
Abstract
The members of the Poly(ADP‐ribose) polymerase (PARP) superfamily are involved in several biological processes and, in particular, in the DNA damage response (DDR). The most studied members, PARP1, PARP2 and PARP3, act as sensors of DNA damages, in order to activate different intracellular repair pathways, including single-strand repair, homologous recombination, conventional and alternative non-homologous end joining. This review recapitulates the functional role of PARPs in the DDR pathways, also in relationship with the cell cycle phases, which drives our knowledge of the mechanisms of action of PARP inhibitors (PARPi), encompassing inhibition of single-strand breaks and base excision repair, PARP trapping and sensitization to antileukemia immune responses. Several studies have demonstrated a preclinical activity of the current available PARPi, olaparib, rucaparib, niraparib, veliparib and talazoparib, as single agent and/or in combination with cytotoxic, hypomethylating or targeted drugs in acute leukemia, thus encouraging the development of clinical trials. We here summarize the most recent preclinical and clinical findings and discuss the synthetic lethal interactions of PARPi in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). Despite the low frequency of genomic alterations of PARP and other DDR-related genes in acute leukemia, selective vulnerabilities have been reported in several disease subgroups, along with a “BRCAness phenotype.” AML carrying the RUNX1-RUNX1T1 or PML-RARA fusion genes or mutations in signaling genes (FLT3-ITD in combination with TET2 or TET2 and DNMT3A deficiency), cohesin complex members (STAG2), TP53 and BCOR as co-occurring lesions, IDH1/2 and ALL cases expressing the TCF3-HLF chimera or TET1 was highly sensitive to PARPi in preclinical studies. These data, along with the warning coming from the observation of cases of therapy-related myeloid malignancies among patients receiving PARPi for solid tumors treatment, indicate that PARPi represents a promising strategy in a personalized medicine setting. The characterization of the clonal and subclonal genetic background and of the DDR functionality is crucial to select acute leukemia patients that will likely benefit of PARPi-based therapeutic regimens.
Similar content being viewed by others

Background
The Poly(ADP‐ribose) polymerase (PARP) superfamily is composed by 17 proteins involved in crucial biological processes such as DNA damage response (DDR), transcription, chromatin structure stabilization, cell metabolism, telomere length maintenance, antiviral response and cell signaling (Table 1) [1]. So far, eight PARP proteins (PARP1, PARP2, PARP3, PARP5, PARP5b, PARP6, PARP9 and PARP14) have been recognized as key factors involved in the maintenance of genetic stability as they control DNA damage repair and cell cycle regulation [2]. PARP proteins act as mediator of the initial phases of the response to DNA damages. Indeed, they interact with DNA damaged sites [3, 4] and promote the recruitment of additional DNA repair proteins [5]. To facilitate the localization of DDR proteins to the site of damage, PARP proteins transfer ADP-ribose residues from NAD+ to target substrates (DNA or proteins) [6]. The majority of PARPs enzymes (n = 12) create mono-(ADP-ribose) modification on their targets (Mono-ADP-Ribose [MAR]ylation), and four of them extend the initial modification site to form poly(ADP-ribose), or PAR chains (PARylation) [7]. This post-translational modification regulates the conformation, stability and activity of the targeted proteins. The generation of PAR chains is crucial to promote DNA damage repair thorough two different mechanisms: (1) the rapid and efficient PAR-dependent recruitment of DNA repair factors and histones on the site of the damage [5], (2) the PARylation of DNA repair factors that consequently promotes their association to the DNA and their interaction with other proteins involved in the repair cascade [8].
This review aims to discuss the potentials of PARP inhibitors (PARPi)-based therapeutic strategies in acute leukemia. To this aim, we first summarize the role of PARP proteins in the DDR and the mechanisms of action of PARPi, which helps understanding their preclinical and clinical successes and failures in acute leukemias. We then focus on synthetic lethal interactions of PARPi in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), which opens a promising therapeutic window for specific disease subgroups.
PARP enzymes in the response to DNA damages
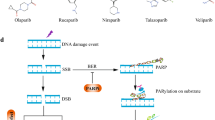
PARP1, PARP2 and PARP3 are the most studied enzymes for their involvement in the DDR. In mammalian cells, they act as DDR sensors in response to different types of DNA damages. In particular, PARP1 responds to single-strand breaks (SSBs), DNA cross-links, stalled replication forks and double-strand breaks (DSBs) [25]. PARP2 seems to recognize more specifically gaps and flap structures [26], while PARP3 was described to respond more selectively to DSBs [9, 27]. Briefly, PARP1, PARP2 and PARP3 cooperate with several mediators in the DDR through the activation of different intracellular repair pathways such as single-strand repair (SSR), homologous recombination (HR), conventional non-homologous end joining (cNHEJ) and alternative non-homologous end join (aNHEJ). The following sections summarize the role of PARP proteins and, in particular of PARP1, in the response to SSBs and DSBs and their role in the activation of different repair pathways.
PARPs in base excision repair and single-strand breaks repair
Base modifications can arise by endogenous sources or errors during DNA replication and both PARP1 and PARP2 enzymes play a central role in their repair throughout base excision repair/single-strand break repair process (BER/SSBR) [28]. PARP1 PARylates a variety of substrates, thus promoting the accumulation repair factors, which in turn interact with single-strand DNA (ssDNA) and act as scaffold for other repair factors [29,30,31]. At first, the mismatched base is cleaved by a DNA glycosylase generating an apurinic/apyrimidinic site (AP, Fig. 1A). Then, apurinic/apyrimidinic Endonuclease 1 (APE1) removes the AP site generating a ssDNA nick which is recognized and processed as a SSB by PARP1[32]. In details, after PARylation, PARP1 interacts with proteins such as DNA polymerase β (Polβ), DNA ligase III (LIG3), X‐ray repair cross‐complementing protein 1 (XRCC1) and bifunctional polynucleotide kinase 3′-phosphatase (PNKP) which are recruited at the SSB site in the repair process[33]. Another type of single base DNA lesion, ssDNA nicks, require the activity of PARP1 to be repaired. ssDNA nicks are generated by the deregulated activity of DNA topoisomerase 1 (TOP1) [34]. Indeed, TOP1 relaxes topological stress in the DNA structure by generating a cut on one DNA strand, controlling its rotation around the intact strand and, finally, removing the nick (Fig. 1B). The entire process can be interrupted, resulting in the generation of abortive TOP1–DNA complexes (TOP1 cleavage complexes, TOP1cc). TOP1cc is removed from the DNA by tyrosyl-DNA phosphodiesterase 1 (TDP1) which is a target of PARP1 [35, 36]. The exposed nick can be a substrate for SSBR [36].

Mechanism of action of PARP1 in base excision repair (A) and single-strand DNA nick repair (B)
PARPs in double-strand breaks repair
In eukaryotic cells, DSBs are repaired through HR, cNHEJ or aNHEJ repair pathways, according to the cell cycle phase [37]. PARP1 plays a critical role in DSB sensing and its recruitment and activation occur within 100 ms after introduction of DSBs [38]. Thus, PARP-1 activation is one of the earliest events in the sensing of DSBs. Independently of the downstream pathway, PARP1 recognizes DSBs and responds to it by recruiting the initial mediators of the DSBs repair response. The choice between the different DNA repair pathways is related to the cell cycle phases (as already mentioned), type of DNA damages, organism and to the proficiency of these pathways [39, 40]. It has been showed that cNHEJ and aNHEJ repair systems repair more efficiently DSBs in comparison with HR [39]. In eukaryotic cells with complex genome, the HR system is preferentially use to repair DSBs that can be generated during DNA replication [41]. The following sections briefly summarize the role of PARP1 in these three repair pathways (Fig. 2).

Mechanism of action of PARP1 in cNHEJ, aNHEJ and HR repair according to the cell cycle phases
PARP1 in conventional non-homologous end joining repair
cNHEJ is the chosen mechanism of DDR during interphase (G0/G1 in particular) [42]. PARP1 PARylates the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), which is a crucial NHEJ factor [43] (Fig. 2A). PARylation stimulates the kinase activity of DNA-PKcs and the requirement of the KU70–KU80 complex [44]. KU70-KU80 complex promotes the localization and activation of the DNA ligase IV/XRCC4 complex on DNA end. Before DNA ligase IV ligation, DNA ends need to be processed by the concomitant action of the nuclease Artemis and aprataxin-polynucleotide kinase-like factor (APLF). After this process, DNA ligase IV complex with XRCC4 and XRCC4-like factor (XLF) to rejoin the DNA ends [45].
PARP1 in alternative non-homologous end joining repair
PARP1 has a crucial role in the regulation of aNHEJ repair, that is mostly active in S and G2 phases of cell cycle [46, 47] (Fig. 2B). In addition, the cell may also use this pathway during G1 phase to respond to DSBs [48, 49]. PARP1 is involved in the first steps of the repair process and, in particular, it promotes the localization of the MRN-CtIP complex on the DNA ends [50]. The CtIP promotes the endo/exonuclease activity of MRN resulting in exposition of microhomology sequences of ssDNA [51]. Then, PARP1, MRN complex and the DNA polymerase θ (Polθ) promote the bridging and alignment of the DNA single strands via the microhomology sequences. The 3' sequences that have no homology are digested by ERCC1 and XPF nucleases. Finally, the generated gaps are filled by Polθ-mediated DNA synthesis and the remaining nicks are repaired by the LIG3-XRCC1 complex [52, 53].
PARP1 in homologous recombination repair
After DNA replication, DSs are preferentially repaired through the HR repair [54]. PARP1 promotes the recruitment and activation of different downstream partners during the process (Fig. 2C), including the MRE11-RAD50- NBS1 (MRN) complexes, which is involved in the response to DSBs via HR [55]. PARP1 is important for the early and rapid recruitment of BRCA1 to DSBs. BRCA1 function is to promote DSB ends resection [56,57,58]. Different studies have highlighted that BRCA1 can be loaded to DSBs independently of PARP1 activity and, mainly, following DNA damage-mediated ubiquitylation [59]. The localization of BRCA1 to DSBs is enhanced by the PARylation of BRCA1-associated RING domain protein 1 (BARD1) on the site of damage. BRCA1-BARD1 complex promotes the endonuclease activity of MRE11 which generates single-strand DNA on the site of damages [60, 61]. Resected DNA is a substrate for RAD51 binding, but it is initially bound by the replication protein A (RPA), requiring mediator proteins to assist RAD51 loading onto single-strand DNA. RAD51 promotes strand-exchange of single-strand DNA filament that invades an unbroken homologous DNA which is typically the sister chromatid [62]. The generation of single-strand DNA is fundamental for the final steps of HR repair and in particular for the mechanism of strand invasion which promotes DSBs repair [63]. The resection activity of the complex BRCA1-BARD1-MRE11 is limited by the activity of PARP1 which promotes the PARylation of BRCA1 and the association of the receptor-associated protein 80 (RAP80). The interaction with RAP80 stabilizes BRCA1 and suppresses HR, which results in limited strand invasion and in the subsequent repair of the DSB.
Genomic alterations of PARPs and DDR genes interactors in acute leukemia
PARP genomic alterations in acute leukemia
According to public genomic data available on acute leukemias on the cBio portal [64] (TCGA-LAML [65] and Beat AML [66] datasets) and our cohort (NGS-PTL [67, 68]), there is no evidence of genomic alterations of PARP1 in adult patients and few cases have been reported in pediatric cohorts [64] (2/295 in TARGET-AML [69], 0.7%; 8/819 in TARGET-ALL [70], 1.0%, Fig. 3A and Table 2). Conversely, alterations have been reported in PARP2: 2.7% of pediatric ALL cases showed gene amplifications (22/819 patients, 2.7%) or deletions (1/819 patients, 0.1%), while they accounted for 1.4% of pediatric AML cases (2/295 patients with deletions, 0.7% and 2/295 patients with amplification, 0.7%). In adults, PARP2 was found mutated in one case and amplified in another one in the TCGA-LAML cohort (2/200 patients, 1.0%). PARP3 was deleted in two cases of pediatric AML and ALL (1/819 ALL TARGET and 1/295 AML-TARGET, 0.1% and 0.3%, respectively) and mutated in one patient from the Beat AML cohort (1/622, 0.2%).

Genomic alterations of PARP1/2/3 and BRCA1/2 in pediatric AML and ALL TARGET study and adult AML cohorts (Beat AML, TCGA-LAML and NGS-PTL). A Oncoprint of genomic alterations of PARP1, PARP2, PARP3, BRCA1 and BRCA2 acute leukemia cohorts. B Frequency and type of genomic alterations
Overall, the genomic lesions preferentially include amplifications of the PARP2 gene (Fig. 3B), with potential gain of function consequences providing a selective advantage to the malignant cells in terms of ability to maintain a tolerable dose of genetic instability.
Genomic alterations of BRCAs and other DDR genes in acute leukemia
Mutations, copy number alterations and/or polymorphisms of other DDR genes have been described, that may result in the deregulation of the HR pathway activity. Mutations in the PARP1/2/3 downstream partners BRCA1/2 genes have been reported in adult AML, with a frequency around 1.9% in the NGS-PTL AML cohort (BRCA1: 3/162 cases, BRCA2: 4/162 cases) and below 1.0% in the Beat AML study (BRCA1: 1/622 cases; BRCA2: 2/200). Deletions, missense and truncating mutations, that likely induce loss of function phenotypes, are the most recurrent alterations in BRCA1 and BRCA2 genes (Table 2), as expected based on the role of the encoded proteins [71, 72]. The presence of polymorphisms in the HR genes RAD51 (135C) and its paralog XRCC3 (241M) has been associated with an increased risk of de novo and t-AML [73]. Deletion of Mre11A and ATM on chromosome 11 has been described in t-AML patients and leads to alterations in both NHEJ and HR as Mre11A is an early factor in these two pathways [74]. Alterations in the Fanconi Anemia (FA) pathway have also been observed, which, as first consequence, results in the development of FA, but about 9% of patients subsequently develop AML with a high incidence of chromosomal breakage [75]. Heterozygous deletions and distinct point mutations in the FANCA gene were found in a small percentage of AML patients [76] and have been associated with those cases carrying chromothripsis [77]. In T-ALL, a FANCC point mutation was identified [78]. Genomic alterations in other DDR genes such as ATM, PRKDC, ATR, RPA1, DSS1, NBN, RAD51, RAD54, CHEK1, CHEK2, ERCC1, POLB, FEN1 and CDK12 have shown synthetic lethality in combination with PARPi [79] and are altered at a frequency ≤ 1% in acute leukemias [64].
Moreover, we have recently reported that about 5% of AML cases carry heterozygous copy number loss of PALB2, a gene of the FA pathway that interacts with BRCA1 and BRCA2 in the DNA damage repair [80]. Patients with PALB2-mutated solid tumors have been reported to be responsive to PARPi [81,82,83], and a phase II clinical trial showed that olaparib is an effective treatment for patients with metastatic breast cancers carrying germline mutations in PALB2 [84]. In addition, another phase II trial is enrolling patients with advanced HER2-negative breast cancer and other solid tumors with mutations in HR genes (excluding BRCA1/2) for the evaluation of the treatment with talazoparib (NCT02401347). Therefore, PALB2 deletion is a potential marker of HR defects, that may also suggest a potential vulnerability to PARP inhibition in AML.
PARP inhibitors: mechanism of actions
Inhibition of single-strand breaks and base excision repair
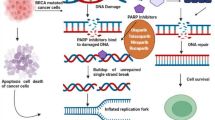
As already mentioned, SSBs arise frequently in proliferating cells and they are repaired through PARP-dependent DNA repair mechanisms (e.g., BER). Efficient SSB repair is essential for the survival of proliferating cells (Fig. 4A). All the developed PARPi act on the catalytic activity of PARP enzymes and, thus, on the repair of SSBs by BER. As a consequence, unrepaired SSBs can be converted to the more cytotoxic DSBs that, if not repaired, cause cell death [85] (Fig. 4B).

Schematic representation of PARPi mechanism of action. A SSBs are normally identified and repaired by PARP1 and B the addition of a PARPi compromises the repair and the SSB is converted in DSBs. C PARP1 inhibitors can generate PARP-DNA complexes that during DNA replication can promote DNA replication forks collapse and, consequently, the generation of DSBs. The fate of DSBs depends on HR proficiency of the cancer cells. If the cells are HR proficient, DSBs are repaired and the cells survive, on the contrary, DSBs are not repaired, damages accumulate during replication until cancer cells’ death
PARP trapping and generation of PARP-DNA complexes
As an additional mechanism of action, PARPi can trap PARP1 and PARP2 enzymes on DNA [86] (Fig. 4C). It has been showed that PARP1 is the crucial factor in DNA trapping by PARPi, as its depletion, rather than PARP2 depletion, reduces the sensitivity to PARP inhibition [86]. PARP entrapment can occur on DNA strand break sites as well as TOP1-processed ribonucleotides or on Okazaki-fragment intermediates during DNA replication [87]. Once trapped, PARP1 cannot dissociate from DNA due to inhibition of its catalytic activity, which is required to release PARylated PARP1 from the DNA. PARP1-DNA complexes are highly toxic for replicating cells. Firstly, they block DNA replication by interfering with the protein complexes at replication forks [88]. This event can culminate in the collapse of replicative forks and the generation of DSBs. Moreover, these processes prevent the accessibility of other repair proteins to the damaged sites [89,90,91]. While catalytic inhibitory effects of the clinically available PARPi are similar, their ability in trapping PARP–DNA complexes varies considerably. Talazoparib exhibits the highest trapping efficiency while Veliparib the lowest (Table 3).
Immunomodulatory activity of PARPi in acute leukemia
In addition to a direct activity on the DDR pathway, PARPi exert an immunomodulatory function, as demonstrated in multiple cancer types. Numerous lines of evidence now suggest that the DDR plays an important role in driving sensitivity and response to immune checkpoint blockade [92]. First, DDR dysfunction, and in particular HR deficiency, can increase the tumor mutational burden [93], which in turn can lead to the generation of neoantigens, and in particular mutation-associated neoantigens can enhance anticancer T cell activity [94]. Second, S phase-specific DNA damages in cells with DDR-related gene alterations or under treatment with PARPi result in the activation of the stimulator of interferon genes pathway, that promotes T cell infiltration and activity [95, 96], but is also associated with upregulation of immunosuppressive PD-L1 expression [97]. Therefore, PARPi, by enhancing tumor immunogenicity through increased tumor mutational burden, neoantigen release and PD-L1 expression, create the ideal microenvironment for combined treatments with immunostimulatory drugs [98]. This evidence, which comes from solid tumors, deserve further investigation in acute leukemias. Moreover, PARPi can counteract immune escape in AML. Malignant cells in monocytic leukemia, but not in myeloblastic and immature disease subtypes, produce reactive oxygen species (ROS) through NADPH oxidase and in turn induce PARP1-dependent apoptosis of natural killer (NK), CD4+ T and CD8+ T cells [99]. In parallel, genotoxic stress can induce the upregulation of NKG2D ligands on tumor cells, a process mediated by ATM [100]. Binding of NKG2D ligands to the NKG2D receptor on the surface of NK cells and activated CD8+ T cells functions as a costimulatory signal. Paczulla and colleagues showed that PARP1 represses NKG2D ligands expression in leukemia stem cells, thus contributing to their selective escape from immune surveillance by NK cells [101]. Accordingly, PARPi induced the expression of NKG2DLs on the surface of AML cells, but not on healthy hematopoietic stem and progenitor cells. Therefore, the inhibition of PARP1 followed by NK cells transplant suppressed leukemogenesis in PDX models. In addition, PARPi were reported to sensitize AML cell lines and primary cells to human tumor necrosis factor- α-related apoptosis-inducing ligand (TRAIL), a key effector molecule of NK cells, by activating the expression of FAS and TNFRSF10B proteins through enhanced the binding of the transcription factor Sp1 to the promoter) [102]. These data open a new scenario for a rationale usage of PARPi in AML in order to maximize the patients’ benefit, including conditions of functional immune response, like minimal residual disease, or in combination with immunotherapies, as immune checkpoint inhibitors, allogeneic stem cell transplantation, NK cell infusion, recombinant TRAIL, small molecules or monoclonal antibodies functioning as receptor agonists that are currently under investigation.
Preclinical data of currently available PARP inhibitors in acute leukemias
This section summarizes the main preclinical data available regarding the efficacy of different PARPi in acute leukemias.
Olaparib
Olaparib (AZD-2281, Ku-0059436) is an orally bioavailable and well-tolerated PARPi. Olaparib binds four members of the PARP super family, PARP1-PARP4 [103] and is the most studied PARP inhibitor in acute leukemia cells. Several selective vulnerabilities to olaparib have been demonstrated in AML and ALL molecular subgroups, that will be better described in “Synthetic lethality and PARP inhibitors: a new therapeutic window for acute leukemia?” section. Based on the evidence that olaparib induces NF-κB-mediated upregulation of FAS [104] and that IKKβ inhibition suppresses HR, PARylation and NHEJ [105] in AML cells, the combined inhibition of the two molecules has been tested. Inactivation of the NF-κB pathway through IKKβ inhibition and olaparib potentiated the efficacy of danunorubin in AML cells (RUNX1-RUNX1T1 rearranged AML cell line, KASUMI1; erythroleukemia cell line, KG1a) and xenograft models (mice transplanted with KG1a cells), both in monotherapy and when used in combination, by increasing apoptosis and reducing cell proliferation [105]. Moreover, olaparib caused synthetic lethality in combination with decitabine in AML models (KG1a; APL cell line, HL-60; KMT2A-AFF1 rearranged AML cell line, MV4-11; FLT3-ITD AML cell line, PL21) by disrupting BER, which was required for repairing decitabine-induced DNA lesions through recruitment of XRCC1 at DNMT1 foci and repair of trapped DNMT1 [106]. Impairment of HR by WEE1 inhibition was instead responsible for the synergism with olaparib in AML (MV4-11; KMT2A-MLLT3 rearranged AML cell line, MOLM13; NPM1/DNMT3A double mut AML cell line, OCI-AML3) and ALL (ETV6-RUNX1 rearranged pre B-ALL cell line, REH) cell lines, resulting in DNA damage accumulation and cell death by apoptosis, as also confirmed in an AML murine model [107]. The combination also reduced the proliferative and clonogenic capacity of primary AML cells and induced cell death by apoptosis.
Rucaparib
Rucaparib (Rubraca/AG-0146991) is an orally bioavailable PARPi, with cancer cytotoxicity levels comparable to olaparib [108]. A recent comprehensive characterization of the target kinase landscape of four FDA-approved PARP drugs found that rucaparib inhibits PARP1, PARP2, PARP3, PARP4, TNSK1 and TNSK2 at nanomolar concentrations [109]. Few preclinical studies have evaluated the efficacy of rucaparib in hematological malignancies. Recently, rucaparib has been tested as single agent or in combination with the antimetabolite 5-fluoro-Uracile (5FU) in vitro (DNMT3A mut OCI-AML2 and T-ALL RPMI-8402 cells) and in vivo in AML (mice transplanted with KMT2A-MLLT3 AML primary cells) and ALL (mice transplanted with pre-T-ALL primary cells) showing elevated efficacy [110]. Upon rucaparib treatment, leukemic cells accumulated low levels of DNA damage and delayed cell cycle progression until S phase arrest. The addition of 5FU in S phase-arrested leukemic cells caused the accumulation of massive DNA damages, probably as a consequence of replicative forks collapse [110].
Niraparib
Niraparib (MK-4827) is an orally bioavailable PARP1/2 inhibitor, also showing cancer cytotoxicity levels comparable to olaparib [108]. Few studies are available on the efficacy of niraparib in acute leukemias. Niraparib, as well as other PARPs inhibitors, was effective against arsenic trioxide (ATO)-resistant acute promyelocytic leukemia (APL) primary cells alone and in combination with hypomethylating agents (azacitidine and decitabine) or high-dose vitamin C (ascorbate) [111]. The synergism was dependent on the role of PARP1 protein in the processing of DNA demethylation. The triple combination of niraparib, decitabine and histone deacetylase inhibitors (HDACi) also showed a synergistic effect on AML cell lines (FLT3-ITD cells MOLM-14) with activation of the ATM pathway, increased production of ROS, decreased mitochondrial membrane potential, and induction of apoptosis. The efficacy of the triple combination was confirmed also on primary leukemic cells (de novo AML, secondary AML, T cell prolymphocytic leukemia and Mixed phenotypes ALL) in which the combination induced DNA damages (H2AX marker) and triggered apoptosis (cleaved PARP1 and Caspase 9) [112]. The mechanism behind the efficacy of the combination consists of trapping of PARP1 and DNMT1 to chromatin, acetylation of DNA repair proteins, and downregulation of the nucleosome-remodeling deacetylase (NuRD) complex that converged toward induction of DSBs, resulting in leukemic cell death.
Veliparib
Veliparib showed modest cytotoxic activity in ALL and AML cell lines in single agent, obtaining a significant reduction in the cell viability only at concentration 5- to 33-fold higher than plasma concentrations achieved in either animals or humans [113,114,115]. However, it was shown to potentiate the efficacy of the alkylating agent temozolomide (TMZ) in different ALL and AML cell lines and, in particular, in mismatch repair (MMR)-deficient ALL (T-ALL: MOLT4 and HBS2; REH) cell lines with low O6-methylguanine-DNA methyltransferase (MGMT) activity. Indeed, MMR and MGMT system promote the elimination of the modified nucleotides generated by TMZ. In primary leukemic cells proficient for MMR and with heterogeneous MGMT activity, the authors found that the efficacy of veliparib and TMZ depended to the leukemia subtype. Indeed, veliparib did not significantly potentiate TMZ activity in primary ALL leukemia cells but enhanced the growth-inhibitory effects of TMZ in AML primary leukemias [113].
Talazoparib
Talazoparib is nowadays the most potent PARP inhibitor due to its ability to induce strong PARP trapping [116]. The number of preclinical and clinical studies evaluating the efficacy of talazoparib against acute leukemia is constantly growing. Kohl and colleagues demonstrated a significant antileukemic activity of talazoparib as monotherapy and in combination with decitabine in primary CD34+ AML, MDS and CMML samples [117]. The strong efficacy of the drug combination might be related to the generation of highly cytotoxic PARP1-DNA, DNMT1-DNA and PARP1-DNMT1-DNA complexes. Moreover, hypomethylating agents induced downregulation of RAD51, BRCA1, BRCA2, or HR-related genes, FEN1 or FANCD2 [118], which increased PARPi efficacy in AML, including FLT3-ITD and complex karyotype leukemic cells [119]. Strong synergism was also found when combining talazoparib with the novel SAHA-bendamustine hybrid, NL101, in AML, both in vitro (MV4-11; HL-60; RUNX1-RUNX1T1 rearranged AML cell line, KASUMI1) and in the xenograft model (MV4-11 cells). The authors demonstrated that the combination induced cell apoptosis and cell cycle arrest in G2/M phase and promoted DNA damage [120]. Moreover, HDAC inhibitors were shown to enhance PARP1 binding to DSBs and PARP trapping was further exacerbated by combined treatment with talazoparib, resulting in increased apoptosis [121].
Synthetic lethality and PARP inhibitors: a new therapeutic window for acute leukemia?
PARPi can act through synthetic lethality, whereby genetic DNA repair defects are enhanced by drug-induced defects in a compensatory pathway [122]. Today, it is well established that PARPi are specifically effective in HR-deficient cells harboring, for example, inactivating mutation of the BRCA1/2 genes [119, 122, 123]. Indeed, PARP inhibition results in an increase in SSBs, that in turn degenerate into DSBs during DNA replication and become highly cytotoxic in BRCA1/2-deficient cells owing to their reduced capacity for DSB repair. Despite the low frequency of BRCA1/2 mutations in acute leukemia, expression changes have been reported. Scardocci and colleagues showed that the median expression of BRCA1 mRNA in AML samples is lower compared to normal bone marrow [124]. BRCA1 downregulation was mediated by promoter hypermethylation and was associated with chromosomal aberrations or therapy-related AML. Of note, PARP1 overexpression and BRCA1 proficiency are predictive of resistance to ex vivo olaparib treatment in AML blasts, whereas formation of γH2AX foci is a marker of sensitivity [125].
During the last decades, it has become clear that oxidative stress and genomic instability, due to mutations in DNA repair genes but also to replication stress, sensitize cancer cells to PARP inhibition [126,127,128,129]. In particular, a “BRCAness phenotype,” has been defined for those tumors that share with BRCA1/2 germline-mutated cases an impaired functionality of the HR pathway, despite carrying functional BRCA1 and BRCA2 [130]. The “BRCAness phenotype” has been also described in acute leukemia. Moreover, heterozygous deletion of PALB2 is a potential marker of HR defects, that may also suggest a potential vulnerability to PARP inhibition [81].
Selective vulnerabilities of PARP inhibitors in acute myeloid leukemia
This section reports data on sensitivity and resistance biomarkers to PARP inhibitors, which are summarized in Table 4.
Fusion genes
Several AML-related molecular alterations, including fusion proteins and mutations, were demonstrated to induce a selective vulnerability to PARPi, through deregulation of the DDR pathway (Fig. 5A and B). Fusion genes are detected in almost 20% of AML patients and usually drive leukemogenesis [145, 146]. Different studies investigated the perturbations deriving from the expression of chimeras in AML and described a relationship between the expression of fusion genes and the response to PARPi. In human CD34+ umbilical cord blood cells expressing RUNX1-RUNX1T1, genes involved in the FA pathway (BRCA2, FANCA, FANCL, FANCF), together with genes in the ATM and ATR and the BER pathways were downregulated, compared with control cells [147]. Consistently, transformed murine hematopoietic cells expressing the RUNX1-RUNX1T1 chimera displayed a compromised functionality of the HR pathway, as proved by the reduced levels of RAD51, ATM, BRCA1 and BRCA2 [131]. Both murine and human AML expressing the chimera responded to olaparib treatment in vitro and in vivo. Moreover, RUNX1-RUNX1T1-driven leukemia is characterized by a mutator phenotype, which implies a predisposition to the acquisition of mutations, both spontaneously and under the pressure of genotoxic agents [148]. This observation has two major consequences: the downside is that chemotherapy can induce the evolution of resistant clones by accumulation of novel mutations, while the upside is that, given their high level of immunogenicity, residual cells may be targeted by combined treatment with PARPi and immunotherapy.

Selective vulnerabilities of PARP inhibitors in AML and ALL. A The known molecular alterations that modify the sensitivity of AML cells to PARPi are represented on the two sides of the balance. Green and red indicate molecular alterations enhancing and hampering PARPi sensitivity, respectively. B Schematic representation of HR and NHEJ repair pathways in response to DSBs. In the scheme, red and green arrows represent the level of expression of different genes associated with AML subtypes (low and high expression, respectively). C The known molecular alterations that modify the sensitivity of ALL cells to PARPi are represented on the two sides of the balance. Green indicates molecular alterations enhancing PARPi sensitivity. To date, no molecular alterations hampering PARP sensitivity have been identified in ALL
High sensitivity to PARPi was also reported in human and murine models expressing the PML-RARA fusion gene [131]. The specificity of the driver fusion protein toward susceptibility to DNA damage accumulation is further supported by the evidence that a KMT2A rearrangement induced a modest sensitivity to PARPi. Two independent studies investigated the effect of PARPi inhibition on KMT2A-MLLT3 rearranged leukemia encoded by the t(9;11)(p22;q23) translocation [131, 133]. Both studies demonstrated a limited sensitivity to PARPi alone. However, Maifrede and colleagues showed that olaparib (in vitro) and talazoparib (in vivo) enhanced the sensitivity of AML cells expressing the KMT2A-MLLT3 chimera to doxorubicin and cytarabine [133]. From a biological point of view, cell proliferation and differentiation arrest in KMT2A-MLLT3 leukemia seems to be strictly dependent on resolution of DSBs originated from oxidative stress [149], causing a modest response to PARPi [131]. Indeed, HOXA9-mediated induction of HR genes in KMT2A-MLLT3-transformed cells led to the upregulation of DDR target genes such as RAD51 and BRCA2 and enhanced resistance to PARP inhibition, that could be reversed by HOXA9 suppression [131].The pressure exerted by antineoplastic agents as combination therapy may cause an accumulation of DSBs until a point of no return [133]. This preclinical evidence suggests that KMT2A-MLLT3 rearranged patients should not be candidate to PARPi in single agent, as their benefit will likely be modest. In addition, treatment with PARPi was synthetic lethal in proliferating and quiescent AML and ALL cells deficient for DNA-PK–mediated NHEJ [132]. These cases, which can be identified by combined gene expression and mutation analysis, included BCR-ABL1-driven leukemias, due to tyrosine kinase-mediated downregulation of BRCA1 and DNA-PKcs protein expression. Taken together, these findings are relevant to the design of personalized therapeutic approaches aided to the eradication of quiescent leukemia stem cells, that are spared by chemotherapy regimens.
Activated signaling pathways
Besides chimeric proteins, activating mutations and/or genomic alterations in the signaling pathway, that affect 59% of AML cases [146], were also associated to an augmented HR activity. In FLT3-ITD expressing cells, the increased activity of the HR pathway was mediated by the overexpression of RAD51 [150, 151] and the production of ROS [152]. These data were also recapitulated in AML patients carrying FLT3-ITD mutation, where the treatment with the FLT3 inhibitor AC220 induced the BRCAness phenotype in FLT3-ITD-mutated cells, with downregulation of HR and NHEJ proteins including BRCA1, BRCA2, PALB2, RAD51, and LIG4 and enhanced sensitivity to talazoparib and olaparib [136]. The combination of AC220 and PARPi caused accumulation of lethal DSBs and leukemia cell death. Moreover, it was able to eliminate FLT3-ITD quiescent and proliferating leukemia stem cells, as well as leukemic progenitors. A recent study showed that the co-occurrence of TET2 or DNMT3A mutations with FLT3-ITD had opposite effect on the response to PARPi. In particular, the co-occurrence of FLT3-ITD and TET2 mutations exacerbated the sensitivity of Lin−CD34+ primary AML cells to olaparib. On the contrary, cells expressing FLT3-ITD alteration alone or in combination with DNMT3A mutations were resistant to the treatment with the inhibitor [135]. In line with data form primary samples, mouse models expressing activated-tyrosine kinases (FLT3-ITD, JAK2V617F, MPLW515L, NRASG12D) showed that Tet2-deficient cells were sensitive to PARPi, while Dnmt3a-deficient cells were not. The combination of talazoparib and quizartinib exerted a strong in vivo inhibitory effect against FLT3-ITDITD;Tet2−/− leukemia, while it was neither effective nor enhanced the effect of the combination in FLT3-ITDITD;Dnmt3a−/− cells. The sensitivity was associated to an increase in lethal DSBs linked to accelerated forks progression and correlated with a reduced expression of BRCA1 and LIG4 proteins in FLT3-ITD;Tet2−/− and FLT3-ITD;Tet2−/−;Dnmt3a−/− compared with FLT3-ITD and FLT3-ITD;Dnmt3a−/− cells. On the contrary, PARP1 was downregulated in FLT3-ITD;Dnmt3a−/− cells, consistently with reduced HR/cNHEJ and aNHEJ activity, respectively. Finally, regarding FLT3-ITD AML, Le et al. described the protecting role of the TGF-β1-TGFβR kinase-SMAD3 pathway in the presence of PARPi in the bone marrow. The use of TGFβR inhibitors in combination with FLT3i in FLT3-ITD;Tet2−/− mice enhanced the effect of the combination and prolonged the survival of treated mice [137].
The same synthetic lethality effect linked to protein kinase inhibition was also described in KIT-mutated AML expressing the RUNX1-RUNX1T1 fusion gene [138]. The inhibition of c-KIT was associated with downregulation of BRCA1 and BRCA2 and the DNA-PK catalytic subunit (cNHEJ pathway), but not PARP1 (aNHEJ pathway). These led to the restoration of the sensitivity to PARPi. On the same line of reasoning, the combination of veliparib and busulfan was effective in myeloproliferative neoplasm (MPN)/AML xenotransplanted models carrying the activating signaling mutation in JAK2V617F, where the pharmacological treatment caused G2/M arrest associated with activation of the ATR-CHK1 pathway [139]. Regarding the signaling pathways, the disruption of the tumor suppressor Pten, which is targeted by deletions in about 3.0% and 1.0% of pediatric and adult AML, respectively [64], causes centromeric instability and favors spontaneous DSBs [153] which is a marker of PARPi sensitivity.
Other genetic subgroups: cohesin or TP53 or IDH1/2 mutant AML
A synthetic lethal effect of PARPi was recorded in presence of STAG2 mutations, a gene encoding for a subunit of the cohesin complex, which regulates sister chromatids during cell division. About 6% of myeloid neoplasm is characterized by the presence of loss-of-function mutations in STAG2, representing the most altered gene (51% of cases) among the cohesin family members [154]. In particular, STAG2 mutations promoted high levels of DNA damage and sensitivity to PARP inhibition [140] in AML. Genome-scale CRISPR/Cas9 screening on the U937 line showed that STAG2-mutated cells were preferentially dependent on members of the base excision repair (PARP1), homologous recombination (BRIP1, RAD51B, RAD51C, RAD54L2, XRCC2, XRCC3, PARP1), mismatch repair machinery (MSH2, POLD3, EXO1) and DNA replication (RPA2, POLD3), in order to avoid a massive accumulation of genomic instability. The loss of STAG2 was associated with an increase in stalled replication fork and, therefore, sensitivity to PARPi, that was confirmed in vitro and in vivo. Notably, U937 and K562 cells bearing inactivating mutations in SMC1 or RAD21, other two members of the cohesin complex, responded to PARPi to a similar extent of STAG2-mutated cells [140]. The synthetic lethal interaction between STAG2-mutant cells and DNA damage repair genes was common to other cancer types carrying STAG2 mutation [155].
Sensitivity to PARPi was also reported in the presence of TP53 mutations, thus opening a novel potential therapeutic window for a subgroup of high-risk patients [141]. In acute erythroblastic leukemia, data from multiplexed genome editing of mouse hematopoietic stem and progenitor cells and transplant assay showed that Trp53/Bcor-mutant tumors (carrying wildtype Dnmt3a and Tet2) were highly sensitive to talazoparib and veliparib, independently of BRCA1/2 status.
Finally, it has been demonstrated that 2-hydroxyglutarate, the oncometabolite produced by IDH1/2-mutated enzymes impaired the HR pathway. Indeed HEL cells expressing mutant IDH1/2 exhibited levels of DSBs repair defects comparable to those detected in BRCA1/2-mutated cell lines and were sensitive to olaparib [143] and talazoparib [142]. Further studies linked the HR defects to the signaling downstream ATM, which was impaired in IDH1R132-mutated AML [156]. In details, ATM expression was downregulated, due to the elevated methylation of the repressive histone mark H3K9 that may rely on inhibition of the histone demethylases KDM4A and/or KDM4B by 2-hydroxyglutarate [143, 156]. Moreover, daunorubicin enhanced the efficacy of PARPi in IDH1/2-mutated AML, while IDH1/2 inhibitors antagonized with PARPi and daunorubicin [142].
Selective vulnerabilities of PARP inhibitors in acute lymphoblastic leukemia
Recent studies revealed that PARP enzymes are highly expressed in T-ALL patients and regulate the expression and post-transcriptional modification of the TET1 gene [144, 157, 158]. For this reason, TET1 expressing T-ALL cells are highly sensitive to olaparib (Fig. 5C). From a biological point of view, olaparib abrogated leukemic growth of T-ALL cells in vivo by antagonizing TET1 activity [144]. Selective vulnerabilities driven by fusion genes have been also identified in ALL. In addition to BCR-ABL1-mediated suppression of HR and NHEJ [132], Piao and colleagues showed that ALL cells carrying the TCF3-HLF chimera were hypersensitive to olaparib, both in vitro and in vivo in combination with temozolomide (Fig. 5C). This sensitivity is related to HR deficiency in TCF3-HLF expressing ALL cells. Indeed, the Microcephalin 1 (MCPH1) gene, that encodes for a G2/M cell cycle checkpoint regulator, is downregulated by the TCF3-HLF fusion protein resulting in the attenuation of HR activity and in the upregulation of the anti-apoptotic factor BCL2, which suppresses HR activity by interfering with BRCA1 [134].
Clinical activity of PARP inhibitors in acute leukemia
Overall, in the setting of acute leukemia, only preliminary clinical data on PARPi exist. Ongoing or finished clinical trials are summarized in Table 5.
Talazoparib was investigated in adult patients with advanced hematological malignancies, including AML, in a single-agent Phase 1 study (NCT01399840). Thirty-three participants were enrolled: Twenty-one of them were affected by AML and four by myelodysplastic syndromes (MDS). For AML, dose-limiting toxicity (DLT) for single-agent talazoparib was demonstrated at a dose of 200 mg/day; no responses were seen in AML, and a hematological improvement was reported in one patient with MDS. Stable disease was reported in 12 of 24 evaluable patients with AML or MDS [159].
Veliparib has been evaluated in two combinatorial regimens in AML. The combination therapy with temozolomide was tested on 48 patients (NCT01139970). DLT level was reached at 200 mg of veliparib, and the phase 2 trial explored the efficacy of the combination between veliparib 150 mg and temozolomide. Complete remission (CR) was documented in 8/48 patients (16.6%). Of note, responders exhibited a veliparib-induced increase in γH2AX in CD34+ cells, as marker of DNA damage accumulation [160]. In an umbrella trial on ALL, AML, and chronic myelomonocytic leukemia patients, veliparib was tested in combination with carboplatin and topotecan (NCT00588991). The selected dose of veliparib for the phase 2 part of the trial was 100 mg body mass index. In the entire cohort, 33% of patients achieved at least partial remission, but the overall response rate was higher in the subset of patients with associated or antecedent MPNs or CMML, reaching 64% (14/22) [161]. DNA damage induction in CD34+ leukemia cells was again confirmed by the increase in H2AX phosphorylation. Moreover, FANCD2 monoubiquitination was a positive prognostic factor with the combination proposed in the trial.
Of note, in the investigational clinical trials with PARPi, blood count was ever used as an exclusion criterion, for the intrinsic nature of AML and MDS. PARPi have the class effect of lowering platelets production, being PARP an essential enzyme in the physiology of platelet formation [162]. However, low platelet, that was for the aforementioned reason an adverse event of extreme interest, was never reported as a DLT in studies with available results. AML patients usually have a complete deficiency in platelet formation for bone marrow dysfunction, and their platelet level depends on transfusion. It is important to note that PARPi does not affect platelet function, and thus, it does not augment the risks in transfusion-dependent patients.
Seminal results, that will drive future development, are expected from the combination of PARPi with decitabine (NCT02878785), particularly in the sub-cohort of TET2 and TET2/DNMT3A-mutant AML for their high sensitivity to DNA damage [135], and from target-restricted studies in IDH1/2 mutant (NCT03953898) and in cohesin mutant (NCT03974217) AML. The preclinical evidence reported in IDH1/2 [142, 143, 156] or cohesin-mutated AML patients [140, 155], as well as other subgroups, seems to promise success for molecularly directed trial designs for PARPi single agent and combination regimens.
Despite the clinical efficacy of PARPi in oncology, the scientific community is constantly evaluating the risk/benefit ratio of the inhibition DDR pathways for cancer patients. Indeed, the main raised concern is related to the potential genotoxic effect of PARPi in healthy normal cells. The development of secondary myeloid neoplasm (t-MNs) following the use of PARPi in breast, ovarian and pancreatic cancers highlighted the potential toxicity of PARP1 inhibition in healthy hematopoietic precursors [163, 164]. Indeed, the overall incidence of t-MNs shifts from 0.3 to 1% of patients depending on primary tumor subtypes and PARPi used in the study [165]. Nevertheless, the incidence of t-MNs after PARPi is significantly lower than the number cases of t-MNs following the use of conventional chemotherapy (overall incidence 0.8% to 6.3%) [166, 167].
Conclusions
Olaparib (2014), Rucaparib (2016), Niraparib (2017) and Talazoparib (2018) are currently FDA approved, each with specific indications, for the treatment of advance ovarian, fallopian tube, peritoneal and Her-negative metastatic breast cancer.
The available data on acute leukemia suggest potential windows for successful PARPi treatment in disease subgroups, defined on the basis of molecular markers and/or DDR activity, and/or in combination with genotoxic or targeted agents or immunotherapy, with a particular attention to the disease stage (e.g., minimal residual disease positivity rather than frank leukemia).
However, we have to take into account the evidence that in solid tumors treatment with PARPi is associated with a significantly higher risk of therapy-related MDS and AML [168] (0.73%; 95% confidence interval [CI]: 0·50–1·07) compared with placebo treatment (0.47%; 95% CI: 0·26–0·85; p = 0.026), with a median latency between first PARPi exposure and disease development of 17.8 months (8.4–29.2) [169]. Accordingly, treatment of relapsed disease in a high-grade serous ovarian carcinoma patient affected by a synchronous AML with olaparib-based maintenance therapy and the antileukemic agent azacitidine resulted in a dramatic expansion of malignant myeloid cells after two cycles, which was fatal to the patient [170]. Recent data also suggest that mutations of the DDR genes TP53, PPM1D and CHEK2, that are involved in clonal hematopoiesis, occur with increased frequency in cancer patients that were exposed to treatment, and in particular to platinum or topoisomerase II inhibitors or radiation therapy [171], indicating that DDR gene alterations improve the competitive fitness of the cells under these conditions. Taken together, this evidence indicates that a detailed characterization of the genetic background (including mutations, copy number alterations and translocations) and its subclonal architecture, and of the DDR functionality is crucial to personalize therapy and define acute leukemia patients that will likely benefit of PARPi-based therapeutic regimens.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- aNHEJ:
-
Alternative non-homologous end joining
- APL:
-
Acute promyelocytic leukemia
- BER:
-
Base excision repair
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- cNHEJ:
-
Conventional non-homologous end joining
- CR:
-
Complete remission
- DLT:
-
Dose-limiting toxicity
- DDR:
-
DNA damage response
- DSB:
-
Double-strand break
- FA:
-
Fanconi anemia
- HR:
-
Homologous recombination
- MRN:
-
Mre11-Rad50-Nbs1
- MDS:
-
Myelodysplastic syndromes
- MMR:
-
Mismatch repair
- MPN:
-
Myeloproliferative neoplasms
- NK:
-
Natural killer
- PARP:
-
Poly(ADP‐ribose) polymerase
- PARPi:
-
PARP inhibitors
- ROS:
-
Reactive oxygen species
- SSB:
-
Single-strand break
- ssDNA:
-
Single-strand DNA
- SSR:
-
Single-strand repair
- T-MNs:
-
Therapy-related myeloid neoplasm
- TRAIL:
-
TNF-related apoptosis-inducing ligand
References
Jubin T, Kadam A, Jariwala M, Bhatt S, Sutariya S, Gani AR, et al. The PARP family: insights into functional aspects of Poly(ADP-ribose) polymerase-1 in cell growth and survival. Cell Prolif. 2016;49(4):421–37.
Slade D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem Pharmacol. 2019;167:33–43.
Eustermann S, Wu WF, Langelier MF, Yang JC, Easton LE, Riccio AA, et al. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol Cell. 2015;60(5):742–54.
Lilyestrom W, van der Woerd MJ, Clark N, Luger K. Structural and biophysical studies of human PARP-1 in complex with damaged DNA. J Mol Biol. 2010;395(5):983–94.
Pascal JM. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst). 2018;71:177–82.
Alemasova EE, Lavrik OI. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019;47(8):3811–27.
Lüscher B, Bütepage M, Eckei L, Krieg S, Verheugd P, Shilton BH. ADP-ribosylation, a multifaceted posttranslational modification involved in the control of cell physiology in health and disease. Chem Rev. 2018;118(3):1092–136.
Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329(1):18–25.
Boehler C, Gauthier LR, Mortusewicz O, Biard DS, Saliou JM, Bresson A, et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci USA. 2011;108(7):2783–8.
Daugherty MD, Young JM, Kerns JA, Malik HS. Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet. 2014;10(5):e1004403.
Kim MK. Novel insight into the function of Tankyrase (Review). Oncol Lett. 2018;16(6):6895–902.
Qiu W, Lam R, Voytyuk O, Romanov V, Gordon R, Gebremeskel S, et al. Insights into the binding of PARP inhibitors to the catalytic domain of human tankyrase-2. Acta Crystallogr D Biol Crystallogr. 2014;70(Pt 10):2740–53.
Tuncel H, Tanaka S, Oka S, Nakai S, Fukutomi R, Okamoto M, et al. PARP6, a mono(ADP-ribosyl) transferase and a negative regulator of cell proliferation, is involved in colorectal cancer development. Int J Oncol. 2012;41(6):2079–86.
Gozgit JM, Vasbinder MM, Abo RP, Kunii K, Kuplast-Barr KG, Gui B, et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell. 2021;39(9):1214-1226.e10.
Yang CS, Jividen K, Spencer A, Dworak N, Ni L, Oostdyk LT, et al. Ubiquitin modification by the E3 ligase/ADP-ribosyltransferase Dtx3L/Parp9. Mol Cell. 2017;66(4):503–516.e5.
Yu M, Schreek S, Cerni C, Schamberger C, Lesniewicz K, Poreba E, et al. PARP-10, a novel Myc-interacting protein with poly(ADP-ribose) polymerase activity, inhibits transformation. Oncogene. 2005;24(12):1982–93.
Verheugd P, Forst AH, Milke L, Herzog N, Feijs KLH, Kremmer E, et al. Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nat Commun. 2013;4:1683.
Leung AKL, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell. 2011;42(4):489–99.
Li L, Zhao H, Liu P, Li C, Quanquin N, Ji X, et al. PARP12 suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Sci Signal. 2018;11(535):eaas9332.
Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, et al. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe. 2013;14(4):435–45.
Cho SH, Goenka S, Henttinen T, Gudapati P, Reinikainen A, Eischen CM, et al. PARP-14, a member of the B aggressive lymphoma family, transduces survival signals in primary B cells. Blood. 2009;113(11):2416–25.
Schuller M, Riedel K, Gibbs-Seymour I, Uth K, Sieg C, Gehring AP, et al. Discovery of a selective allosteric inhibitor targeting macrodomain 2 of polyadenosine-diphosphate-ribose polymerase 14. ACS Chem Biol. 2017;12(11):2866–74.
Jwa M, Chang P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK-and IRE1α-mediated unfolded protein response. Nat Cell Biol. 2012;14(11):1223–30.
Di Paola S, Micaroni M, Di Tullio G, Buccione R, Di Girolamo M. PARP16/ARTD15 is a novel endoplasmic-reticulum-associated mono-ADP-ribosyltransferase that interacts with, and modifies karyopherin-ß1. PLoS ONE. 2012;7(6):e37352.
Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8–24.
Yélamos J, Schreiber V, Dantzer F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med. 2008;14(4):169–78.
Rulten SL, Fisher AEO, Robert I, Zuma MC, Rouleau M, Ju L, et al. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41(1):33–45.
Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9(8):619–31.
El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31(19):5526–33.
Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, et al. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat Commun. 2018;9(1):746.
Caldecott KW, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase β and possibly poly(ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Nucleic Acids Res. 1996;24(22):4387–94.
Lebedeva NA, Rechkunova NI, Endutkin AV, Lavrik OI. Apurinic/apyrimidinic endonuclease 1 and Tyrosyl-DNA phosphodiesterase 1 prevent suicidal covalent DNA-protein crosslink at apurinic/apyrimidinic site. Front Cell Dev Biol. 2021;8:617301.
Abbotts R, Wilson DM. Coordination of DNA single strand break Repair. Free Radic Biol Med. 2017;107:228–44.
Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802.
Yang SW, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci USA. 1996;93(21):11534–9.
Das BB, Huang SN, Murai J, Rehman I, Amé JC, Sengupta S, et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014;42(7):4435–49.
Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510.
Caron MC, Sharma AK, O’Sullivan J, Myler LR, Ferreira MT, Rodrigue A, et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat Commun. 2019;10(1):2954.
Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst). 2008;7(10):1765–71.
Lamarche BJ, Orazio NI, Weitzman MD. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010;584(17):3682–95.
Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47(2):320–9.
Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair HHS Public Access. Nat Rev Mol Cell Biol. 2017;18(8):495–506.
Han Y, Jin F, Xie Y, Liu Y, Hu S, Liu XD, et al. DNA-PKcs PARylation regulates DNA-PK kinase activity in the DNA damage response. Mol Med Rep. 2019;20(4):3609–16.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu Rev Biochem. 2010;79:181–211.
Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2(3):130–43.
Yu W, Lescale C, Babin L, Bedora-Faure M, Lenden-Hasse H, Baron L, et al. Repair of G1 induced DNA double-strand breaks in S-G2/M by alternative NHEJ. Nat Commun. 2020;11(1):5239.
Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, et al. Essential roles for polymerase θ-mediated end joining in the repair of chromosome breaks. Mol Cell. 2016;63(4):662–73.
Xiong X, Du Z, Wang Y, Feng Z, Fan P, Yan C, et al. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015;43(3):1659–70.
Caracciolo D, Montesano M, Tagliaferri P, Tassone P. Alternative non-homologous end joining repair: A master regulator of genomic instability in cancer. Precis. Cancer Med. 2019; 2. https://pcm.amegroups.com/article/view/4925. Accessed 11 Jan 2022.
Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16(8):814–8.
Anand R, Ranjha L, Cannavo E, Cejka P. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol Cell. 2016;64(5):940–50.
Kent T, Chandramouly G, Mcdevitt SM, Ozdemir AY, Pomerantz RT. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat Struct Mol Biol. 2015;22(3):230–7.
Okano S, Lan L, Tomkinson AE, Yasui A. Translocation of XRCC1 and DNA ligase IIIα from centrosomes to chromosomes in response to DNA damage in mitotic human cells. Nucleic Acids Res. 2005;33(1):422–9.
Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196–207.
Haince J-F, McDonald D, Rodrigue A, Déry U, Masson J-Y, Hendzel MJ, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283(2):1197–208.
Cruz-García A, López-Saavedra A, Huertas P. BRCA1 accelerates CtIP-ediated DNA-end resection. Cell Rep. 2014;9(2):451–9.
Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–75.
Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell. 2013;23(5):693–704.
Wu J, Huen MSY, Lu L-Y, Ye L, Dou Y, Ljungman M, et al. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009;29(3):849–60.
Tarsounas M, Sung P. The antitumorigenic roles of BRCA1–BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol. 2020;21(5):284–99.
Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, et al. PARP1-driven Poly-ADP-ribosylation regulates BRCA1 function in homologous recombination mediated DNA repair. Cancer Discov. 2014;4(12):1430–47.
Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7(4):016600.
Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113.
cBioPortal for Cancer Genomics. [cited 2021 Dec 30]. https://www.cbioportal.org/.
Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6(3):271-281.e7.
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–31.
Simonetti G, Padella A, do Valle IF, Fontana MC, Fonzi E, Bruno S, et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer. 2019;125(5):712–25.
Simonetti G, Mengucci C, Padella A, Fonzi E, Picone G, Delpino C, et al. Integrated genomic-metabolic classification of acute myeloid leukemia defines a subgroup with NPM1 and cohesin/DNA damage mutations. Leukemia. 2021;35(10):2813–26.
Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2019;25(3):530.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15.
Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576–9.
Zámborszky J, Szikriszt B, Gervai JZ, Pipek O, Póti Á, Krzystanek M, et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene. 2017;36(6):746–55.
Seedhouse C, Faulkner R, Ashraf N, Das-Gupta E, Russell N. Polymorphisms in genes involved in homologous recombination repair interact to increase the risk of developing acute myeloid leukemia. Clin Cancer Res. 2004;10(8):2675–80.
Jacoby MA, De RE, Pizarro J, Shao J, Koboldt DC, Fulton RS, et al. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia. 2014;28(6):1242–51.
Alter BP. Fanconi’s anemia and malignancies. Am J Hematol. 1996;53(2):99–110.
Tischkowitz MD, Morgan NV, Grimwade D, Eddy C, Ball S, Vorechovsky I, et al. Deletion and reduced expression of the Fanconi anemia FANCA gene in sporadic acute myeloid leukemia. Leukemia. 2004;18(3):420–5.
Fontana MC, Marconi G, Feenstra JDM, Fonzi E, Papayannidis C, Ghelli Luserna Di Rorá A, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia. 2018;32(7):1609–20.
Rischewski JR, Clausen H, Leber V, Niemeyer C, Ritter J, Schindler D, et al. A heterozygous frameshift mutation in the Fanconi Anemia C gene in familiary T-aLL and secondary malignancy. Klin Padiatr. 2000;212(4):174–6.
Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34(5–6):360–94.
Padella A, Fontana MC, Marconi G, Fonzi E, Petracci E, Ferrari A, et al. Loss of PALB2 predicts poor prognosis in acute myeloid leukemia and suggests novel therapeutic strategies targeting the DNA repair pathway. Blood Cancer J. 2021;11(1):7.
Grellety T, Peyraud F, Sevenet N, Tredan O, Dohollou N, Barouk-Simonet E, et al. Dramatic response to PARP inhibition in a PALB2-mutated breast cancer: moving beyond BRCA. Ann Oncol. 2020;31(6):822–3.
Kuemmel S, Harrach H, Schmutzler RK, Kostara A, Ziegler-Löhr K, Dyson MH, et al. Olaparib for metastatic breast cancer in a patient with a germline PALB2 variant. NPJ Breast Cancer. 2020;6:31.
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(1):162–74.
Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. 2020;38(36):4274–82.
Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed Pharmacother. 2020;123:109661.
Murai J, Huang SYN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99.
Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol Cell. 2018;71(2):319-331.e3.
Hanzlikova H, Caldecott KW. Perspectives on PARPs in S phase. Trends Genet. 2019;35(6):412–22.
Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. 2016;109(1):djw199.
Pommier Y, O’Connor MJ, De Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016 26;8(362):362ps17.
Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18(10):610–21.
Sun W, Zhang Q, Wang R, Li Y, Sun Y, Yang L. Targeting DNA damage repair for immune checkpoint inhibition: mechanisms and potential clinical applications. Front Oncol. 2021;11:648687.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23(1):239–254.e6.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–9.
Pantelidou C, Sonzogni O, Taveira MDO, Mehta AK, Kothari A, Wang D, et al. Parp inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral sting pathway activation in brca-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9(6):722–37.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–20.
Mosely SIS, Prime JE, Sainson RCA, Koopmann JO, Wang DYQ, Greenawalt DM, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5(1):29–41.
Aurelius J, Thorén FB, Akhiani AA, Brune M, Palmqvist L, Hansson M, et al. Monocytic AML cells inactivate antileukemic lymphocytes: role of NADPH oxidase/gp91phox expression and the PARP-1/PAR pathway of apoptosis. Blood. 2012;119(24):5832–7.
Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436(7054):1186–90.
Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572(7768):254–9.
Meng XW, Koh BD, Zhang JS, Flatten KS, Schneider PA, Billadeau DD, et al. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J Biol Chem. 2014;289(30):20543–58.
Casorelli I, Tenedini E, Tagliafico E, Blasi MF, Giuliani A, Crescenzi M, et al. Identification of a molecular signature for leukemic promyelocytes and their normal counterparts: Focus on DNA repair genes. Leukemia. 2006;20(11):1978–88.
Faraoni I, Aloisio F, De Gabrieli A, Consalvo MI, Lavorgna S, Voso MT, et al. The poly(ADP-ribose) polymerase inhibitor olaparib induces up-regulation of death receptors in primary acute myeloid leukemia blasts by NF-κB activation. Cancer Lett. 2018;423:127–38.
Li D, Luo Y, Chen X, Zhang LY, Wang T, Zhuang Y, et al. NF-kB and Poly(ADP-ribose) polymerase 1 form a positive feedback loop that regulates DNA repair in acute myeloid leukemia cells. Mol Cancer Res. 2019;17(3):761–72.
Orta ML, Höglund A, Calderón-Montaño JM, Domínguez I, Burgos-Morón E, Visnes T, et al. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2′-deoxycytidine lesions. Nucleic Acids Res. 2014;42(14):9108–20.
Garcia TB, Snedeker JC, Baturin D, Gardner L, Fosmire SP, Zhou C, et al. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16(10):2058–68.
Shen Y, Aoyagi-Scharber M, Wang B. Trapping poly(ADP-ribose) polymerase. J Pharmacol Exp Ther. 2015;353(3):446–57.
Antolin AA, Ameratunga M, Banerji U, Clarke PA, Workman P, Al-Lazikani B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci Rep. 2020;10(1):2585.
Falzacappa MV, Ronchini C, Faretta M, Iacobucci I, Di Rorà AG, Martinelli G, et al. The Combination of the PARP inhibitor rucaparib and 5FU is an effective strategy for treating acute leukemias. Mol Cancer Ther. 2015;14(4):889–98.
Giansanti M, De Gabrieli A, Prete SP, Ottone T, Divona MD, Karimi T, et al. Poly(ADP-ribose) polymerase inhibitors for arsenic trioxide-resistant acute promyelocytic leukemia: synergistic in vitro antitumor effects with hypomethylating agents or high-dose vitamin C. J Pharmacol Exp Ther. 2021;377(3):385–97.
Valdez BC, Li Y, Murray D, Liu Y, Nieto Y, Champlin RE, et al. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget. 2017;9(3):3908–21.
Horton TM, Jenkins G, Pati D, Zhang L, Dolan ME, Ribes-Zamora A, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: Influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther. 2009;8(8):2232–42.
Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–37.
Kummar S, Kinders R, Gutierrez M, Rubinstein L, Parchment RE, Phillips LR, et al. Inhibition of Poly(ADP-ribose) polymerase (PARP) by ABT-888 in patients with advanced malignancies: Results of a phase 0 trial. J Clin Oncol. 2009;27(16):2705–11.
Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, et al. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents—a potential therapy for cancer. Cancer Cell. 2016;30(4):637–50.
Kohl V, Flach J, Naumann N, Brendel S, Kleiner H, Weiss C, et al. Antileukemic efficacy in vitro of talazoparib and APE1 inhibitor III combined with decitabine in myeloid malignancies. Cancers (Basel). 2019;11(10):1493.
Kogan AA, Mclaughlin LJ, Topper M, Muvarak N, Stojanovic L, Creed TM, et al. DNA demethylating agents generate a brcaness effect in multiple sporadic tumor types: prediction for sensitivity to PARP inhibitors in AML. Blood. 2017;130(Suppl 1):3347.
Farmer H, McCabe H, Lord CJ, Tutt AHJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Li X, Li C, Jin J, Wang J, Huang J, Ma Z, et al. High PARP-1 expression predicts poor survival in acute myeloid leukemia and PARP-1 inhibitor and SAHA-bendamustine hybrid inhibitor combination treatment synergistically enhances anti-tumor effects. EBioMedicine. 2018;38:47–56.
Robert C, Nagaria PK, Pawar N, Adewuyi A, Gojo I, Meyers DJ, et al. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. 2016;45:14–23.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Scardocci A, Guidi F, D’Alo F, Gumiero D, Fabiani E, Diruscio A, et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br J Cancer. 2006;95(8):1108–13.
Faraoni I, Compagnone M, Lavorgna S, Angelini DF, Cencioni MT, Piras E, et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: role as biomarkers of response to the PARP inhibitor olaparib. Biochim Biophys Acta Mol Basis Dis. 2015;1852(3):462–72.
Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Ž, et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature. 2018;559(7713):285–9.
Chen SH, Yu X. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci Adv. 2019;5(4):eaav4340.
Giovannini S, Weller MC, Repmann S, Moch H, Jiricny J. Synthetic lethality between BRCA1 deficiency and poly(ADP-ribose) polymerase inhibition is modulated by processing of endogenous oxidative DNA damage. Nucleic Acids Res. 2019;47(17):9132–43.
Pillay N, Tighe A, Nelson L, Littler S, Coulson-Gilmer C, Bah N, et al. DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP-ribose) glycohydrolase inhibitors. Cancer Cell. 2019;35(3):519-533.e8.
Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9.
Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21(12):1481–90.
Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest. 2017;127(6):2392–406.
Maifrede S, Martinez E, Nieborowska-Skorska M, Di Marcantonio D, Hulse M, Le BV, et al. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017;1(19):1467–72.
Piao J, Takai S, Kamiya T, Inukai T, Sugita K, Ohyashiki K, et al. Poly(ADP-ribose) polymerase inhibitors selectively induce cytotoxicity in TCF3-HLF–positive leukemic cells. Cancer Lett. 2017;386:131–40.
Maifrede S, Le BV, Nieborowska-Skorska M, Golovine K, Sullivan-Reed K, Dunuwille WM, et al. TET2 and DNMT3A mutations exert divergent effects on DNA repair and sensitivity of leukemia cells to PARP inhibitors. Cancer Res. 2021;81(19):5089–101.
Maifrede S, Nieborowska-Skorska M, Sullivan-Reed K, Dasgupta Y, Podszywalow-Bartnicka P, Le BV, et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood. 2018;132(1):67–77.
Le BV, Podszywalow-Bartnicka P, Maifrede S, Sullivan-Reed K, Nieborowska-Skorska M, Golovine K, et al. TGFβR-SMAD3 signaling induces resistance to PARP inhibitors in the bone marrow microenvironment. Cell Rep. 2020;33(1):108221.
Nieborowska-Skorska M, Paietta EM, Levine RL, Fernandez HF, Tallman MS, Litzow MR, et al. Inhibition of the mutated c-KIT kinase in AML1-ETO–positive leukemia cells restores sensitivity to PARP inhibitor. Blood Adv. 2019;3(23):4050–4.
Patel PR, Senyuk V, Rodriguez NS, Oh AL, Bonetti E, Mahmud D, et al. Synergistic cytotoxic effect of busulfan and the PARP inhibitor veliparib in myeloproliferative neoplasms. Biol Blood Marrow Transplant. 2019;25(5):855–60.
Tothova Z, Valton AL, Gorelov RA, Vallurupalli M, Krill-Burger JM, Holmes A, et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight. 2021;6(3):e142149.
Iacobucci I, Qu C, Varotto E, Janke LJ, Yang X, Seth A, et al. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood. 2021;137(12):1628–40.
Molenaar RJ, Radivoyevitch T, Nagata Y, Khurshed M, Przychodzen B, Makishima H, et al. Idh1/2 mutations sensitize acute myeloid leukemia to parp inhibition and this is reversed by idh1/2-mutant inhibitors. Clin Cancer Res. 2018;24(7):1705–15.
Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375):eaal2463.
Bamezai S, Demir D, Pulikkottil AJ, Ciccarone F, Fischbein E, Sinha A, et al. TET1 promotes growth of T-cell acute lymphoblastic leukemia and can be antagonized via PARP inhibition. Leukemia. 2021;35(2):389–403.
Padella A, Simonetti G, Paciello G, Giotopoulos G, Baldazzi C, Righi S, et al. Novel and rare fusion transcripts involving transcription factors and tumor suppressor genes in acute myeloid leukemia. Cancers (Basel). 2019;11(12):1951.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson G, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Krejci O, Wunderlich M, Geiger H, Chou F-S, Schleimer D, Jansen M, et al. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood. 2008;111(4):2190–9.
Forster VJ, Nahari MH, Martinez-Soria N, Bradburn AK, Ptasinska A, Assi SA, et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia. 2015;30(1):250–3.
Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514(7520):107–11.
Bagrintseva K, Geisenhof S, Kern R, Eichenlaub S, Reindl C, Ellwart JW, et al. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl-x(L). Blood. 2005;105(9):3679–85.
Seedhouse CH, Hunter HM, Lloyd-Lewis B, Massip AM, Pallis M, Carter GI, et al. DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia. 2006;20(12):2130–6.
Gaymes TJ, Mohamedali A, Eiliazadeh AL, Darling D, Mufti GJ. FLT3 and JAK2 mutations in acute myeloid leukemia promote interchromosomal homologous recombination and the potential for copy neutral loss of heterozygosity. Cancer Res. 2017;77(7):1697–708.
Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70.
Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood. 2014;124(11):1790–8.
Mondal G, Stevers M, Goode B, Ashworth A, Solomon DA. A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat Commun. 2019;10(1):1686.
Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell. 2016;30(2):337–48.
Ciccarone F, Valentini E, Zampieri M, Caiafa P. 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget. 2015;6(27):24333–47.
Roper SJ, Chrysanthou S, Senner CE, Sienerth A, Gnan S, Murray A, et al. ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 2014;42(14):8914–27.
Klisovic RB, Tricot G, Coutre S, Kovacsovics T, Giles F, Genna T, et al. A phase I trial of AVN944 in patients with advanced hematologic malignancies. J Clin Oncol. 2007;25:14026–14026.
Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, et al. A phase 1 study of the PARP inhibitor veliparib in combination with temozolomide in acute myeloid leukemia. Clin Cancer Res. 2017;23(3):697–706.
Pratz KW, Rudek MA, Gojo I, Litzow MR, McDevitt MA, Ji J, et al. A phase I study of topotecan, carboplatin and the PARP inhibitor veliparib in acute leukemias, aggressive myeloproliferative neoplasms, and chronic myelomonocytic leukemia. Clin Cancer Res. 2017;23(4):899–907.
De Botton S, Sabri S, Daugas E, Zermati Y, Guidotti JE, Hermine O, et al. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100(4):1310–7.
Mirza MR, Benigno B, Dørum A, Mahner S, Bessette P, Barceló IB, et al. Long-term safety in patients with recurrent ovarian cancer treated with niraparib versus placebo: Results from the phase III ENGOT-OV16/NOVA trial. Gynecol Oncol. 2020;159(2):442–8.
Yarchoan M, Myzak MC, Johnson BA, De Jesus-Acosta AD, Le DT, Jaffee EM, et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget. 2017;8(27):44073–81.
Skelding KA, Lincz LF. PARP inhibitors and haematological malignancies—friend or foe? Cancers (Basel). 2021;13(21):5328.
Morton LM, Dores GM, Schonfeld SJ, Linet MS, Sigel BS, Lam CJK, et al. Association of chemotherapy for solid tumors with development of therapy-related myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019;5(3):318–25.
Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. 2013;40:666–75.
Ball S, Sultan A, Zaw MH, Thein KZ. Secondary hematologic malignancies with poly adenosine diphosphate ribose polymerase inhibitors: Is the buzz real? Insights from a meta-analysis of phase 3 randomized controlled trials. J Geriatr Oncol. 2019;10(3):518–20.
Morice PM, Leary A, Dolladille C, Chrétien B, Poulain L, González-Martín A, et al. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: a safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021;8(2):e122–34.
Iluta S, Pasca S, Gafencu G, Jurj A, Terec A, Teodorescu P, et al. Azacytidine plus olaparib for relapsed acute myeloid leukaemia, ineligible for intensive chemotherapy, diagnosed with a synchronous malignancy. J Cell Mol Med. 2021;25(13):6094–102.
Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219–26.
Mufti G, Estey E, Popat R, Mattison R, Menne T, Azar J, et al. Results of a phase 1 study of BMN 673, a potent and specific PARP-1/2 inhibitor, in patients with advanced hematological malignancies. Haematologica. 2014;99:33–4.
Acknowledgements
Not applicable.
Funding
This work was supported by TrevisoAIL and by ERA-Per-Med (Reference Number: ERAPERMED2018-275).
Author information
Authors and Affiliations
Contributions
AP, AGLDR and GS drafted the first version of the manuscript and created the figures. GM and GMa contributed to the clinical sections. MG contributed to the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
G.M. has competing interests with Menarini/Stemline Therapeutics, Pfizer, Astellas, Abbvie, Astrazeneca. G.Ma. has competing interests with Ariad/Incyte, Pfizer, Celgene/BMS, Amgen, Roche, AbbVie, GlaxoSmithKline, Astellas, Daiichi Sankyo, Takeda, Gilead, Astellas, Janssen, Novartis, MSD.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Padella, A., Ghelli Luserna Di Rorà, A., Marconi, G. et al. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J Hematol Oncol 15, 10 (2022). https://doi.org/10.1186/s13045-022-01228-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01228-0