape of waldenstrom macroglobulinemia and its impact on treatment strategies. J Clin Oncol Off J Am Soc Clin Oncol. 2020;38(11):1198–208.
Ennishi D, Healy S, Bashashati A, Saberi S, Hother C, Mottok A, Chan FC, Chong L, Abraham L, Kridel R, et al. TMEM30A loss-of-function mutations drive lymphomagenesis and confer therapeutically exploitable vulnerability in B-cell lymphoma. Nat Med. 2020;26(4):577–88.
Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71(4):1418–30.
Webb MS, Tortora N, Cremese M, Kozlowska H, Blaquiere M, Devine DV, Kornbrust DJ. Toxicity and toxicokinetics of a phosphorothioate oligonucleotide against the c-myc oncogene in cynomolgus monkeys. Antisense Nucleic Acid Drug Dev. 2001;11(3):155–63.
Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, Puvvada S, Kipps TJ, Anderson MA, Salem AH, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35(8):826–33.
de Vos S, Swinnen LJ, Wang D, Reid E, Fowler N, Cordero J, Dunbar M, Enschede SH, Nolan C, Petrich AM, et al. Venetoclax, bendamustine, and rituximab in patients with relapsed or refractory NHL: a phase Ib dose-finding study. Ann Oncol. 2018;29(9):1932–8.
Morschhauser F, Feugier P, Flinn IW, Gasiorowski R, Greil R, Illes A, Johnson NA, Larouche JF, Lugtenburg PJ, Patti C, et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood. 2021;137(5):600–9.
Bojarczuk K, Wienand K, Ryan JA, Chen L, Villalobos-Ortiz M, Mandato E, Stachura J, Letai A, Lawton LN, Chapuy B, et al. Targeted inhibition of PI3Kalpha/delta is synergistic with BCL2 blockade in genetically defined subtypes of DLBCL. Blood. 2019;133(1):70–80.
Sasi BK, Martines C, Xerxa E, Porro F, Kalkan H, Fazio R, Turkalj S, Bojnik E, Pyrzynska B, Stachura J, et al. Inhibition of SYK or BTK augments venetoclax sensitivity in SHP1-negative/BCL2-positive diffuse large B-cell lymphoma. Leukemia. 2019;33(10):2416–28.
Scholze H, Stephenson RE, Reynolds R, Shah S, Puri R, Butler SD, Trujillo-Alonso V, Teater MR, van Besien H, Gibbs-Curtis D, et al. Combined EZH2 and BCL2 inhibitors as precision therapy for genetically defined DLBCL subtypes. Blood Adv. 2020;4(20):5226–31.
Runckel K, Barth MJ, Mavis C, Gu JJ, Hernandez-Ilizaliturri FJ. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018;2(23):3516–25.
Cheng C, Wang T, Song Z, Peng L, Gao M, Hermine O, Rousseaux S, Khochbin S, Mi JQ, Wang J. Induction of autophagy and autophagy-dependent apoptosis in diffuse large B-cell lymphoma by a new antimalarial artemisinin derivative, SM1044. Cancer Med. 2018;7(2):380–96.
Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19(5):649–59.
Salles G, Adib D, Navia SB, Rajarethinam A, Whalen J, Radford J, Opat S, McKay P, Jurczak W, Gandhi Laurent D, et al. Phase 2 multicenter study of tazemetostat, an EZH2 inhibitor, in patients with relapsed or refractory follicular lymphoma. Blood. 2019;134(Supp 1):123–123.
Pagel J, Morschhauser F, Herve T, Chaidos A, Phillips T, Ribrag V, Campbell P, Fruchart C, Jurczak W, McKay P, et al. Interim update from a phase 2 multicenter study of tazemetostat, an EZH2 Inhibitor, in patients with relapsed or refractory (R/R) follicular lymphoma (FL). Clin Lymphoma Myeloma Leuk. 2018;18:S278–9.
Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, Shiroishi M, Takizawa T, Yasumatsu I, Horiuchi T, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108(10):2069–78.
Harb W, Abramson J, Lunning M, Goy A, Maddocks K, Lebedinsky C, Senderowicz A, Trojer P, Bradley WD, Flinn I. A phase 1 study of CPI-1205, a small molecule inhibitor of EZH2, preliminary safety in patients with B-cell lymphomas. Ann Oncol. 2018;29:7.
Ogura M. Corrigenda. Br J Haematol. 2014;166(4):629–629.
Assouline SE, Nielsen TH, Yu S, Alcaide M, Chong L, MacDonald D, Tosikyan A, Kukreti V, Kezouh A, Petrogiannis-Haliotis T, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128(2):185–94.
Guan X-W, Hua-Qing W, Jia L, Liu F-T. The novel HDAC inhibitor chidamide synergizes with rituximab to inhibit DLBCL tumor growth in vitro and in vivo by up-regulating CD20 expression. Blood. 2018;132(Supplement 1):2947–2947.
Lue JK, Prabhu SA, Liu Y, Gonzalez Y, Verma A, Mundi PS, Abshiru N, Camarillo JM, Mehta S, Chen EI, et al. Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin Cancer Res. 2019;25(17):5271–83.
Batlevi CL, Crump M, Andreadis C, Rizzieri D, Assouline SE, Fox S, van der Jagt RHC, Copeland A, Potvin D, Chao R, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol. 2017;178(3):434–41.
Younes A, Berdeja JG, Patel MR, Flinn I, Gerecitano JF, Neelapu SS, Kelly KR, Copeland AR, Akins A, Clancy MS, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016;17(5):622–31.
Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3(4):e196-204.
Esteve-Arenys A, Valero JG, Chamorro-Jorganes A, Gonzalez D, Rodriguez V, Dlouhy I, Salaverria I, Campo E, Colomer D, Martinez A, et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene. 2018;37(14):1830–44.
Cummin TEC, Cox KL, Murray TD, Turaj AH, Dunning L, English VL, Fell R, Packham G, Ma Y, Powell B, et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL2 expression. Blood Adv. 2020;4(14):3316–28.
Abramson JS, Blum KA, Flinn IW, Gutierrez M, Goy A, Maris M, Cooper M, O’Meara M, Borger D, Mertz J, et al. BET inhibitor CPI-0610 is well tolerated and induces responses in diffuse large B-cell lymphoma and follicular lymphoma: preliminary analysis of an ongoing phase 1 study. Blood. 2015;126(23):1491.
Hogg SJ, Newbold A, Vervoort SJ, Cluse LA, Martin BP, Gregory GP, Lefebure M, Vidacs E, Tothill RW, Bradner JE, et al. BET inhibition induces apoptosis in aggressive B-cell lymphoma via epigenetic regulation of BCL2 family members. Mol Cancer Ther. 2016;15(9):2030–41.
Martin P, Bartlett NL, Rivera IIR, Revuelta M, Chavez JC, Reagan JL, Smith SM, LaCasce AS, Zhang L, Zhai M, et al. A phase I, open label, multicenter trial of oral azacitidine (CC-486) Plus R-CHOP in patients with high-risk, previously untreated diffuse large B-cell lymphoma, grade 3B follicular lymphoma, or transformed lymphoma. Blood. 2017;130:192.
Magagnoli M, Carlo-Stella C, Santoro A. Copanlisib for the treatment of adults with relapsed follicular lymphoma. Expert Rev Clin Pharmacol. 2020;13(8):813–23.
Whitfield JR, Beaulieu ME, Soucek L. Strategies to inhibit Myc and their clinical applicability. Front Cell Dev Biol. 2017;5:10.
Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci USA. 2002;99(6):3830–5.
Chen BJ, Wu YL, Tanaka Y, Zhang W. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int J Biol Sci. 2014;10(10):1084–96.
Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, Sagar V, Luan Y, Chalmers ZR, Unno K, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer cell. 2019;36(5):483–97.
Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074–G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther. 2010;335(3):715–27.
Wang H, Chauhan J, Hu A, Pendleton K, Yap JL, Sabato PE, Jones JW, Perri M, Yu J, Cione E, Kane MA. Disruption of Myc-Max heterodimerization with improved cell-penetrating analogs of the small molecule 10074–G5. Oncotarget. 2013;4(6):936–49.
Mo H, Henriksson M. Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc Natl Acad Sci USA. 2005;103(16):6344–9.
Mo H, Vita M, Crespin M, Henriksson M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle. 2006;5(19):2191–4.
Neidle S. Quadruplex nucleic acids as novel therapeutic targets. J Med Chem. 2016;59:5987–6011.
Brown ZZ, Mapelli C, Farasat I, Shoultz AV, Johnson SA, Orvieto F, Santoprete A, Bianchi E, McCracken AB, Chen K, et al. Multiple synthetic routes to the mini-protein omomyc and coiled-coil domain truncations. J Org Chem. 2020;85(3):1466–75.
Masso-Valles D, Soucek L. Blocking Myc to treat cancer: reflecting on two decades of omomyc. Cells. 2020;9(4):883.
Zhang X, Bi C, Lu T, Zhang W, Yue T, Wang C, Tian T, Zhang X, Huang Y, Lunning M, et al. Targeting translation initiation by synthetic rocaglates for treating MYC-driven lymphomas. Leukemia. 2020;34(1):138–50.
Chu J, Zhang W, Cencic R, O’Connor PBF, Robert F, Devine WG, Selznick A, Henkel T, Merrick WC, Brown LE, et al. Rocaglates induce gain-of-function alterations to eIF4A and eIF4F. Cell Rep. 2020;30(8):2481–8.
Ren Y, Bi C, Zhao X, Lwin T, Wang C, Yuan J, Silva AS, Shah BD, Fang B, Li T, et al. PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J Clin Invest. 2018;128(12):5517–30.
Wang J, Liu Z, Wang Z, Wang S, Chen Z, Li Z, Zhang M, Zou J, Dong B, Gao J, et al. Targeting c-Myc: JQ1 as a promising option for c-Myc-amplified esophageal squamous cell carcinoma. Cancer Lett. 2018;419:64–74.
Wu Z, Hu Z, Han X, Li Z, Zhu Q, Wang Y, Zheng Q, Yan J. The BET-Bromodomain Inhibitor JQ1 synergized ABT-263 against colorectal cancer cells through suppressing c-Myc-induced miR-1271-5p expression. Biomed Pharmacother. 2017;95:1574–9.
Devaiah BN, Mu J, Akman B, Uppal S, Weissman JD, Cheng D, Baranello L, Nie Z, Levens D, Singer DS. MYC protein stability is negatively regulated by BRD4. Proc Natl Acad Sci USA. 2020;117(24):13457–67.
Zelenetz AD, Salles G, Mason KD, Casulo C, Le Gouill S, Sehn LH, Tilly H, Cartron G, Chamuleau MED, Goy A, et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: results from the CAVALLI phase 1b trial. Blood. 2019;133(18):1964–76.
O’Steen S, Green DJ, Gopal AK, Orozco JJ, Kenoyer AL, Lin Y, Wilbur DS, Hamlin DK, Fisher DR, Hylarides MD, et al. Venetoclax synergizes with radiotherapy for treatment of B-cell lymphomas. Cancer Res. 2017;77(14):3885–93.
Copie-Bergman C. Double-hit DLBCL: should we limit FISH testing? Blood. 2018;131(18):1997–8.
Pham LV, Huang S, Zhang H, Zhang J, Bell T, Zhou S, Pogue E, Ding Z, Lam L, Westin J, et al. Strategic therapeutic targeting to overcome venetoclax resistance in aggressive b-cell lymphomas. Clin Cancer Res. 2018;24(16):3967–80.
Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115(16):3304–13.
Haselager MV, Kielbassa K, Ter Burg J, Bax DJC, Fernandes SM, Borst J, Tam C, Forconi F, Chiodin G, Brown JR, et al. Changes in BCL2 members after ibrutinib or venetoclax uncover functional hierarchy in determining resistance to venetoclax in CLL. Blood. 2020;136(25):2918–26.
Li L, Pongtornpipat P, Tiutan T, Kendrick SL, Park S, Persky DO, Rimsza LM, Puvvada SD, Schatz JH. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia. 2015;29(8):1702–12.
Klanova M, Andera L, Brazina J, Svadlenka J, Benesova S, Soukup J, Prukova D, Vejmelkova D, Jaksa R, Helman K, et al. Targeting of BCL2 Family Proteins with ABT-199 and Homoharringtonine Reveals BCL2- and MCL1-Dependent Subgroups of Diffuse Large B-Cell Lymphoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2016;22(5):1138–49.
Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, Whittington DA, Huang X, Poppe L, Cheng AC, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582–97.
Michie J, Kearney CJ, Hawkins ED, Silke J, Oliaro J. The immuno-modulatory effects of inhibitor of apoptosis protein antagonists in cancer immunotherapy. Cells. 2020;9(1):207.
Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12.
Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, Alexander MD, Kumar PT, Hendi MS, Lee YH, et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem. 2014;57(9):3666–77.
Cong H, Xu L, Wu Y, Qu Z, Bian T, Zhang W, Xing C, Zhuang C. Inhibitor of Apoptosis Protein (IAP) antagonists in anticancer agent discovery: current status and perspectives. J Med Chem. 2019;62(12):5750–72.
Perry AM, Mitrovic Z, Chan WC. Biological prognostic markers in diffuse large B-cell lymphoma.pdf. Cancer Control. 2012;19:214–26.
Rauch A, Hennig D, Schafer C, Wirth M, Marx C, Heinzel T, Schneider G, Kramer OH. Survivin and YM155: How faithful is the liaison? Biochim Biophys Acta. 2014;1845(2):202–20.
Kita A, Mitsuoka K, Kaneko N, Nakata M, Yamanaka K, Jitsuoka M, Miyoshi S, Noda A, Mori M, Nakahara T, et al. Sepantronium bromide (YM155) enhances response of human B-cell non-Hodgkin lymphoma to rituximab. J Pharmacol Exp Ther. 2012;343(1):178–83.
Kaneko N, Mitsuoka K, Amino N, Yamanaka K, Kita A, Mori M, Miyoshi S, Kuromitsu S. Combination of YM155, a survivin suppressant, with bendamustine and rituximab: a new combination therapy to treat relapsed/refractory diffuse large B-cell lymphoma. Clin Cancer Res. 2014;20(7):1814–22.
Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R, et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem. 2011;54(8):2714–26.
DiPersio JF, Erba HP, Larson RA, Luger SM, Tallman MS, Brill JM, Vuagniaux G, Rouits E, Sorensen JM, Zanna C. Oral Debio1143 (AT406), an antagonist of inhibitor of apoptosis proteins, combined with daunorubicin and cytarabine in patients with poor-risk acute myeloid leukemia–results of a phase I dose-escalation study. Clin Lymphoma Myeloma Leuk. 2015;15(7):443–9.
Zinngrebe J, Schlichtig F, Kraus JM, Meyer M, Boldrin E, Kestler HA, Meyer LH, Fischer-Posovszky P, Debatin KM. Biomarker profile for prediction of response to SMAC mimetic monotherapy in pediatric precursor B-cell acute lymphoblastic leukemia. Int J Cancer. 2020;146(11):3219–31.
Vaque JP, Martinez N, Batlle-Lopez A, Perez C, Montes-Moreno S, Sanchez-Beato M, Piris MA. B-cell lymphoma mutations: improving diagnostics and enabling targeted therapies. Haematologica. 2014;99(2):222–31.
Beauchamp E, Yap MC, Iyer A, Perinpanayagam MA, Gamma JM, Vincent KM, Lakshmanan M, Raju A, Tergaonkar V, Tan SY, et al. Targeting N-myristoylation for therapy of B-cell lymphomas. Nat Commun. 2020;11(1):5348.
Lv S, Wen H, Shan X, Li J, Wu Y, Yu X, Huang W, Wei Q. Loss of KMT2D induces prostate cancer ROS-mediated DNA damage by suppressing the enhancer activity and DNA binding of antioxidant transcription factor FOXO3. Epigenetics. 2019;14(12):1194–208.
Sermer D, Pasqualucci L, Wendel HG, Melnick A, Younes A. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol. 2019;16(8):494–507.
Wang G, Chow RD, Zhu L, Bai Z, Ye L, Zhang F, Renauer PA, Dong MB, Dai X, Zhang X, et al. CRISPR-GEMM pooled mutagenic screening identifies KMT2D as a major modulator of immune checkpoint blockade. Cancer Discov. 2020;10(12):1912–33.
Bereshchenko OR, Gu W, Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002;32(4):606–13.
Guan XW, Wang HQ, Ban WW, Chang Z, Chen HZ, Jia L, Liu FT. Novel HDAC inhibitor Chidamide synergizes with Rituximab to inhibit diffuse large B-cell lymphoma tumour growth by upregulating CD20. Cell Death Dis. 2020;11(1):20.
Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282(18):13141–5.
Devaiah BN, Gegonne A, Singer DS. Bromodomain 4: a cellular Swiss army knife. J Leukoc Biol. 2016;100(4):679–86.
Najafova Z, Tirado-Magallanes R, Subramaniam M, Hossan T, Schmidt G, Nagarajan S, Baumgart SJ, Mishra VK, Bedi U, Hesse E, et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017;45(1):127–41.
Ozer HG, El-Gamal D, Powell B, Hing ZA, Blachly JS, Harrington B, Mitchell S, Grieselhuber NR, Williams K, Lai TH, et al. BRD4 profiling identifies critical chronic lymphocytic leukemia oncogenic circuits and reveals sensitivity to PLX51107, a novel structurally distinct BET inhibitor. Cancer Discov. 2018;8(4):458–77.
Vazquez R, Riveiro ME, Astorgues-Xerri L, Odore E, Rezai K, Erba E, Panini N, Rinaldi A, Kwee I, Beltrame L, et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget. 2017;8(5):7598–613.
Kurniyati K, Kelly JF, Vinogradov E, Robotham A, Tu Y, Wang J, Liu J, Logan SM, Li C. A novel glycan modifies the flagellar filament proteins of the oral bacterium Treponema denticola. Mol Microbiol. 2017;103(1):67–85.
Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. 2017;23(4):493–500.
Li W, Gupta SK, Han W, Kundson RA, Nelson S, Knutson D, Greipp PT, Elsawa SF, Sotomayor EM, Gupta M. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J Hematol Oncol. 2019;12(1):73.
Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov. 2017;16(4):241–63.
Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178(5):2623–9.
Tamma R, Ingravallo G, Gaudio F, Annese T, Albano F, Ruggieri S, Dicataldo M, Maiorano E, Specchia G, Ribatti D. STAT3, tumor microenvironment, and microvessel density in diffuse large B cell lymphomas. Leuk Lymphoma. 2020;61(3):567–74.
Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D, Chinnaswamy K, Wen B, Dai L, Kumar P, et al. Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein. J Med Chem. 2019;62(24):11280–300.
Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, Chen J, Yang CY, Liu Z, Wang M, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer cell. 2019;36(5):498–511.
Shastri A, Choudhary G, Teixeira M, Gordon-Mitchell S, Ramachandra N, Bernard L, Bhattacharyya S, Lopez R, Pradhan K, Giricz O, et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J Clin Invest. 2018;128(12):5479–88.
Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6(1):119.
Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7:314ra185.
Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomark Res. 2013;1(1):5.
Rancea M, Will A, Borchmann P, Monsef I, Engert A, Skoetz N. Sixteenth biannual report of the Cochrane Haematological Malignancies Group: focus on Non-Hodgkin’s lymphoma. J Natl Cancer Inst. 2014;106(8):dju170.
Doheny D, Sirkisoon S, Carpenter RL, Aguayo NR, Regua AT, Anguelov M, Manore SG, Arrigo A, Jalboush SA, Wong GL, et al. Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene. 2020;39(42):6589–605.
Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, Szoke A, Drummond M, Pristupa A, Granston T, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4(5):652–9.
Vajpayee N, Thakral C, Gopaluni S, Newman N, Gajra A. Activation of mammalian target of rapamycin in diffuse large B-cell lymphoma: a clinicopathological study. Leuk Res. 2012;36(11):1403–9.
Graf SA, Gopal AK. Idelalisib for the treatment of non-Hodgkin lymphoma. Expert Opin Pharmacother. 2016;17(2):265–74.
Kapoor I, Li Y, Sharma A, Zhu H, Bodo J, Xu W, Hsi ED, Hill BT, Almasan A. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis. 2019;10(12):924.
Yahiaoui A, Meadows SA, Sorensen RA, Cui ZH, Keegan KS, Brockett R, Chen G, Queva C, Li L, Tannheimer SL. PI3Kdelta inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kdelta and BTK inhibitors. PLoS ONE. 2017;12(2):e017221.
Krause G, Hassenruck F, Hallek M. Copanlisib for treatment of B-cell malignancies: the development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des Devel Ther. 2018;12:2577–90.
Ysebaert L, Morschhauser F. Enzastaurin hydrochloride for lymphoma: reassessing the results of clinical trials in light of recent advances in the biology of B-cell malignancies. Expert Opin Investig Drugs. 2011;20(8):1167–74.
Robertson MJ, Kahl BS, Vose JM, de Vos S, Laughlin M, Flynn PJ, Rowland K, Cruz JC, Goldberg SL, Musib L, et al. Phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(13):1741–6.
Civallero M, Cosenza M, Bari A, Sacchi S. Rational combinations of enzastaurin with novel targeted agents for patients with B-cell non-Hodgkin’s lymphoma. Expert Opin Investig Drugs. 2011;20(8):1029–31.
Petrich AM, Leshchenko V, Kuo PY, Xia B, Thirukonda VK, Ulahannan N, Gordon S, Fazzari MJ, Ye BH, Sparano JA, et al. Akt inhibitors MK-2206 and nelfinavir overcome mTOR inhibitor resistance in diffuse large B-cell lymphoma. Clin Cancer Res. 2012;18(9):2534–44.
Wang J, Xu-Monette ZY, Jabbar KJ, Shen Q, Manyam GC, Tzankov A, Visco C, Wang J, Montes-Moreno S, Dybkaer K, et al. AKT hyperactivation and the potential of AKT-targeted therapy in diffuse large B-cell lymphoma. Am J Pathol. 2017;187(8):1700–16.
Yee KW, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, Ferrajoli A, Thomas D, Wierda W, Apostolidou E, Albitar M, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2006;12(17):5165–73.
Merli M, Ferrario A, Maffioli M, Arcaini L, Passamonti F. Everolimus in diffuse large B-cell lymphomas. Future Oncol. 2015;11(3):373–83.
Ansell SM, Tang H, Kurtin PJ, Koenig PA, Inwards DJ, Shah K, Ziesmer SC, Feldman AL, Rao R, Gupta M, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol. 2011;12(4):361–8.
Zoellner AK, Bayerl S, Hutter G, Zimmermann Y, Hiddemann W, Dreyling M. Temsirolimus inhibits cell growth in combination with inhibitors of the B-cell receptor pathway. Leuk Lymphoma. 2015;56(12):3393–400.
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, Nagel ZD, Zou J, Wang C, Kapoor P, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556–62.
Meng X, Matlawska-Wasowska K, Girodon F, Mazel T, Willman CL, Atlas S, Chen IM, Harvey RC, Hunger SP, Ness SA, et al. GSI-I (Z-LLNle-CHO) inhibits gamma-secretase and the proteosome to trigger cell death in precursor-B acute lymphoblastic leukemia. Leukemia. 2011;25(7):1135–46.
Tohda S, Sato T, Kogoshi H, Fu L, Sakano S, Nara N. Establishment of a novel B-cell lymphoma cell line with suppressed growth by gamma-secretase inhibitors. Leuk Res. 2006;30(11):1385–90.
Godfrey JK, Nabhan C, Karrison T, Kline JP, Cohen KS, Bishop MR, Stadler WM, Karmali R, Venugopal P, Rapoport AP, et al. Phase 1 study of lenalidomide plus dose-adjusted EPOCH-R in patients with aggressive B-cell lymphomas with deregulated MYC and BCL2. Cancer. 2019;125(11):1830–6.
Nowakowski GS, LaPlant B, Macon WR, Reeder CB, Foran JM, Nelson GD, Thompson CA, Rivera CE, Inwards DJ, Micallef IN, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33(3):251–7.
Vitolo U, Chiappella A, Franceschetti S, Carella AM, Baldi I, Inghirami G, Spina M, Pavone V, Ladetto M, Liberati AM, et al. Lenalidomide plus R-CHOP21 in elderly patients with untreated diffuse large B-cell lymphoma: results of the REAL07 open-label, multicentre, phase 2 trial. Lancet Oncol. 2014;15(7):730–7.
Wang M, Fowler N, Wagner-Bartak N, Feng L, Romaguera J, Neelapu SS, Hagemeister F, Fanale M, Oki Y, Pro B, et al. Oral lenalidomide with rituximab in relapsed or refractory diffuse large cell, follicular and transformed lymphoma: a phase II clinical trial. Leukemia. 2013;27(9):1902–9.
Goy A, Ramchandren R, Ghosh N, Munoz J, Morgan DS, Dang NH, Knapp M, Delioukina M, Kingsley E, Ping J, et al. Ibrutinib plus lenalidomide and rituximab has promising activity in relapsed/refractory non-germinal center B-cell-like DLBCL. Blood. 2019;134(13):1024–36.
Thieblemont C, Tilly H, Gomes da Silva M, Casasnovas RO, Fruchart C, Morschhauser F, Haioun C, Lazarovici J, Grosicka A, Perrot A, et al. Lenalidomide maintenance compared with placebo in responding elderly patients with diffuse large B-cell lymphoma treated with first-line rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35(22):2473–81.
Jiang L, Sun Y, Wang J, He Q, Chen X, Lan X, Chen J, Dou QP, Shi X, Liu J. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J Exp Clin Cancer Res. 2019;38(1):453.
Kasamon YL, Price LSL, Okusanya OO, Richardson NC, Li RJ, Ma L, Wu YT, Theoret M, Pazdur R, Gormley NJ. FDA approval summary: selinexor for relapsed or refractory diffuse large B-cell lymphoma. Oncologist. 2021;99:1–12.
Dey J, Deckwerth TL, Kerwin WS, Casalini JR, Merrell AJ, Grenley MO, Burns C, Ditzler SH, Dixon CP, Beirne E, et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk diffuse large B-cell lymphoma to BCL2 inhibition. Sci Rep. 2017;7(1):18007.
Liu SH, Gu Y, Pascual B, Yan Z, Hallin M, Zhang C, Fan C, Wang W, Lam J, Spilker ME, et al. A novel CXCR4 antagonist IgG1 antibody (PF-06747143) for the treatment of hematologic malignancies. Blood Adv. 2017;1(15):1088–100.
Falgas A, Pallares V, Unzueta U, Nunez Y, Sierra J, Gallardo A, Alba-Castellon L, Mangues MA, Alamo P, Villaverde A, et al. Specific cytotoxic effect of an auristatin nanoconjugate towards CXCR4(+) diffuse large B-cell lymphoma cells. Int J Nanomed. 2021;16:1869–88.
Lapa C, Hanscheid H, Kircher M, Schirbel A, Wunderlich G, Werner RA, Samnick S, Kotzerke J, Einsele H, Buck AK, et al. Feasibility of CXCR4-directed radioligand therapy in advanced diffuse large B-Cell lymphoma. J Nucl Med. 2019;60(1):60–4.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, Millenson MM, Cohen AD, Schuster SJ, Lebovic D, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol Off J Am Soc Clin Oncol. 2016;34(23):2698–704.
Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, Rodig SJ, Ligon AH, Roemer MGM, Reddy N, et al. Nivolumab for relapsed/refractory diffuse large B-cell lymphoma in patients ineligible for or having failed autologous transplantation: a single-arm, phase II study. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37(6):481–9.
Godfrey J, Tumuluru S, Bao R, Leukam M, Venkataraman G, Phillip J, Fitzpatrick C, McElherne J, MacNabb BW, Orlowski R, et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T-cell-inflamed phenotype. Blood. 2019;133(21):2279–90.
Smith SD, Till BG, Shadman MS, Lynch RC, Cowan AJ, Wu QV, Voutsinas J, Rasmussen HA, Blue K, Ujjani CS, et al. Pembrolizumab with R-CHOP in previously untreated diffuse large B-cell lymphoma: potential for biomarker driven therapy. Br J Haematol. 2020;189(6):1119–26.
Gharaibeh L, Elmadany N, Alwosaibai K, Alshaer W. Notch1 in cancer therapy: possible clinical implications and challenges. Mol Pharmacol. 2020;98(5):559–76.
Chen F, Pisklakova A, Li M, Baz R, Sullivan DM, Nefedova Y. Gamma-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr). 2011;34(6):545–51.
Saltarella I, Frassanito MA, Lamanuzzi A, Brevi A, Leone P, Desantis V, Di Marzo L, Bellone M, Derudas D, Ribatti D, et al. Homotypic and heterotypic activation of the notch pathway in multiple myeloma-enhanced angiogenesis: a novel therapeutic target? Neoplasia. 2019;21(1):93–105.
Aster JC, Blacklow SC, Pear WS. Notch signalling in T-cell lymphoblastic leukaemia/lymphoma and other haematological malignancies. J Pathol. 2011;223(2):262–73.
Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, Hudecek M, Comstock ML, Rajan A, Patel BKR, et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019;134(19):1585–97.
Fabbro D, Bauer M, Murone M, Lehal R. Notch inhibition in cancer: challenges and opportunities. Chimia (Aarau). 2020;74(10):779–83.
Lehal R, Zaric J, Vigolo M, Urech C, Frismantas V, Zangger N, Cao L, Berger A, Chicote I, Loubery S, et al. Pharmacological disruption of the Notch transcription factor complex. Proc Natl Acad Sci USA. 2020;117(28):16292–301.
Miao Y, Medeiros LJ, Li Y, Li J, Young KH. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol. 2019;16(10):634–52.
Karmali R, Gordon LI. Molecular subtyping in diffuse large B cell lymphoma: closer to an approach of precision therapy. Curr Treat Opt Oncol. 2017;18(2):1–17.
Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014;10(6):957–67.
Al-Juhaishi T, McKay J, Sindel A, Yazbeck V. Perspectives on chemotherapy for the management of double-hit lymphoma. Expert Opin Pharmacother. 2020;21(6):653–61.
Suljagic M, Longo PG, Bennardo S, Perlas E, Leone G, Laurenti L, Efremov DG. The Syk inhibitor fostamatinib disodium (R788) inhibits tumor growth in the Emu- TCL1 transgenic mouse model of CLL by blocking antigen-dependent B-cell receptor signaling. Blood. 2010;116(23):4894–905.
Choi MY, Kipps TJ. Inhibitors of B-cell receptor signaling for patients with B-cell malignancies. Cancer J. 2012;18(5):404–10.
Coffey G, Betz A, DeGuzman F, Pak Y, Inagaki M, Baker DC, Hollenbach SJ, Pandey A, Sinha U. The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer. J Pharmacol Exp Ther. 2014;351(3):538–48.
Ma J, Xing W, Coffey G, Dresser K, Lu K, Guo A, Raca G, Pandey A, Conley P, Yu H, Wang YL, et al. Cerdulatinib, a novel dual SYK/JAK kinase inhibitor, has broad anti-tumor activity in both ABC and GCB types of diffuse large B cell lymphoma. Oncotarget. 2015;6(41):43881–96.
Chen Y-B, LaCasce AS. Nzastaurin. Expert Opin Investig Drugs. 2008;17:939–44.
Young RM, Shaffer AL 3rd, Phelan JD, Staudt LM. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin Hematol. 2015;52(2):77–85.
Ferch U, Kloo B, Gewies A, Pfander V, Duwel M, Peschel C, Krappmann D, Ruland J. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2009;206(11):2313–20.
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–9.
Karmali R, Gordon LI. Molecular subtyping in diffuse large B cell lymphoma: closer to an approach of precision therapy. Curr Treat Options Oncol. 2017;18(2):11.
Zhang LH, Kosek J, Wang M, Heise C, Schafer PH, Chopra R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br J Haematol. 2013;160(4):487–502.
Friedberg JW. How I treat double-hit lymphoma. Blood. 2017;130(5):590–6.
Salati M, Tarantino V, Maiorana A, Bettelli S, Luminari S. Durable remission in a patient with leptomeningeal relapse of a MYC/BCL6-positive double-hit DLBCL treated with lenalidomide monotherapy. Hematol Oncol. 2017;35(4):861–3.
Zhu M. Inhibitory effects of bortezomib in a subcutaneous tumor model of H22 mouse hepatocarcinoma cells. Pathol Res Pract. 2019;215(6):152388.
Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009;113(24):6069–76.
Cengiz Seval G, Beksac M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin Drug Saf. 2018;17(9):953–62.
Yazbeck V, Shafer D, Perkins EB, Coppola D, Sokol L, Richards KL, Shea T, Ruan J, Parekh S, Strair R, et al. A phase II trial of bortezomib and vorinostat in mantle cell lymphoma and diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(9):569–75.
Camicia R, Winkler HC, Hassa PO. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Mol Cancer. 2015;14:207.
Tian Z, Zhao JJ, Tai YT, Amin SB, Hu Y, Berger AJ, Richardson P, Chauhan D, Anderson KC. Investigational agent MLN9708/2238 targets tumor-suppressor miR33b in MM cells. Blood. 2012;120(19):3958–67.
Liu WCJ, Tamayo AT, Ruan C, Li L, Zhou S, Shen C, Young KH, Westin J, Davis RE, Hu S, Medeiros LJ, Ford RJ, Pham LV. Preclinical efficacy and biological effects of the oral proteasome inhibitor ixazomib in diffuse large B-cell lymphoma.pdf. Oncotarget. 2017;9:346.
Chitta K, Paulus A, Akhtar S, Blake MK, Caulfield TR, Novak AJ, Ansell SM, Advani P, Ailawadhi S, Sher T, et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenstrom macroglobulinaemia tumour cells. Br J Haematol. 2015;169(3):377–90.
Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921–46.
Luo Q, Pan W, Zhou S, Wang G, Yi H, Zhang L, Yan X, Yuan L, Liu Z, Wang J, et al. A novel BCL2 inhibitor APG-2575 exerts synthetic lethality with BTK or MDM2-p53 inhibitor in diffuse large B-cell lymphoma. Oncol Res. 2020;28(4):331–44.
Ceder S, Eriksson SE, Cheteh EH, Dawar S, Corrales Benitez M, Bykov VJN, Fujihara KM, Grandin M, Li X, Ramm S, et al. A thiol-bound drug reservoir enhances APR-246-induced mutant p53 tumor cell death. EMBO Mol Med. 2021;13(2):852.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24(17):2899–908.
Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, Cluzeau T, Sweet KL, McLemore A, McGraw KL, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol Off J Am Soc Clin Oncol. 2021;39(14):1584–94.
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979–83.
Montesinos PBB, Catalani O, Esteve J, Gamel K, Konopleva MY, Martinelli G, Monnet A, Papayannidis C, Park A, Récher C, Rodríguez-Veiga R, Röllig C, Vey N, Wei AH, Yoon SS, Fenaux P. MIRROS_ a randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Fut Oncol. 2020;16:807–15.
Azizian NG, Li Y. XPO1-dependent nuclear export as a target for cancer therapy. J Hematol Oncol. 2020;13(1):61.
Luo B, Huang L, Gu Y, Li C, Lu H, Chen G, Peng Z, Feng Z. Expression of exportin-1 in diffuse large B-cell lymphoma: immunohistochemistry and TCGA analyses. Int J Clin Exp Pathol. 2018;11(12):5547–60.
Deng M, Zhang M, Xu-Monette ZY, Pham LV, Tzankov A, Visco C, Fang X, Bhagat G, Zhu F, Dybkaer K, et al. XPO1 expression worsens the prognosis of unfavorable DLBCL that can be effectively targeted by selinexor in the absence of mutant p53. J Hematol Oncol. 2020;13(1):148.
Liu Y, Azizian NG, Dou Y, Pham LV, Li Y. Simultaneous targeting of XPO1 and BCL2 as an effective treatment strategy for double-hit lymphoma. J Hematol Oncol. 2019;12(1):119.
Ben-Barouch S, Kuruvilla J. Selinexor (KTP-330) - a selective inhibitor of nuclear export (SINE): anti-tumor activity in diffuse large B-cell lymphoma (DLBCL). Expert Opin Investig Drugs. 2020;29(1):15–21.
Toyokuni S. Mysterious link between iron overload and CDKN2A/2B. J Clin Biochem Nutr. 2011;48(1):46–9.
Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, Do H, Mitchell C, Cullinane C, Liu W, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27(4):590–600.
Rhee K, Bresnahan W, Hirai A, Hirai M, Thompson EA. c-Myc and cyclin D3 (CcnD3) genes are independent targets for glucocorticoid inhibition of lymphoid cell proliferation. Cancer Res. 1995;55(18):4188–95.
Cheng W, Yang Z, Wang S, Li Y, Wei H, Tian X, Kan Q. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur J Med Chem. 2019;164:615–39.
Tanaka Y, Momose S, Tabayashi T, Sawada K, Yamashita T, Higashi M, Sagawa M, Tokuhira M, Rosenwald A, Kizaki M, et al. Abemaciclib, a CDK4/6 inhibitor, exerts preclinical activity against aggressive germinal center-derived B-cell lymphomas. Cancer Sci. 2020;111(2):749–59.
Lacrima K, Rinaldi A, Vignati S, Martin V, Tibiletti MG, Gaidano G, Catapano CV, Bertoni F. Cyclin-dependent kinase inhibitor seliciclib shows in vitro activity in diffuse large B-cell lymphomas. Leuk Lymphoma. 2007;48(1):158–67.
Miljkovic MD, Roschewski M, Dunleavy K, Wilson WH. Hybrid dosing of the cyclin-dependent kinase (CDK) inhibitor flavopiridol in relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma. Leuk Lymphoma. 2019;60(13):3320–3.
Wei M, Zhao R, Cao Y, Wei Y, Li M, Dong Z, Liu Y, Ruan H, Li Y, Cao S, et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur J Med Chem. 2021;209:112.
Jiang B, Gao Y, Che J, Lu W, Kaltheuner IH, Dries R, Kalocsay M, Berberich MJ, Jiang J, You I, et al. Discovery and resistance mechanism of a selective CDK12 degrader. Nat Chem Biol. 2021;17(6):675–83.
Falgas A, Pallares V, Unzueta U, Cespedes MV, Arroyo-Solera I, Moreno MJ, Sierra J, Gallardo A, Mangues MA, Vazquez E, et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica. 2020;105(3):741–53.
Falgas A, Pallares V, Serna N, Sanchez-Garcia L, Sierra J, Gallardo A, Alba-Castellon L, Alamo P, Unzueta U, Villaverde A, et al. Selective delivery of T22-PE24-H6 to CXCR4(+) diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics. 2020;10(12):5169–80.
Nayak L, Iwamoto FM, LaCasce A, Mukundan S, Roemer MGM, Chapuy B, Armand P, Rodig SJ, Shipp MA. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood. 2017;129(23):3071–3.
Rutherford SC, Abramson JS, Bartlett NL, Barta SK, Khan N, Joyce R, Maddocks K, Ali-Shaw T, Senese S, Yuan Y, et al. Venetoclax with dose-adjusted EPOCH-R as initial therapy for patients with aggressive B-cell lymphoma: a single-arm, multicentre, phase 1 study. Lancet Haematol. 2021;8(11):e818–27.
Davies A, Cummin TE, Barrans S, Maishman T, Mamot C, Novak U, Caddy J, Stanton L, Kazmi-Stokes S, McMillan A, et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): an open-label, randomised, phase 3 trial. Lancet Oncol. 2019;20(5):649–62.
Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A, Parker J, Hafeez A, Tuck D, Younes A. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica. 2017;102(11):1923–30.
Ji J, Liu Z, Kuang P, Dong T, Chen X, Li J, Zhang C, Liu J, Zhang L, Shen K, et al. A new conditioning regimen with chidamide, cladribine, gemcitabine and busulfan significantly improve the outcome of high-risk or relapsed/refractory non-Hodgkin’s lymphomas. Int J Cancer. 2021;149(12):2075–82.
Brem EA, Li H, Beaven AW, Caimi PF, Cerchietti L, Alizadeh AA, Olin R, Henry NL, Dillon H, Little RF, et al. SWOG 1918: A phase II/III randomized study of R-miniCHOP with or without oral azacitidine (CC-486) in participants age 75 years or older with newly diagnosed aggressive non-Hodgkin lymphomas—aiming to improve therapy, outcomes, and validate a prospective frailty tool. J Geriatr Oncol. 2022;13(2):258–64.
Martin P, Bartlett NL, Chavez JC, Reagan JL, Smith SM, LaCasce AS, Jones J, Drew J, Wu C, Mulvey E, et al. Phase 1 study of oral azacitidine (CC-486) plus R-CHOP in previously untreated intermediate- to high-risk DLBCL. Blood. 2022;139(8):1147–59.
Seymour EK, Khan HY, Li Y, Chaker M, Muqbil I, Aboukameel A, Ramchandren R, Houde C, Sterbis G, Yang J, et al. Selinexor in combination with R-CHOP for frontline treatment of non-hodgkin lymphoma: results of a phase I study. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27(12):3307–16.
Schoch LK, Asiama A, Zahurak M, Shanbhag S, Hurtt J, Sawyer K, Swinnen LJ, Wagner-Johnston N, Jones RJ, Ambinder RF, et al. Pharmacokinetically-targeted dosed everolimus maintenance therapy in lymphoma patients. Cancer Chemother Pharmacol. 2018;81(2):347–54.
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92.
Dubois S, Mareschal S, Cornic M, Picquenot J-M, Bertrand P, Bohers E, Maingonnat C, Viailly P-J, Ruminy P, Alcantara M, et al. Targeted EZH2 inhibitors in diffuse large B-cell lymphoma (DLBCL): immunohistochemical and mutational profiles of patients may determine candidates for treatment. Blood. 2014;124(21):1656–1656.
Acknowledgements
Not applicable.
Funding
The study was supported by the National Natural Science Foundation of China (81973172, 82003579), Natural Science Foundation of Zhejiang Province (LR21H300003, LQ21H300005), Science and Technology Program of Traditional Chinese Medicine, Zhejiang (2021ZB038).
Author information
Authors and Affiliations
Contributions
Y. Z., J. C., M. W., Y. G. and Y. X. contributed to the collection of references and manuscript preparation. X. D. and H. Y. contributed to the plan and modification of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The content of this manuscript has not been previously published and is not under consideration for publication elsewhere. All the authors are aware of and agree with the content of the paper and are listed as co-authors of the paper.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhuang, Y., Che, J., Wu, M. et al. Altered pathways and targeted therapy in double hit lymphoma. J Hematol Oncol 15, 26 (2022). https://doi.org/10.1186/s13045-022-01249-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01249-9