
Abstract
RNA-binding protein (RBP) has a highly dynamic spatiotemporal regulation process and important biological functions. They are critical to maintain the transcriptome through post-transcriptionally controlling the processing and transportation of RNA, including regulating RNA splicing, polyadenylation, mRNA stability, mRNA localization, and translation. Alteration of each process will affect the RNA life cycle, produce abnormal protein phenotypes, and thus lead to the occurrence and development of tumors. Here, we summarize RBPs involved in tumor progression and the underlying molecular mechanisms whereby they are regulated and exert their effects. This analysis is an important step towards the comprehensive characterization of post-transcriptional gene regulation involved in tumor progression.
Similar content being viewed by others

Introduction
It is found that there are approximately 1914 human RNA-binding proteins (RBPs), accounting for 7.5% of protein-coding genes [1,2,3]. RBPs are highly species-conservative and play a key role in maintaining homeostasis of gene expression [4, 5]. Mounting evidences have shown that RBPs are involved in various important cellular processes, for instance, cell transport, localization, development, differentiation, and metabolism. Additionally, RBPs engage in almost every step of post-transcriptional regulation, supervise the formation and function of transcripts, and maintain cell homeostasis. Mechanistically, RBPs regulate RNA splicing, polyadenylation, mRNA stability, mRNA localization, and translation through interacting with coding and non-coding RNAs (ncRNAs) and other proteins [6, 7]. Increasing studies have shown that RBP-mediated RNA modifications are critical for cancer progression [8]. Furthermore, RBPs are abnormally expressed in different types of cancer and regulate the expression and function of oncogenes and tumor suppressor genes [9]. Therefore, it will provide new ideas or methods for finding novel targets of cancer treatment by revealing the mechanisms underlying RBP expression and the interaction between RBPs and their target RNAs.
Structure of RBPs
Many RBPs consist of multiple repetitive sequences that contain only a few specific basic domains. Structurally, common RNA-binding domains mainly include RNA-recognition motif (RRM), K homology (KH) domain, double-stranded RBD (dsRBD), cold-shock domain (CSD), arginine-glycine-glycine (RGG) motif, tyrosine-rich domain, and zinc fingers (ZnF) of the CCHC, CCCH, ZZ type etc. [10]. According to the different functions of RBPs in cells, RBPs can be divided into epithelial splicing regulatory proteins (ESRP1), cytoplasmic polyadenylation element binding protein family (CPEB1/2), Hu-antigen R (HuR), heterogeneous nuclear ribonucleoprotein family members (hnRNP A/D/H/K/M/E/L), insulin-like growth factor 2 mRNA family members (IMP1/2/3), zfh family of transcription factors (ZEB1/2), KH-type splicing regulatory protein (KHSRP), La ribonucleoprotein domain family members (LARP1/6/7), Lin-28 homolog proteins (Lin28), Musashi protein family (MSI1/2), Pumilio protein family (PUM1/2), Quaking (QK), RNA-binding motif protein family (4/10/38/47), Src-associated substrate during mitosis of 68 kDa (SAM68), serine and arginine rich splicing factor (SRSF1/3), T cell intracellular antigens (TIA1/TIAR), and Upstream of N-Ras (UNR) [10]. Figure 1 summarizes the basic domains of RNA-binding proteins (TRBP, LIN28, RBM38, ZEB1, HnRNPA1, SAM68, CPEB4) in certain tumors. In order to realize the diversity of RBP functions, these repetitive sequences can be arranged in different combinations for a specific RNA, and precise recognition of proteins can be achieved by rearranging these basic domains, which confers the diversity of RBP functions. Each basic domain recognizes RNA, but the domains of most of these proteins required multiple copies to comply with their functions (Fig. 2) [11]. This unique structure makes the binding specificity and affinity of RBPs be extremely improved. However, almost half of the RBPs have no specific binding sequences and structural elements. To explain this “non-specific” phenomenon, Jankowsky et al. [12] establish a model that integrates various parameters relating to the binding ability of RBPs, for example, the concentration ratio of RBPs to RNA in cells, the affinity distribution coefficient of RBPs, the constants of RNA substrate binding and dissociation rates, and the synergistic effect of RBPs and cofactors. Based on the above characteristics, RBPs can form a huge molecular interaction network and have a considerable impact on cell functions. Therefore, a systematic and functional study of RBPs will help us discover its role in tumors [13].

Currently, more than 50 domains of RBPs have been discovered. Here, we select the common RBP domains. Different domains are represented by colored boxes: RNA recognition motif (RRM), K homology (KH) domain, tyrosine-rich domain, arginine-glycine-glycine (RGG) motif, cold-shock domain (CSD), zinc fingers of the CCCH, CCHC, ZZ type etc

RBPs can interact with rRNAs, ncRNAs, snRNAs, miRNAs, mRNAs, tRNAs, and snoRNAs by binding to specific RNA-binding domains to perform specific biological functions
At present, the research methods of RBPs mainly include homopolymer binding method, ultraviolet cross-linking, SELEX, EMSA, whole-genome in vivo immunoprecipitation, and protein affinity purification [14]. In addition, there is an online database (RBPDB: http://rbpdb.ccbr.utoronto.ca/) including 1171 known RBPs, in which the users can browse by domain and species. And the Cancer Genome Atlas (TCGA) database can be used to download RNA high-throughput sequencing and clinical pathology data for determining the abnormal expression of RBPs between cancerous and normal samples [14]. Subsequently, the KEGG pathway and GO enrichment analysis of abnormal expression of RBPs can be used to systematically explore their potential functions and molecular mechanisms (Fig. 3) [15]. The discovered RBPs will promote our understanding of the molecular mechanism underlying tumor progression and provide potential biomarkers for clinical diagnosis and prognosis [16].

The whole process of RBP analysis
Abnormality of RBPs in cancer
Tumor is a complex heterogeneous disease and its pathogenesis is difficult to determine. Tumor is always accompanied with genetic mutations, which disturb the homeostasis of carcinogenic or tumor suppressor signaling pathways [10]. Perspectively, RBP holds structural and functional diversity essential for regulating several necessary cellular processes, for example, RNA splicing, modification, transport, localization, stability, degradation, and translation [17]. Some RBPs are commonly expressed and conservatively evolved to maintain the basic functions of cells. RBP disorders can cause various diseases, including cancer (Table 1) [55]. The target RNAs of RBP is very diverse. In addition to binding to exons, introns, and untranslated regions (UTR) of mRNA, RBP can also bind to non-coding RNA, such as microRNA (miRNA), transfer RNA (tRNA), siRNA, telomerase RNA, small nucleolar RNA (snoRNA), and splicing small nucleolar RNA (snRNA). Non-coding RNA forms secondary structures that can bind to RBPs and regulate multiple processes such as splicing, RNA modification, protein localization and secretion, and chromosome remodeling [56]. For example, the expression of long non-coding RNA (lncRNA) NEAT1 is upregulated in human CRC (colorectal cancer) tissues and related to the poor prognosis of CRC patients. NEAT1 interacts with RBP DDX5 and enhances its stability, subsequently activates the Wnt/β-catenin signaling pathway and promotes tumorigenesis [57]; the RBP TRBP is a molecular chaperone required for DICER1 to function. The mutation of TRBP results in abnormal expression of miRNA, and cancer cell proliferation and differentiation [58, 59]; HnRNPL is also a multifunctional RBP that interacts with long non-coding RNA (lncRNA) in different cancers. HnRNPL is mainly located in the nucleus, but it can also exert effects in the cytoplasm via post-transcriptionally regulating lncRNA expression [60, 61]. Additionally, the oncogenic transcription factor MYC can upregulate the mRNA levels of heteronuclear ribonucleoproteins (hnRNP) A1 and A2 in a variety of malignant tumors, and hnRNPs facilitate the synthesis of M-type pyruvate kinase (PKM2), thus strengthening the Warburg effect [24, 25, 62,63,64]. In gastric cancer cells, the PI3K/AKT/NF-κB and METTL3/ZMYM1/E-cadherin signaling pathways can promote the transcription of RBP human antigen R (HuR), which is engaged in increased cell growth and anti-apoptotic capability [28, 30, 65]. Furthermore, epithelial-mesenchymal transition (EMT)—specific transcription factor ZEB1 protein can directly inhibit the mRNA level of epithelial splicing regulatory protein 1 (ESRP1), which leads to the increased expression of various spliceosomes of cell surface antigen CD44 and induces lung, breast, and pancreatic cancer stem-like and aggressive cells [35,36,37]. In breast cancer, RNA-binding motif protein 38 (RBM38) participates in the TGF-β-induced EMT pathway and stabilizes the tumor suppressor gene PTEN transcript to enhance expression [40,41,42]. Moreover, our previous study indicates that the RBM38 can suppress breast cancer metastasis through facilitating STARD13-related competing endogenous RNAs (ceRNA) network (Fig. 4) [66]. In hepatocellular carcinoma (HCC), dysregulation of RBPs disrupts the transcriptome balance within tumor cells, thereby driving tumorigenicity. For example, NELFE and PTBP3, as a kind of RBP, promote the malignant growth and metastasis of HCC cells by regulating the binding of MYC and splice variants (NEAT1_1, NEAT1_2, and miR-612) to the target promoter and the stability of MYC-regulated mRNAs [46, 50]. In summary, the abnormal function of RBPs has a significant impact on tumor phenotypes.

Signal pathways and metabolic pathways involved in abnormal RBP. HnRNP A1 /A2 is involved in the synthesis of M-type pyruvate kinase (PKM2), thereby enhancing the Warburg effect and let-7a-5p/Stat3/hnRNP-A1/PKM2 forms a cyclically regulated aerobic glycolysis. HuR regulates the PI3K/AKT/NF-κB signaling pathway. RNPC1 promotes the STARD13-mediated ceRNA network, thereby inhibiting the occurrence of EMT. The transcription factor ZEB1 protein can inhibit the mRNA level of epithelial splicing regulator protein 1 (ESRP1), resulting in the upregulation of the alternative spliceosome in the cell surface antigen CD44
NcRNA-mediated RBP disorders
NcRNA-mediated post-transcriptional regulation is closely related to the expression of RBPs. For example, LIN28 is an RBP mainly composing of two subunits, LIN 28A and LIN 28B, both of which have a similar structure and function. LIN28 is often dysregulated in tumor cells, and the upregulation of LIN28 is inversely related to the poor prognosis of advanced cancer. LIN28 inhibits the processing of let-7 precursor miRNA into mature miRNA in poorly differentiated cells, increases PD-L1 transcription and translation, and then promotes the growth and immune escape of various cancer cells [67]. Conversely, the let-7 family members can inhibit LIN28 expression [68]. Meanwhile, let-7 can bind to PD-L1 3′UTR, thereby inhibiting PD-L1 expression and enhancing the immune response of cells [51]. Therefore, there is a mutual negative feedback regulation between LIN28 and let-7 families. Furthermore, there are numerous evidences showing that this LIN28/let-7 two-way negative feedback can be combined with some cytokines (such as the SCR family, MYC family, and NF-κB) to form a complex regulatory system involving in tumor development (Fig. 5) [69,70,71,72]. In addition, RBP Musashi1 (MSI1) is restrained by a class of tumor suppressor miRNAs, including miR-34a, miR-101, miR-128, miR-137, and miR-138, and this inhibition reduces glioblastoma progression [73, 74]. What’s more, lncRNA is also engaged in the regulation of RBPs. For instance, NEAT1 is a lncRNA contributing to the formation of paraspeckle nuclear structures. Knockdown of NEAT1 results in the decreased expression of RBPs including hnRNP A2 and IGFBP3, both of which can inhibit tumor proliferation, migration, and invasion in liver cancer cells [75]. Moreover, lncRNA MT1JP enhances the p53 translation by combining with RBP TIAR, thereby regulating p53-related pathways, such as cell cycle, apoptosis, and proliferation [76]. In glioblastoma, LINC00470, a long non-coding RNA located on the chromosome, belongs to an oncogene RNA. LINC00470 directly binds to FUS and anchors FUS in the cytoplasm, activates FUS protein and forms a stable complex with AKT, activates AKT nuclear translocation and thereby promotes tumorigenesis [77].

The LIN28/let-7 two-way negative feedback mechanism is involved in the processing of let-7 precursor miRNA into mature miRNA and combines with some cytokines (such as the SCR family, MYC family, and NF-κB) to form a complex factor regulatory system involved in cancer occurrence
Post-translational modification-mediated RBP disorders
The abnormal function of RBP is also caused by abnormal post-translational modifications (PTM) in tumors, originates from multiple signaling pathways and activates the spatial conformation of enzymes, thereby inducing changes in its functional chemical groups, including acetylation, phosphorylation, methylation, ubiquitination, and N6-methyladenosine methylation (m6A) (Fig. 6) [78]. PTM can change the binding ability of RBP and play a key role in regulating protein activity, stability, positioning, interaction, or folding [79, 80]. It is worth noting that the PTM of RNA-binding elements in RBP are particularly prominent. The abnormal modification of this site may be one of the main factors leading to RBP dysfunction in tumors [81]. For example, as a member of the TAR family, the acetylation level of RBP Sam68 is higher than that of normal cells, thereby enhancing the binding ability of Sam68 to target genes whose expression is closely related to tumor cell proliferation [82, 83]. In non-invasive breast epithelial cells, activation of the TGFβ/AKT2 pathway phosphorylates serine 43 of hnRNPE1, which triggers translation of EMT-related mRNAs (DAB2 and ILEI) and facilitates EMT, metastasis, and invasion [84,85,86]. GSK3 can mediate phosphorylation of RBM38 at serine 195, causing RBM38 conformation to change, which prevents it from binding to eukaryotic translation initiation factor 4E (eIF4E) on p53 mRNA in breast and colon cancer cells [87,88,89]. In acute megakaryoblastic leukemia, the protein arginine methyltransferase 1 (PRMT1) is highly expressed and can methylate R578 residue on RBM15 and promotes PRMT1 ubiquitination-mediated degradation by E3 ligase (CNOT4). Notably, RBM15 can regulate a series of genes related to megakaryocyte production [90, 91]. RBP’s PTMs are involved in important biological processes, including carcinogenesis, apoptosis, tumorigenesis and cancer progression, cell division and cell response to stress drugs. Different types of RBP have different effects on protein transport, secretion, function and elimination. RBP can dynamically change the regionalization, transport and physical interaction of key molecules that regulate different cellular processes. Therefore, PTM enables RBP to cause rapid changes in gene expression programs. For example, the affinity of RBP for target RNA or DNA and change the interaction of RBP with other proteins. Different PTMs regulate RBP abundance, subcellular localization, and multiple protein kinases and signaling pathways. Through the comprehensive integration of genetic, biochemical, molecular biology, and structural analysis of RBP protein-protein and protein-RNA complexes, we hope to find new targets for therapeutic intervention.

RBP post-translational modifications (PTMs) (acetylation, phosphorylation, methylation, and ubiquitination) are involved in important biological processes. Different PTMs regulate the abundance of RBP, subcellular localization, and different protein kinases and signal transduction pathways. Abnormal PTM promotes carcinogenesis, apoptosis, tumorigenesis and cancer progression by changing the binding ability of IGF2BPs to mRNA, and regulating the protein activity, stability, and localization of hnRNPE1, RBM15 and Sam68
Transcriptional regulation-mediated RBP disorders
In addition to produce protein-coding mRNA, mammalian gene expression also produces many ncRNAs, including lncRNA, many of which are directly involved in transcription control [92]. The occurrence of RNA transcriptional regulation involves multiple RBPs participating in the recruitment of specific RNA-binding proteins (RBPs). The transcriptional regulation of RBPs is a fast and effective method that can continuously adapt transcription and translation of the proteome to the environment. Indeed, many RBPs have a direct role in transcription, and their ectopic regulation can lead to tumorigenesis. For example, the abnormal function of typical splicing regulators in transcription is closely linked to the pathogenesis of cancer. Importantly, these cancer-specific splicing variants are usually upregulated in tumors, which help tumor cell survival and cancer progression and also predict the survival of cancer patients (Fig. 7). Common specific splicing variants are SRSF2, RBFox2, NONO, etc. [93, 94]. SRSF2 belongs to SR protein and is composed of RRM domain and RS domain. Its main functions are splicing activator, transcriptional activation, RNA stability, mRNA transport and translation [95]. SRSF2 protein is highly expressed in hepatocellular carcinoma and predicts a poor prognosis of patients. SRSF2 can stimulate the expression of clinical HCC-related splice variant GCH1 at the transcription level [93]. GCH1 splice variants play a key role in cancer cell survival and tumorigenic potential [96]. Therefore, SRSF2 regulates the tumorigenesis of liver cancer cells by controlling the expression of cancer-associated splice variants. Stress particles (SGs) are stagnant translation initiation complexes that contain untranslated mRNA and RNA-binding protein (RBP). RBP fox-1 homolog 2 (Rbfox2) is part of SG and belongs to typical RBP [97]. It regulates variable pre-mRNA splicing in the nucleus and binds to retinoblastoma 1 (Rb1) mRNA [98]. Rbfox2 migrates to the cytoplasm when subjected to external pressure [98]. The RNA recognition motif domain of the Rbfox family protein binds to the 3′UTR region of the target mRNA related to cancer development, which contributes to the stability of the mRNA and thus promotes the progression of cancer [99, 100]. NONO is a versatile RBP that mainly plays a biological role in the nucleus [101]. In addition to the characteristics of nucleic acid binding, NONO can also interact with the terminal end region of the RNA polymerase II subunit to participate in pre-mRNA synthesis and treatment [102]. NONO expression is abnormal in prostate cancer, colorectal cancer, breast cancer and melanoma, which indicates that NONO may cause tumors [103,104,105,106]. In addition, at the transcription level, upstream-of-N-Ras (UNR) also plays an important role. UNR named CSDE1, belongs to a conservative RBP consisting of five cold-shock domains (CSDs). The specific domains of UNR can bind to mRNA (IRES) and thus change its structure to a more powerful translation [107]. UNR has been reported to regulate the expression of c-fos, c-Myc and other proto-oncogenes [108]. UNR is mainly located in the cytoplasm to regulate the transcription, translation and stability of mRNA [109]. In melanoma, UNR is usually highly expressed in melanoma and associated with invasion and metastasis by activating the extension of VIM and RAC1 mRNA [110]. Similarly, UNR is overexpressed in colorectal cancer (CRC) cells and patients, and closely related to cell survival, invasion, anti-apoptosis and poor prognosis by regulating EMT and c-MYC expression [108]. Clinically, the expression of UNR is related to the poor prognosis of resection PCDA patients. The expression of UNR is of great significance to clinicopathological features such as vascular infiltration, neuropathological infiltration, pT, and stage [111]. Based on the above research result, the UNR/CSDE1 axis might be as a prognostic biomarker for resection PCDA patients.

In addition to producing protein-encoding mRNA, genomic DNA also produces many non-coding RNAs, including long non-coding RNA (lncRNA) and miRNA, many of which are directly involved in transcription control. The occurrence of RNA transcription regulation involves multiple RNA-binding proteins (SRSF2, RBFox2, NONO). The abnormal function of typical splicing regulators in transcription is closely related to the pathogenesis of cancer
The molecular mechanisms underlying RBP roles in cancer progression
There are various mechanisms contributing to RBP-mediated regulation on cancer progression, including alternative splicing, polyadenylation, stability, subcellular localization and translation.
Alternative splicing
Alternative splicing is a major post-transcriptional regulatory mechanism contributing to protein diversity and mRNA stability. The main types of aberrant splicing in tumors are constitutive splicing, exon skipped or included, alternative 5_ splice-sites, alternative 3_ splice-sites, intron retention and mutually exclusive exon [112]. Abnormal or erroneous shearing is one of the principal causes of the abnormal function of RBPs and can promote cancer occurrence [113,114,115,116]. Some RBPs can form complexes with core proteins linked to splicing and together control the splicing of molecules in tumor cells. There are also some RBPs whose binding sites are tied to the regulation of exon. These proteins can increase or decrease the activity of spliceosome in various situations (Fig. 8) [103, 117, 118]. For example, SF3B1, hnRNP and serine/arginine (SR)-rich selective splicing regulators that recognize spliceosomes by binding to spliceosome point, which in turn regulates splicing [93, 119,120,121,122]. Additionally, SF3B1 missense mutations are not able to recognize pe-mRNA, leading to differential splicing of transcripts [123,124,125]. The most frequently mutated (SR) protein is SRSF2, whose mutation site is proline 195, which holds the RNA-binding specificity [121, 126,127,128]. The anomalous expression of hnRNPs and SRs in numerous cancers suggests that the splicing function of these two proteins plays a significant role in tumor progression. Additionally, RBP QKI in STAR family members is a splicing factor. Compared with normal tissues, the expression of QKI is significantly reduced in lung cancer, which is associated with a poor prognosis [129,130,131,132]. QKI binds to QKI response element [QRE, 5′-A (C/A) UAA-3] and exerts multifunctional effects on target RNAs, including localization, stability, translation efficiency, and microRNA (miRNA) processing [133, 134]. In normal cells, QKI can compete with the core splicing factor SF1, selectively suppress the splicing of exon 12 of NUMB mRNA, and upregulate the expression of NUMB isoforms, thereby inhibiting cell proliferation and preventing the activation of Notch signaling pathway [135, 136]. RBM10 is another splicing factor. Like QKI, RBM10 expression is significantly reduced in lung cancer cells. By inhibiting the splicing of exon 9 of NUMB mRNA, RBM10 increases the expression of NUMB. RBM10 can also suppress cell proliferation though regulating the Notch signaling pathway [137,138,139]. HnRNPA1/2 and PTB splicing factors hold the opposite functions and their inhibition of splicing is related to tumorigenesis [62]. The expression of hnRNPA1/2 is relatively high in gliomas and associated with a poor prognosis [140]. By binding to the promoter of hnRNPA1/2 and PTB, c-Myc upregulates PKM to promote tumor cell proliferation and transformation [24, 62, 140, 141]. After hnRNPA2 expression is upregulated and the exon 7 of CFLAR mRNA, exon 12a of BIN1 mRNA and exons 6-8 of WWOX mRNA cannot be spliced, resulting in the conversion of these tumor suppressor proteins into anti-apoptotic protein isomers in human astrocytoma cells [140]. Similarly, RBP hnRNA1 can inhibit the splicing of exon 11 of RON mRNA, which promotes this conversion of the tyrosine kinase receptor into oncoproteins and controls the occurrence of tumor invasion and migration [140]. In human tumors, defects of alternative splicing often occur, which may be due to mutations in splicing regulatory elements of specific cancer-related genes, or changes in the process of splicing regulation. Alternative splicing can also produce a new class of tumor proteins or tumor suppressors, which promotes disease progression by regulating RNA subtypes associated with tumor signaling pathways. Dysregulation of alternative splicing is a basic process of cancer. Understanding the regulatory factors of the splicing mechanism is a key step in understanding the role of alternative splicing in cancer, and provides new targets and biomarkers for the treatment of tumors.

Alternative splicing contributes to protein diversity and mRNA stability. Abnormal or erroneous splicing of RBPs is one of the causes of cancer. Some RBPs can form complexes with core proteins and combine with splicing, increase or decrease the activity of spliceosomes and jointly control the splicing of tumor cell molecules. SF3B1, hnRNP, and SR proteins are selective splicing regulators. They recognize spliceosomes by binding to spliceosomes, which in turn regulates splicing. Abnormalities in alternative splicing regulators are often associated with tumorigenesis
Alternative polyadenylation (APA)
In most eukaryotes, RBPs attend an important part of pre-mRNA post-processing, adding a poly (A) tails at the 3′ end of the mRNA. This process corresponds to the addition of a long string of adenylate on the 3′ end of the transcript. The pre-mRNA 3′ end processed complexes are mainly composed of CPSF, CSTF, CFI, CFII, CTD and RNAPII. About 85 proteins engage in this process [142, 143]. This process is necessary for mRNA maturation, stability, nuclear transport and efficient translation [144, 145]. The length of the poly (A) tails must be an extremely important biological characteristic and is highly conservative in evolution in diverse species and genes. Similar to splicing, most selective APA is located in the intron region upstream of the last exon and its role is to generate non-coding transcripts or transcripts with truncated coding regions. APA makes multiple different transcripts, and the coding region and 3′untranslated region (3′UTRs) of these transcripts are different. By amending the coding sequence, APA affects the function of the protein. For 3′UTRs, APA can alter its length, thereby regulating the stability, subcellular position and translation efficiency of target mRNAs. In cancer, abnormality at the 3′ end of pre-mRNA often occurs.
Tumor cells express a large number of aberrant mRNAs. The length of these mRNA 3′UTRs is shorter than that in normal cells. Studies have revealed that this phenomenon is usually caused by APA [146,147,148]. Because 3′UTRs contain a large number of binding sites for negative regulators, the shorter 3′UTRs greatly improve the stability of mRNA and the protein expression by more than ten times [146, 149]. It has been indicated that the shortening of 3′UTRs is regulated by a cleavage complex (CFIm), and there are mainly three polypeptides CFIm25, CFIm59, and CFIm68 (Fig. 9) [150,151,152]. Recently, Masamha et al. [153] find that CFIm25 plays a carcinogenic role in gliomas, and knockdown of CFIm25 decreases the expression of terminal polyadenylation of cyclin D1 (CCND1) and increases the proliferation of tumor cells. In cancer, RBPs can also co-regulate mRNA with APA, such as cytosolic polyadenylation element-binding protein 1 (CPEB1), which regulates polyadenylation of transcripts, especially cell cycle-related mRNAs, including extending poly (A) tails and enhancing translation efficiency [154]. Studies have discovered that the binding of CPEB1 to mRNA can assist APA in its 3′UTR region [155]. Furthermore, CPEB1 recognizes the binding site of non-canonical poly (A) polymerase Gld2/4 and noradenylation enzyme PRN, and specifically binds to the cytoplasmic polyadenylation element (CPE), resulting in a shortened length of hundreds of mRNA 3′UTRs and promoting tumorigenesis [156]. For example, in hepatocellular carcinoma, CPEB1 can directly target the 3′UTR of SIRT1, control the poly (A) tail length, inhibit its translation, and negatively regulate the stemness and resistance of liver cancer [157]. In endometrial cancer, miR-183 inhibits CPEB1 expression at the transcription and translation levels, and targets CPEB1 to induce EMT and promote tumorigenesis of EC cells [158]. In addition, CPEB4, another member of the CPEB family, is highly expressed in melanoma, glioblastoma and pancreatic ductal adenocarcinoma [159, 160]. In melanoma cells, MITF and RAB72A are vital regulators of their progression. CPEB4 can control the polyadenylation of MITF and RAB72A, improve their translation efficiency and further enhance the cell proliferation capacity [160]. In pancreatic cancer, CPEB4 can prolong the poly (A) tails of TPA mRNA and activate its translation, thereby enhancing cancer cell growth, invasion and angiogenesis [159]. Recently, reports have shown that CPEB1 and CPEB4 co-regulate the post-transcriptional process of VEGF. First, activation of CPEB1 increases the APA of CPEB4 and VEGF expression and the dual activation of VEGF promotes tumor angiogenesis [161]. These studies indicate that CPEBs are likely to be potential therapeutic targets for angiogenesis-dependent diseases including cancer [154].

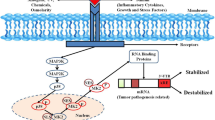
APA makes the miRNA binding site missing by a cleavage complex (CFIm), which makes the mRNA more stable, the translation efficiency is higher, and the transcription of the mRNA is out of control, thereby promoting the migration and invasion of tumor cells
Stability
The transcription, translation, stability and subcellular localization of mRNA are closely related to the highly regulated RNA-binding protein (RBP), thereby exerting a variety of cellular functions. In addition to the poly (A) tail at the 3′ end, the structure that impacts the stability of the eukaryotic mRNA has a cap structure at the 5′ end. There are many ways to degrade mRNA, one of which is to start with the hydrolysis of poly (A) tail, and then process to 5′ end decapping and 5′ → 3′ direction degradation, or after poly (A) tail hydrolysis, degradation in the 3′ → 5′ direction [162]. To date, the structure involves in reverse degradation of mRNA is the AU-rich original (ARE) of 3′UTR, and it is estimated that 16% of the transcripts contain ARE [163]. When cells are not stimulated, ARE-containing mRNAs are degraded at any time by poly (A) tails. After cells are subjected to different stimuli, ARE-binding proteins (ARBPs) may promote mRNA degradation or improve their stability [164]. ARE is the most studied structure related to mRNA stability [165]. The most common RBPs that can stabilize mRNA are human antigen R (HuR), T cell intracellular antigen 1 (TIA-1), and TIA-1-related protein (TIAR) [166]. The abnormal increase and extension of ARE-encoded mRNAs are related to cancer (Fig. 10), including the ARE-containing transcripts, oncoproteins, growth factors, and their receptors and cyclins. Abnormal expression of mediators is closely related to angiogenesis, chemotaxis, and invasiveness, which suggests that mRNA stability plays an important role in tumorigenesis [167, 168].

HUR, PTPB, or other RNA binding proteins enhance the stabilization of BCL2, MCL1, c-myc, cyclin E1, BLxL, and ZEB1 PTEN-related mRNAs by binding to the ARE sequence elements of mRNA in 3′UTR, thereby increasing the expression of cancer-related proteins and promoting tumor angiogenesis, migration, invasion and drug resistance
On the one hand, RBPs can enhance the stability of mRNA to regulate the protein expression of its target genes. For example, the PTBP family belongs to RBPs, and there are three main members PTBP1, PTBP2, and PTBP3. PTBP1 and PTBP2 regulate mRNA splicing, stability, localization and translation [169]. However, PTBP3 can induce epithelial-mesenchymal transition (EMT) of breast tumor cells and promote their aggressive growth and metastasis. PTBP3 regulates the expression of the transcription factor ZEB1 by binding to the 3′UTR of its mRNA, thereby preventing its degradation [47]. Some studies have also indicated that HuR can regulate some apoptosis-related genes, such as stabilizing their mRNAs by binding to the 3′UTRs of MCL1, BCL2 and BLxL. This post-transcriptional regulation promotes the growth of glioma cells and heightens its resistance to multiple drug tolerability, including etoposide, topotecan, and cisplatin [170]. In addition, by binding to the 3′UTR of cyclin E1, which is overexpressed in breast cancer, HuR improves cyclin E1 mRNA stability and thus increases its protein expression [171]. In addition, the IGF2BPs family is important for tumor development through regulating mRNA stability. For example, IGF2BP1 can prevent the degradation of PTEN mRNA and promote the targeted migration of tumor cells [172]. IGF2BP1 can also stabilize c-MYC and MKI67 mRNA and promote their expression, thereby regulating the proliferation and apoptosis of liver cancer cells [173]. IGF2BP2 can stabilize the mRNA of oncogene HMGA1 and facilitate the proliferation of cancer cells [174]. At the same time, IGF2BP2 can suppress the degradation of RAF1 mRNA caused by miR-195 and enhance the viability of colorectal cancer cells [175]. On the other hand, RBPs can also accelerate the degradation of mRNA. For instance, MCPIP1 can selectively expedite the degradation of a series of anti-apoptotic mRNAs, including Bcl2L1, Bcl2A1, RelB, Birc3, and Bcl3, and induce apoptosis in breast cancer cells [176]. In the IGF2BPs family, IGF2BP1 can reduce the stability of HULC mRNA which is specifically and highly expressed in liver cancer [177]. IGF2BP3 can accelerate the degradation of EIF4E-BP2 mRNA and thus promote the proliferation of cervical cancer cells [178].
Subcellular localization
Some mRNAs are transcribed in the nucleus, then transported into the cytoplasm and translated to produce proteins. However, other mRNA transcripts directly target specific regions for local translation or distribution, resulting in an asymmetric distribution of cytoplasmic proteins [179]. The subcellular localization of RNA mainly recognizes the cis motif by RBP, or specific binding elements (zipcodes) in the 3′UTR of the target gene form a secondary structure, which serves as the binding site of RBP to regulate localization in the cell and then mediate the localization of RNA to a specific subcellular compartment [180]. First, hnRNPs in the nucleus recognize and bind to mRNA, and they are consigned into the cytoplasm from the nucleus. One part of the hnRNPs returns to the nucleus and the other forms a complex with the motor protein [179]. This mechanism is crucial for establishing and maintaining cell polarity, disruption of which can result in cancer progression [181] (Fig. 11). For example, The RBP Tia1 binds to a subset of cellular stress-related transcripts (p53 mRNA) and controls p53 mRNA translational silencing and RNA particle localization. When Tia1’s DNA is damaged, Tia1 dissociates from its mRNA target, p53 mRNA is released from the stress particles and binds to polysomes, resulting in the relocation and translation of mRNA [182]. TIA1 interacts with various tumor-related mRNAs and participates in cancer cell proliferation, apoptosis, invasion, metastasis, angiogenesis and immune escape [183, 184]. IGF2BP1, which is a member of the RBPs of the VICKZ family, is mainly composed of two RNA recognition motifs (RRMs) and four K homology (KH) domains. IGF2BP1 plays an important role in the process of carcinogenesis [185]. After transcription, IGF2BP1 can correct the mRNA expression of certain oncogenes, thereby promoting tumor cell proliferation and growth, invasion and chemical metabolism, which is related to a poor overall survival and metastasis of various cancers [186]. In polar cells, such as migrating fibroblasts and neurons, IGF2BP1 promotes the transport of ACTB transcripts to actin-rich processes [172]. In the beginning, IGF2BP1 in the nucleus is linked to the transcript of β-actin. After being transported to the cytoplasm, IGF2BP1 causes translational silencing of the mRNA but has no implication for its stability. Then mRNA is transported to the periphery of the cell; Src-directed tyrosine phosphorylation in the junction region of KF domains 2 and 3 of IGF2BP1 can induce the disassembly of cytoplasmic messenger ribonuclear protein and activate the translation of ACTB mRNA. Finally, the translation of mRNA starts and accelerates the accumulation of β-actin [187, 188]. In addition, IGF2BP1 is overexpressed in primary tumor tissues such as breast cancer, colon cancer and non-small cell lung cancer [189,190,191]. Downregulation of IGF2BP1 can affect the transport of adhesion and migration-related target genes, including α-actin, β-actin, E-cadherin and the Arp2/3 complex subunit Arp-16 [192, 193]. Therefore, the silencing of IGF2BP1 can destroy the connection between cells, decrease its adhesion ability, and promote cell migration and invasion. In addition, adenomatous polyposis protein (APC) is also an RBP whose function is related to cell localization. This protein is a positive microtubule end tracking factor and suppresses colon cancer progression. Recent studies have shown that APCs are not uniform as classical RBPs in their localization function. APC binds to 3′UTR of β2B-tubulin to regulate the location of cytoskeleton-related mRNAs. Studies have shown that the RNA binding region of APC contains cancer-related missense mutations and deletions [194].

DNA is transcribed into RNA. With the help of hnRNA, pre-mRNA is transported to the cytoplasm. RBP binds to pre-mRNA and participates in splicing and splicing to form mRNA. Then hnRNA enters the cytoplasm and becomes mRNPs, which becomes a substrate for mRNA localization, and subsequently to be deadenylated so as to ensure that the mRNA can be effectively located to a specific position and efficiently synthesize protein
Translation
The translation process is intricate and can consist of three stages: initiation, extension and termination. Translational regulation of most mRNAs occurs at the initial stage. RBPs, such as the 5′cap-binding complex eIF4F and poly (A)-binding protein (PABP), are required for cyclization and translational activation of mRNAs. In cancer, almost all key oncogenic signaling pathways are abnormal and cause translational disorders such as the PI3K/AKT/mTOR, RAS/MAPK and Wnt/β-catenin signaling pathways [195,196,197]. It has been reported that dysregulated expression of eIF4E, a component of the eIF4F complex, is associated with approximately 30% of human tumors [198]. In addition, it has been indicated that knockdown of eIF4E in mice not only sustain a normal physiological state but also manifest the significant inhibition of tumors [199]. These studies indicate that in mammalian cells, the expression of eIF4E is higher than normal for protein translation, and eIF4E can induce translation of oncogenic mRNAs and promote cancer development. During the gradual development of cancer, certain structural and sequence-specific regulatory elements can control protein translation [197]. As one of these organizational elements, the 5′UTR has received widespread attention. 5′UTR is an internal ribosome entry site (IRES) and can recruit ribosomes directly in combination with IRES transacting factors (ITAFs) to initiate translation in a hat-dependent manner. The occurrence of cancer is associated with dysregulation of hat structure-dependent translation. Research has shown that La ribonucleoprotein domain family member 3 (LARP3) can induce IRES-mediated translation of two genes, including anti-apoptotic XIAP and EMT-related LAMB1 mRNA, and enhance cancer cell survival and invasion [139,140,141] [200,201,202]. In addition to 5′UTR, 3′UTRs also play a major role in the regulation of the translation process. For example, DAB2 and ILEI genes encoding EMT-related proteins contain TGF-β-activated translation (BAT) stem-loop elements on their 3′UTRs [84, 85]. When the TGF-β signaling pathway is silenced, the BAT element recruits eukaryotic translation elongation factors 1α1 (eEF1A1) and hnRNPE1 to prevent binding to their 3-UTR BAT elements, which can mediate the translational silence of Dab2 and ILEI (two EMT transcripts). Upon the TGF-β signaling pathway is activated, hnRNPE1 is phosphorylated and sheds from the BAT element to promote the occurrence of EMT [85]. In addition, Rumis’ Pumilio (PUM) human ortholog family is also involved in the regulation of protein translation. It also enhances the activity of multiple E2F3 target microRNAs (miRNAs) by binding to E2F3 3′UTR. PUM inhibits the translation of proliferating cell transcription factor E2F3 protein; its overexpression is related to tumor development [203]. Studies have found that the 3′UTR regions of the oncogenes E2F3, JUN and NRAS on the regulatory axis are shortened, leading to the disappearance of the PUM binding site, which in turn triggers the expression of these three genes and promotes cell proliferation (Fig. 12) [204].

The mRNA translation process is mainly the 5′cap structure of the eIF4F cap-binding complex that recognizes and binds to the mRNA. When the 43S pre-initiation complex-mediated cap binding and ribosome binding, translation starts from the AUG. Abnormal mRNA translation of eIF4E, XIAP and LAMB1 is closely related to the occurrence of cancer. In addition, the binding of PUM and miRNAs to 3′UTR can inhibit the translation of E2F3, JUN, and NRAS mRNA and its overexpression is related to tumor development
Chromosomal remodeling and DNA damage
RBPs are increasingly appreciated for their participation in the entire life course of RNA [205]. Recent studies have shown that in addition to participating in traditional RNA processing, RBPs locating at chromatin may also participate in chromatin remodeling and DNA damage repair in mammalian genomes [206, 207]. Chromatin immunoprecipitation sequencing (ChIP-seq) revealed the extensive interaction of RBP RBFox2 with chromatin, RBFox2 interacts with Polycomb complex 2 (PRC2) and H3K27me3 so as to maintain a key signal of gene-stable expression in the mammalian genome [208]. Another interesting phenomenon is that by comparing RBP and TF ChIP-seq signals in HepG2 cells, Xiao et al. found a co-existence by a large number of transcription factors, enhancers and RBP, indicating that they have a coordinated function at the chromatin level [209]. For example, transcription factor YY1 and RBP RBM25 have a coordinated function chromatin remodeling, as evident by the fact that knockout of RBM25 attenuates YY1-mediated effects on chromatin binding, DNA circularization, and transcription [209]. In addition, DNA double-strand damage repair is essential for maintaining genome stability and preventing developmental disorders, neurodegenerative diseases and cancer. There exists two main ways to repair DNA double-strand breaks: non-homologous end joining (NHEJ) and homologous recombination (HR). Increasing evidences show that RBPs are involved in DNA damage response (DDR) and DNA repair [210]. For example, RBMX is a hnRNP protein which regulates alternative splicing through RNA recognition (RRM) and promotes DNA damage repair through HR; this process is independent of ATM signaling and H2AX, but depends on PARP1 [211]; RBP HuR has been reported as a PAR-binding protein in response to DNA damage. HuR binds and stabilizes PARG mRNA, thereby increasing PARG expression and regulating PARP-1-chromatin dynamics, which contributes to DNA repair and the inhibition of PARP inhibitor resistance [212]. In addition, triple-negative breast cancer (TNBC) patients are usually treated with chemotherapy, but the repair of DNA double-strand breaks (DSB) in tumor cells may result in chemoresistance. Studies have shown that IGFBP-3 participates in the repair of DSB damage through NHEJ, responds to DNA-destructive chemotherapy, and forms a nuclear complex with EGFR and DNA-PKCs, promotes the stability of tumor cell genome and leads to the migration and invasion of tumor cells [213]. Moreover, poly (ADP-ribose) polymerase (PARP) interacts with RBPs and regulates various important cellular processes, such as coordinated regulation of single-stranded DNA repair and differentiation, cell proliferation, and tumor transformation [214]. Importantly, PARP1 inhibitors have been widely used for cases of BRCA1 mutation or dysfunction [215]. Notably, PARP inhibitors may inhibit DNA repair through the NHEJ pathway and cause increased DNA double-strand breaks, thereby inhibiting tumor cell proliferation and metastasis [216]. Clinical studies have shown that the PARP inhibitor veliparib (ABT-888) can enhance the effects of radiation therapy through promoting the accumulation of nuclear EGFR translocation and DNA damage [217]. Therefore, we believe that more studies should focus on the interaction of PARP and RBP, which jointly regulates tumorigenesis, and PARP inhibitors might be the critical partners for RBP-targeted therapy in the future.
Conclusions
Notably, in recent years, reversible modification of N6-methyladenosine (m6A) on mRNA has been shown to be a common phenomenon [218]. Both mRNA and lncRNA have m6A modifications in large amounts. m6A can accelerate the processing time of mRNA precursors and the speed of mRNA transport and nucleation in cells [219]. Importantly, RBP is a key regulator of m6A’s biological function by regulating methyltransferase installation. The methyltransferase complex is mainly composed of methyltransferase-like 3 (Mettl3), Mettl14, and Wilms Tumor1-related proteins (WTAP) and subunits, including Virilizer, RNA-binding motif protein 15 and zinc finger protein Zc3h13 [220]. RBP-mediated m6A modification can also cause a series of diseases, including tumors, nervous system diseases and delayed embryonic development [221, 222]. For example, RBP IGF2BP recognizes the K homology (KH) domain through its m6A binding site and acts as an m6A reader. With the help of cofactors, IGF2BPs promote the stability of thousands of potential target mRNAs, hinder miRNA-induced RNA decay and improve the expression of oncogenes (MYC, Actin, Lin28, SRF) to play a carcinogenic role in cancer [223].
This article mainly discusses the structure and abnormal expression of RBPs in tumors and focuses on the molecular mechanism thereby they exert their effects in cancer. Since RBPs are involved in various processes of tumors, suggesting that RBPs participate in a very complex regulatory network and change numerous characteristics of tumors (Fig. 13). RBPs not only regulate the life course from DNA to RNA, but also participate in the energy metabolism program of cancer cells and evading immune surveillance by controlling the stability and/or translation of mRNA. Under carcinogenic conditions, RBP mainly affects cancer development by changing a large number of downstream target genes related to tumors, thereby expanding its impact on tumors through the “chain effect.” Therefore, the potential of RBPs in cancer biology will be unlimited. At present, small molecule inhibitors targeting RBPs are being explored (Table 2). Based on the above research results, more and more researchers have explored onco-RBPs-targeting cancer treatment [230]. In the future, the potential of RBPs as drug targets should be further tapped.

RBPs participate in the entire physiological process of RNA and play a key role in the function of RNA. In tumor cells, the abnormal function of RBPs makes tumor cells be highly heterogeneous
As more and more cancer-related RBPs are discovered, identifying their target genes is needed. A key issue is that the binding of many RBPs to RNA does not depend on classical RBDs [231], so identifying RBP-RNA interactions is a challenging task. In establishing bioinformatics predictions, structural studies of ribonucleoprotein complexes can provide important data for new modeling algorithms created by bioinformatics tools and also point the way to identify the different roles of RBPs in normal and disease environments [232]. In addition, various evolving methodologies base on immunoprecipitation technology (including antibodies for immunoprecipitation) can be used for large-scale identification of RBPs genomes, their targets and binding sites [233]. Although this method is still in its infancy, it is likely to be widely used in a short time. Additionally, combined with the latest research progress, cell models are being used for RBP research, such as human tissue organoids, biomimetic microfluidic culture systems and tumor xenograft of patients. The molecular mechanisms by which RBPs and non-coding RNAs co-regulated a series of mRNAs are being deeply explored [234], which allows us to fully understand RBP-mediated regulatory network in tumors. Therefore, in order to reveal such a huge regulation, the integration of a large number of data resources and technologies is necessary. The advance of such related comprehensive analysis software will be a great challenge.
Availability of data and materials
Not applicable
Abbreviations
- RBPs:
-
RNA-binding proteins
- RRMs:
-
RNA recognition elements
- RBDs:
-
RNA-binding domains
- ncRNAs:
-
Non-coding RNAs
- lncRNA:
-
Long non-coding RNA
- hnRNPL:
-
Heterogeneous nuclear ribonucleoprotein L
- hnRNP:
-
Heteronuclear ribonucleoprotein
- PKM2:
-
M2-type pyruvate kinase
- HuR:
-
Human antigen R
- EMT:
-
Epithelial-mesenchymal transition
- ESRP1:
-
Epithelial splicing regulatory protein 1
- RBM38:
-
RNA-binding motif protein 38
- PD-L1:
-
Programmed cell death-ligand 1
- MSI1:
-
Musashi1
- PTM:
-
Post-translational modifications
- CBP:
-
CREB-binding protein
- PRMT1:
-
Protein arginine methyltransferase 1
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- CNOT4:
-
Ccr4-Not transcription complex subunit 4
- DAB2:
-
Tumor suppressor disabled-2
- ILEI:
-
Interleukin-like EMT inducer
- PRMT1:
-
Protein arginine methyltransferase 1
- SF3B1:
-
Splicing factor 3B subunit 1A
- MDS:
-
Myelodysplastic syndrome
- SR:
-
Serine/arginine
- APA:
-
polyadenylation
- APC:
-
Adenomatous polyposis protein
- 3′UTR:
-
3′untranslated regions
References
Neelamraju Y, Hashemikhabir S, Janga SC. The human RBPome: from genes and proteins to human disease. J Proteomics. 2015;127:61–70.
Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–45.
Beckmann BM, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume AM, Schwarzl T, Curk T, Foehr S, Huber W, et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat Commun. 2015;6:10127.
Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–64.
Matia-González AM, Laing EE, Gerber AP. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat Struct Mol Biol. 2015;22:1027–33.
Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205.
Mitchell SF, Parker R. Principles and properties of eukaryotic mRNPs. Mol Cell. 2014;54:547–58.
Iino K, Mitobe Y, Ikeda K, Takayama KI, Suzuki T, Kawabata H, Suzuki Y, Horie-Inoue K, Inoue S. RNA-binding protein NONO promotes breast cancer proliferation by post-transcriptional regulation of SKP2 and E2F8. Cancer Sci. 2020;111:148–59.
Lujan DA, Ochoa JL, Hartley RS. Cold-inducible RNA binding protein in cancer and inflammation. Wiley Interdiscip Rev RNA. 2018;9.
Pereira B, Billaud M, Almeida R. RNA-binding proteins in cancer: old players and new actors. Trends Cancer. 2017;3:506–28.
Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–90.
Jankowsky E, Harris ME. Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol. 2015;16:533–44.
Li W, Li N, Gao L, You C. Integrated analysis of the roles and prognostic value of RNA binding proteins in lung adenocarcinoma. PeerJ. 2020;8:e8509.
Ramanathan M, Porter DF, Khavari PA. Methods to study RNA-protein interactions. Nat Methods. 2019;16:225–34.
Li W, Li X, Gao LN, You CG. Integrated analysis of the functions and prognostic values of RNA binding proteins in lung squamous cell carcinoma. Front Genet. 2020;11:185.
Li W, Gao L-N, Song P-P, You C-G. Development and validation of a RNA binding protein-associated prognostic model for lung adenocarcinoma. Aging. 2020;12:3558–73.
Wang ZL, Li B, Luo YX, Lin Q, Liu SR, Zhang XQ, Zhou H, Yang JH, Qu LH. Comprehensive genomic characterization of RNA-binding proteins across human cancers. Cell Rep. 2018;22:286–98.
Ding J, Wang DZ. Mystery of Trbp, tale of a RBP in the miRNA pathway. Cell Cycle. 2015;14:3007–8.
Goodarzi H, Zhang S, Buss CG, Fish L, Tavazoie S, Tavazoie SF. Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins. Nature. 2014;513:256–60.
Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, et al. Retraction: a TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2016;48:221.
Zhang T, Huang XH, Dong L, Hu D, Ge C, Zhan YQ, Xu WX, Yu M, Li W, Wang X, et al. PCBP-1 regulates alternative splicing of the CD44 gene and inhibits invasion in human hepatoma cell line HepG2 cells. Mol Cancer. 2010;9:72.
Grelet S, Link LA, Howley B, Obellianne C, Palanisamy V, Gangaraju VK, Diehl JA, Howe PH. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat Cell Biol. 2017;19:1105–15.
Fei T, Chen Y, Xiao T, Li W, Cato L, Zhang P, Cotter MB, Bowden M, Lis RT, Zhao SG, et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci U S A. 2017;114:E5207–e5215.
David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–8.
Yang H, Zhu R, Zhao X, Liu L, Zhou Z, Zhao L, Liang B, Ma W, Zhao J, Liu J, Huang G. Sirtuin-mediated deacetylation of hnRNP A1 suppresses glycolysis and growth in hepatocellular carcinoma. Oncogene. 2019;38:4915–31.
Luan W, Wang Y, Chen X, Shi Y, Oncotarget YYJ: PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. 2015, 6.
Li L, Yan S, Zhang H, Zhang M, Huang G, Chen M. Interaction of hnRNP K with MAP 1B-LC1 promotes TGF-β1-mediated epithelial to mesenchymal transition in lung cancer cells. BMC Cancer. 2019;19:894.
Huang H, Han Y, Zhang C, Wu J, Feng J, Qu L, Shou C. HNRNPC as a candidate biomarker for chemoresistance in gastric cancer. Tumour Biol. 2016;37:3527–34.
Wu Y, Zhao W, Liu Y, Tan X, Li X, Zou Q, Xiao Z, Xu H, Wang Y, Yang X. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. Embo J. 2018;37.
Kang MJ, Ryu BK, Lee MG, Han J, Lee JH, Ha TK, Byun DS, Chae KS, Lee BH, Chun HS, et al: NF-kappaB activates transcription of the RNA-binding factor HuR, via PI3K-AKT signaling, to promote gastric tumorigenesis. Gastroenterology 2008, 135:2030-2042, 2042.e2031-2033.
Yang F, Hu A, Li D, Wang J, Guo Y, Liu Y, Li H, Chen Y, Wang X, Huang K, et al. Circ-HuR suppresses HuR expression and gastric cancer progression by inhibiting CNBP transactivation. Mol Cancer. 2019;18:158.
Al-Haidari A, Algaber A, Madhi R, Syk I, Thorlacius H. MiR-155-5p controls colon cancer cell migration via post-transcriptional regulation of Human Antigen R (HuR). Cancer Lett. 2018;421:145–51.
Wu JI. Lin YP, Tseng CW, Chen HJ, Wang LH: Crabp2 promotes metastasis of lung cancer cells via HuR and integrin β1/FAK/ERK signaling. Sci Rep. 2019;9:845.
Shu C, Yan D, Mo Y, Gu J, Shah N, He J. Long noncoding RNA lncARSR promotes epithelial ovarian cancer cell proliferation and invasion by association with HuR and miR-200 family. Am J Cancer Res. 2018;8:981–92.
Preca BT, Bajdak K, Mock K, Sundararajan V, Pfannstiel J, Maurer J, Wellner U, Hopt UT, Brummer T, Brabletz S, et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int J Cancer. 2015;137:2566–77.
Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, Dospoy PD, Augustyn A, Hight SK, Sato M, et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J Clin Invest. 2016;126:3219–35.
Qu R, Chen X, Zhang C. LncRNA ZEB1-AS1/miR-409-3p/ZEB1 feedback loop is involved in the progression of non-small cell lung cancer. Biochem Biophys Res Commun. 2018;507:450–6.
Cortés M, Sanchez-Moral L, de Barrios O, Fernández-Aceñero MJ, Martínez-Campanario MC, Esteve-Codina A, Darling DS, Győrffy B, Lawrence T, Dean DC, Postigo A. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. Embo j. 2017;36:3336–55.
Caramel J, Ligier M, Puisieux A. Pleiotropic roles for ZEB1 in cancer. Cancer Res. 2018;78:30–5.
Zhou XJ, Wu J, Shi L, Li XX, Zhu L, Sun X, Qian JY, Wang Y, Wei JF, Ding Q. PTEN expression is upregulated by a RNA-binding protein RBM38 via enhancing its mRNA stability in breast cancer. J Exp Clin Cancer Res. 2017;36:149.
Wu J, Zhou XJ, Sun X, Xia TS, Li XX, Shi L, Zhu L, Zhou WB, Wei JF, Ding Q. RBM38 is involved in TGF-β-induced epithelial-to-mesenchymal transition by stabilising zonula occludens-1 mRNA in breast cancer. Br J Cancer. 2017;117:675–84.
Zheng L, Zhang Z, Zhang S, Guo Q, Zhang F, Gao L, Ni H, Guo X, Xiang C, Xi T. RNA binding protein RNPC1 inhibits breast cancer cell metastasis via activating STARD13-correlated ceRNA network. Mol Pharm. 2018;15:2123–32.
Wampfler J, Federzoni EA, Torbett BE, Fey MF, Tschan MP. The RNA binding proteins RBM38 and DND1 are repressed in AML and have a novel function in APL differentiation. Leuk Res. 2016;41:96–102.
Huang W, Wei XL, Ni W, Cao M, Meng L, Yang H. The expression of RNA-binding protein RBM38 decreased in renal cell carcinoma and represses renal cancer cell proliferation, migration, and invasion. Tumour Biol. 2017;39:1010428317701635.
Ye J, Liang R, Bai T, Lin Y, Mai R, Wei M, Ye X, Li L, Wu F. RBM38 plays a tumor-suppressor role via stabilizing the p53-mdm2 loop function in hepatocellular carcinoma. J Exp Clin Cancer Res. 2018;37:212.
Yang X, Qu S, Wang L, Zhang H, Yang Z, Wang J, Dai B, Tao K, Shang R, Liu Z, et al. PTBP3 splicing factor promotes hepatocellular carcinoma by destroying the splicing balance of NEAT1 and pre-miR-612. Oncogene. 2018;37:6399–413.
Hou P, Li L, Chen F, Chen Y, Liu H, Li J, Bai J, Zheng J. PTBP3-mediated regulation of ZEB1 mRNA stability promotes epithelial-mesenchymal transition in breast cancer. Cancer Res. 2018;78:387–98.
Liang X, Chen W, Shi H, Gu X, Li Y, Qi Y, Xu K, Zhao A, Liu J. PTBP3 contributes to the metastasis of gastric cancer by mediating CAV1 alternative splicing. Cell Death Dis. 2018;9:569.
Sun YM, Wang WT, Zeng ZC, Chen TQ, Han C, Pan Q, Huang W, Fang K, Sun LY, Zhou YF, et al. circMYBL2, a circRNA from MYBL2, regulates FLT3 translation by recruiting PTBP1 to promote FLT3-ITD AML progression. Blood. 2019;134:1533–46.
Dang H, Takai A, Forgues M, Pomyen Y, Mou H, Xue W, Ray D, Ha KCH, Morris QD, Hughes TR, Wang XW. Oncogenic activation of the RNA binding protein NELFE and MYC signaling in hepatocellular carcinoma. Cancer Cell. 2017;32:101–114.e108.
Chen Y, Xie C, Zheng X, Nie X, Wang Z, Liu H, Zhao Y. LIN28/let-7/PD-L1 pathway as a target for cancer immunotherapy. Cancer Immunol Res. 2019;7:487–97.
Wang T, Wang G, Hao D, Liu X, Wang D, Ning N, Li X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol Cancer. 2015;14:125.
Song H, Li D, Wang X, Fang E, Yang F, Hu A, Wang J, Guo Y, Liu Y, Li H, et al. HNF4A-AS1/hnRNPU/CTCF axis as a therapeutic target for aerobic glycolysis and neuroblastoma progression. J Hematol Oncol. 2020;13:24.
Zhang L, Song D, Zhu B, Wang X. The role of nuclear matrix protein HNRNPU in maintaining the architecture of 3D genome. Semin Cell Dev Biol. 2019;90:161–7.
Nyati KK, Agarwal RG, Sharma P, Kishimoto T. Arid5a regulation and the roles of Arid5a in the inflammatory response and disease. Front Immunol. 2019;10:2790.
Hentze MW, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018;19:327–41.
Zhang M, Weng W, Zhang Q, Wu Y, Ni S, Tan C, Xu M, Sun H, Liu C, Wei P, Du X. The lncRNA NEAT1 activates Wnt/β-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. 2018;11:113.
Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–33.
Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009;41:365–70.
Sahu A, Singhal U, Chinnaiyan AM. Long noncoding RNAs in cancer: from function to translation. Trends Cancer. 2015;1:93–109.
Gloss BS, Dinger ME. The specificity of long noncoding RNA expression. Biochim Biophys Acta. 2016;1859:16–22.
Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107:1894–9.
Luan W, Wang Y, Chen X, Shi Y, Wang J, Zhang J, Qian J, Li R, Tao T, Wei W, et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget. 2015;6:13006–18.
Yao A, Xiang Y, Si YR, Fan LJ, Li JP, Li H, Guo W, He HX, Liang XJ, Tan Y, et al. PKM2 promotes glucose metabolism through a let-7a-5p/Stat3/hnRNP-A1 regulatory feedback loop in breast cancer cells. J Cell Biochem. 2019;120:6542–54.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, Zhao G. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142.
Li XX, Shi L, Zhou XJ, Wu J, Xia TS, Zhou WB, Sun X, Zhu L, Wei JF, Ding Q. The role of c-Myc-RBM38 loop in the growth suppression in breast cancer. J Exp Clin Cancer Res. 2017;36:49.
Li M, Chen H, Wu T. LIN28: a cancer stem cell promoter for immunotherapy in head and neck squamous cell carcinoma. Oral Oncol. 2019;98:92–5.
Viswanathan SR, Daley GQ. Lin28: a microRNA regulator with a macro role. Cell. 2010;140:445–9.
Peng S, Chen LL, Lei XX, Yang L, Lin H, Carmichael GG, Huang Y. Genome-wide studies reveal that Lin28 enhances the translation of genes important for growth and survival of human embryonic stem cells. Stem Cells. 2011;29:496–504.
Balzeau J, Menezes MR, Cao S, Hagan JP. The LIN28/let-7 pathway in cancer. Front Genet. 2017;8:31.
Jiang S, Baltimore D. RNA-binding protein Lin28 in cancer and immunity. Cancer Lett. 2016;375:108–13.
Manier S, Powers JT, Sacco A, Glavey SV, Huynh D, Reagan MR, Salem KZ, Moschetta M, Shi J, Mishima Y, et al. The LIN28B/let-7 axis is a novel therapeutic pathway in multiple myeloma. Leukemia. 2017;31:853–60.
Vo DT, Qiao M, Smith AD, Burns SC, Brenner AJ, Penalva LO. The oncogenic RNA-binding protein Musashi1 is regulated by tumor suppressor miRNAs. RNA Biol. 2011;8:817–28.
Velasco MX, Kosti A, Guardia GDA, Santos MC, Tegge A, Qiao M, Correa BRS, Hernández G, Kokovay E, Galante PAF, Penalva LOF. Antagonism between the RNA-binding protein Musashi1 and miR-137 and its potential impact on neurogenesis and glioblastoma development. Rna. 2019;25:768–82.
Mang Y, Li L, Ran J, Zhang S, Liu J, Li L, Chen Y, Liu J, Gao Y, Ren G. Long noncoding RNA NEAT1 promotes cell proliferation and invasion by regulating hnRNP A2 expression in hepatocellular carcinoma cells. Onco Targets Ther. 2017;10:1003–16.
Liu L, Yue H, Liu Q, Yuan J, Li J, Wei G, Chen X, Lu Y, Guo M, Luo J, Chen R. LncRNA MT1JP functions as a tumor suppressor by interacting with TIAR to modulate the p53 pathway. Oncotarget. 2016;7:15787–800.
Liu C, Zhang Y, She X, Fan L, Li P, Feng J, Fu H, Liu Q, Liu Q, Zhao C, et al. A cytoplasmic long noncoding RNA LINC00470 as a new AKT activator to mediate glioblastoma cell autophagy. J Hematol Oncol. 2018;11:77.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–200.
Xu Y, Wu W, Han Q, Wang Y, Li C, Zhang P, Xu H. Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol. 2019;9:180239.
Lo Piccolo L, Mochizuki H, Nagai Y. The lncRNA hsrω regulates arginine dimethylation of human FUS to cause its proteasomal degradation in Drosophila. J Cell Sci. 2019;132.
Castello A, Fischer B, Frese CK, Horos R, Alleaume AM, Foehr S, Curk T, Krijgsveld J, Hentze MW. Comprehensive identification of RNA-binding domains in human cells. Mol Cell. 2016;63:696–710.
Nakka KK, Chaudhary N, Joshi S, Bhat J, Singh K, Chatterjee S, Malhotra R, De A, Santra MK, Dilworth FJ, Chattopadhyay S. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc Natl Acad Sci U S A. 2015;112:E3374–83.
Frisone P, Pradella D, Di Matteo A, Belloni E, Ghigna C, Paronetto MP. SAM68: signal transduction and RNA metabolism in human cancer. Biomed Res Int. 2015;2015:528954.
Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol. 2010;12:286–93.
Hussey GS, Chaudhury A, Dawson AE, Lindner DJ, Knudsen CR, Wilce MC, Merrick WC, Howe PH. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell. 2011;41:419–31.
Howley BV, Howe PH. TGF-beta signaling in cancer: post-transcriptional regulation of EMT via hnRNP E1. Cytokine. 2019;118:19–26.
Zhang J, Xu E, Ren C, Yan W, Zhang M, Chen M, Cardiff RD, Imai DM, Wisner E, Chen X. Mice deficient in Rbm38, a target of the p53 family, are susceptible to accelerated aging and spontaneous tumors. Proc Natl Acad Sci U S A. 2014;111:18637–42.
Zhang M, Zhang J, Chen X, Cho SJ, Chen X. Glycogen synthase kinase 3 promotes p53 mRNA translation via phosphorylation of RNPC1. Genes Dev. 2013;27:2246–58.
Zhang Y, Feng X, Sun W, Zhang J, Chen X. Serine 195 phosphorylation in the RNA-binding protein Rbm38 increases p63 expression by modulating Rbm38's interaction with the Ago2-miR203 complex. J Biol Chem. 2019;294:2449–59.
Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, Aoyagi S, Guo A, Khodadadi-Jamayran A, Zhou D, et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife. 2015;4.
Jin S, Mi Y, Song J, Zhang P, Liu Y. PRMT1-RBM15 axis regulates megakaryocytic differentiation of human umbilical cord blood CD34(+) cells. Exp Ther Med. 2018;15:2563–8.
Fu XD. Non-coding RNA: a new frontier in regulatory biology. Natl Sci Rev. 2014;1:190–204.
Luo C, Cheng Y, Liu Y, Chen L, Liu L, Wei N, Xie Z, Wu W, Feng Y. SRSF2 regulates alternative splicing to drive hepatocellular carcinoma development. Cancer Res. 2017;77:1168–78.
Choi S, Sa M, Cho N, Kim KK, Park SH. Rbfox2 dissociation from stress granules suppresses cancer progression. Exp Mol Med. 2019;51:1–12.
Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;153:855–68.
Lötsch J, Klepstad P, Doehring A, Dale O. A GTP cyclohydrolase 1 genetic variant delays cancer pain. Pain. 2010;148:103–6.
Anderson P, Kedersha N, Ivanov P: Stress granules, P-bodies and cancer. Biochim Biophys Acta 2015, 1849:861-870.
Park C, Choi S, Kim YE, Lee S, Park SH, Adelstein RS, Kawamoto S, Kim KK. Stress granules contain Rbfox2 with cell cycle-related mRNAs. Sci Rep. 2017;7:11211.
Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, Martin KC. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron. 2016;89:113–28.
Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al: A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499:172-177.
Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO--multi-functional nuclear proteins. FEBS Lett. 2002;531:109–14.
Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, Ingles CJ. Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. Rna. 2002;8:1102–11.
Takayama KI. Splicing factors have an essential role in prostate cancer progression and androgen receptor signaling. Biomolecules. 2019;9.
Yang P, Chen T, Xu Z, Zhu H, Wang J, He Z. Long noncoding RNA GAPLINC promotes invasion in colorectal cancer by targeting SNAI2 through binding with PSF and NONO. Oncotarget. 2016;7:42183–94.
Zhu Z, Zhao X, Zhao L, Yang H, Liu L, Li J, Wu J, Yang F, Huang G, Liu J. p54(nrb)/NONO regulates lipid metabolism and breast cancer growth through SREBP-1A. Oncogene. 2016;35:1399–410.
McGregor SM, Alikhan MB, John RA, Kotler H, Bridge JA, Mujacic I, Kadri S, Segal J, Krausz T. Melanotic PEComa of the sinonasal mucosa with NONO-TFE3 fusion: an elusive mimic of sinonasal melanoma. Am J Surg Pathol. 2017;41:717–22.
Mitchell SA, Spriggs KA, Coldwell MJ, Jackson RJ, Willis AE. The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol Cell. 2003;11:757–71.
Martinez-Useros J, Garcia-Carbonero N, Li W, Fernandez-Aceñero MJ, Cristobal I, Rincon R, Rodriguez-Remirez M, Borrero-Palacios A, Garcia-Foncillas J. UNR/CSDE1 expression is critical to maintain invasive phenotype of colorectal cancer through regulation of c-MYC and epithelial-to-mesenchymal transition. J Clin Med. 2019;8.
Mihailovich M, Militti C, Gabaldón T, Gebauer F. Eukaryotic cold shock domain proteins: highly versatile regulators of gene expression. Bioessays. 2010;32:109–18.
Wurth L, Papasaikas P, Olmeda D, Bley N, Calvo GT, Guerrero S, Cerezo-Wallis D, Martinez-Useros J, García-Fernández M, Hüttelmaier S, et al. UNR/CSDE1 drives a post-transcriptional program to promote melanoma invasion and metastasis. Cancer Cell. 2016;30:694–707.
Martinez-Useros J, Georgiev-Hristov T, Fernández-Aceñero MJ, Borrero-Palacios A, Indacochea A, Guerrero S, Li W, Cebrián A, Gómez Del Pulgar T, Puime-Otin A, et al. UNR/CDSE1 expression as prognosis biomarker in resectable pancreatic ductal adenocarcinoma patients: A proof-of-concept. PLoS One. 2017;12:e0182044.
Frankiw L, Baltimore D, Li G. Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol. 2019;19:675–87.
David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–64.
Chabot B, Shkreta L. Defective control of pre-messenger RNA splicing in human disease. J Cell Biol. 2016;212:13–27.
Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976–86.
Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689–701.
Coltri PP, Dos Santos MGP, da Silva GHG. Splicing and cancer: challenges and opportunities. Wiley Interdiscip Rev RNA. 2019;10:e1527.
Wang Y, Bao Y, Zhang S, Wang Z. Splicing dysregulation in cancer: from mechanistic understanding to a new class of therapeutic targets. Sci China Life Sci. 2020;63:469–84.
Gonçalves V, Henriques AF, Pereira JF, Costa AN, Moyer MP, Moita LF, Gama-Carvalho M, Matos P, Jordan P. Corrigendum: phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. Rna. 2016;22:166.
Chen L, Luo C, Shen L, Liu Y, Wang Q, Zhang C, Guo R, Zhang Y, Xie Z, Wei N, et al. SRSF1 Prevents DNA damage and promotes tumorigenesis through regulation of DBF4B Pre-mRNA splicing. Cell Rep. 2017;21:3406–13.
Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, Chen L, Monterroza DD, Li H, DeKelver RC, et al. SRSF2 is essential for hematopoiesis, and its myelodysplastic syndrome-related mutations dysregulate alternative pre-mRNA splicing. Mol Cell Biol. 2015;35:3071–82.
Kędzierska H, Popławski P, Hoser G, Rybicka B, Rodzik K, Sokół E, Bogusławska J, Tański Z, Fogtman A, Koblowska M, Piekiełko-Witkowska A. Decreased expression of SRSF2 splicing factor inhibits apoptotic pathways in renal cancer. Int J Mol Sci. 2016;17.
Maji D, Grossfield A, Kielkopf CL. Structures of SF3b1 reveal a dynamic Achilles heel of spliceosome assembly: implications for cancer-associated abnormalities and drug discovery. Biochim Biophys Acta Gene Regul Mech. 2019;1862:194440.
Hosono N. Genetic abnormalities and pathophysiology of MDS. Int J Clin Oncol. 2019;24:885–92.
Foy A, McMullin MF. Somatic SF3B1 mutations in myelodysplastic syndrome with ring sideroblasts and chronic lymphocytic leukaemia. J Clin Pathol. 2019;72:778–82.
Smeets MF, Tan SY, Xu JJ, Anande G, Unnikrishnan A, Chalk AM, Taylor SR, Pimanda JE, Wall M, Purton LE, Walkley CR. Srsf2(P95H) initiates myeloid bias and myelodysplastic/myeloproliferative syndrome from hemopoietic stem cells. Blood. 2018;132:608–21.
Skrdlant L, Stark JM, Lin RJ. Myelodysplasia-associated mutations in serine/arginine-rich splicing factor SRSF2 lead to alternative splicing of CDC25C. BMC Mol Biol. 2016;17:18.
Stoehr A, Yang Y, Patel S, Evangelista AM, Aponte A, Wang G, Liu P, Boylston J, Kloner PH, Lin Y, et al. Prolyl hydroxylation regulates protein degradation, synthesis, and splicing in human induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc Res. 2016;110:346–58.
Liang G, Meng W, Huang X, Zhu W, Yin C, Wang C, Fassan M, Yu Y, Kudo M. Xiao S, et al: miR-196b-5p-mediated downregulation of TSPAN12 and GATA6 promotes tumor progression in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2020;117:4347–57.
Qin M, Wei G, Sun X. Circ-UBR5: an exonic circular RNA and novel small nuclear RNA involved in RNA splicing. Biochem Biophys Res Commun. 2018;503:1027–34.
Kim EJ, Kim JS, Lee S, Lee H, Yoon JS, Hong JH, Chun SH, Sun S, Won HS, Hong SA, et al. QKI, a miR-200 target gene, suppresses epithelial-to-mesenchymal transition and tumor growth. Int J Cancer. 2019;145:1585–95.
de Miguel FJ, Pajares MJ, Martínez-Terroba E, Ajona D, Morales X, Sharma RD, Pardo FJ, Rouzaut A, Rubio A, Montuenga LM, Pio R. A large-scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Mol Oncol. 2016;10:1437–49.
Chénard CA, Richard S. New implications for the QUAKING RNA binding protein in human disease. J Neurosci Res. 2008;86:233–42.
Hojo H, Yashiro Y, Noda Y, Ogami K, Yamagishi R, Okada S, Hoshino SI, Suzuki T. The RNA-binding protein QKI-7 recruits the poly(A) polymerase GLD-2 for 3′ adenylation and selective stabilization of microRNA-122. J Biol Chem. 2020;295:390–402.
Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, Fang Z, Fang R, Lu D, Ji H, Hui J. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014;10:e1004289.
Coomer AO, Black F, Greystoke A, Munkley J, Elliott DJ. Alternative splicing in lung cancer. Biochim Biophys Acta Gene Regul Mech. 2019;1862:194388.
Bechara EG, Sebestyén E, Bernardis I, Eyras E, Valcárcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell. 2013;52:720–33.
Hernández J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcárcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13:466–72.
Collins KM, Kainov YA, Christodolou E, Ray D, Morris Q, Hughes T, Taylor IA, Makeyev EV, Ramos A. An RRM-ZnF RNA recognition module targets RBM10 to exonic sequences to promote exon exclusion. Nucleic Acids Res. 2017;45:6761–74.
Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakàcs A, Coppola L, Karni R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71:4464–72.
Li L, Yang Y, Wu M, Yu Z, Wang C, Dou G, He H, Wang H, Yang N, Qi H, Xu X. β-Asarone induces apoptosis and cell cycle arrest of human glioma U251 cells via suppression of HnRNP A2/B1-mediated pathway in vitro and in vivo. Molecules. 2018;23.
Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 2009;33:365–76.
Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18:18–30.
Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506.
Xu C, Zhang J. A different perspective on alternative cleavage and polyadenylation. Nat Rev Genet. 2020;21:63.
Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–84.
Singh P, Alley TL, Wright SM, Kamdar S, Schott W, Wilpan RY, Mills KD, Graber JH. Global changes in processing of mRNA 3′ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009;69:9422–30.
Gruber AR, Martin G, Müller P, Schmidt A, Gruber AJ, Gumienny R, Mittal N, Jayachandran R, Pieters J, Keller W, et al. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nat Commun. 2014;5:5465.
Hoffman Y, Bublik DR, Ugalde AP, Elkon R, Biniashvili T, Agami R, Oren M, Pilpel Y. 3′UTR shortening potentiates microRNA-based repression of pro-differentiation genes in proliferating human cells. PLoS Genet. 2016;12:e1005879.
Gruber AR, Martin G, Keller W, Zavolan M. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 2012;9:1405–12.
Martin G, Gruber AR, Keller W, Zavolan M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3' UTR length. Cell Rep. 2012;1:753–63.
Hardy JG, Norbury CJ. Cleavage factor Im (CFIm) as a regulator of alternative polyadenylation. Biochem Soc Trans. 2016;44:1051–7.
Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510:412–6.
D'Ambrogio A, Nagaoka K, Richter JD. Translational control of cell growth and malignancy by the CPEBs. Nat Rev Cancer. 2013;13:283–90.
Bava FA, Eliscovich C, Ferreira PG, Miñana B, Ben-Dov C, Guigó R, Valcárcel J, Méndez R. CPEB1 coordinates alternative 3′-UTR formation with translational regulation. Nature. 2013;495:121–5.
Kronja I, Orr-Weaver TL. Translational regulation of the cell cycle: when, where, how and why? Philos Trans R Soc Lond B Biol Sci. 2011;366:3638–52.
Xu M, Fang S, Song J, Chen M, Zhang Q, Weng Q, Fan X, Chen W, Wu X, Wu F, et al. CPEB1 mediates hepatocellular carcinoma cancer stemness and chemoresistance. Cell Death Dis. 2018;9:957.
Xiong H, Chen R, Liu S, Lin Q, Chen H, Jiang Q. MicroRNA-183 induces epithelial-mesenchymal transition and promotes endometrial cancer cell migration and invasion in by targeting CPEB1. J Cell Biochem. 2018;119:8123–37.
Ortiz-Zapater E, Pineda D, Martínez-Bosch N, Fernández-Miranda G, Iglesias M, Alameda F, Moreno M, Eliscovich C, Eyras E, Real FX, et al. Key contribution of CPEB4-mediated translational control to cancer progression. Nat Med. 2011;18:83–90.
Pérez-Guijarro E, Karras P, Cifdaloz M, Martínez-Herranz R, Cañón E, Graña O, Horcajada-Reales C, Alonso-Curbelo D, Calvo TG, Gómez-López G, et al. Lineage-specific roles of the cytoplasmic polyadenylation factor CPEB4 in the regulation of melanoma drivers. Nat Commun. 2016;7:13418.
Calderone V, Gallego J, Fernandez-Miranda G, Garcia-Pras E, Maillo C, Berzigotti A, Mejias M, Bava FA, Angulo-Urarte A, Graupera M, et al. Sequential functions of CPEB1 and CPEB4 regulate pathologic expression of vascular endothelial growth factor and angiogenesis in chronic liver disease. Gastroenterology. 2016;150:982–997.e930.
Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–26.
Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2011;39:D66–9.
Fallmann J, Sedlyarov V, Tanzer A, Kovarik P, Hofacker IL. AREsite2: an enhanced database for the comprehensive investigation of AU/GU/U-rich elements. Nucleic Acids Res. 2016;44:D90–5.
Stoecklin G, Anderson P. Posttranscriptional mechanisms regulating the inflammatory response. Adv Immunol. 2006;89:1–37.
Waris S, García-Mauriño SM, Sivakumaran A, Beckham SA, Loughlin FE, Gorospe M, Díaz-Moreno I, Wilce MCJ, Wilce JA. TIA-1 RRM23 binding and recognition of target oligonucleotides. Nucleic Acids Res. 2017;45:4944–57.
Hitti E, Bakheet T, Al-Souhibani N, Moghrabi W, Al-Yahya S, Al-Ghamdi M, Al-Saif M, Shoukri MM, Lánczky A, Grépin R, et al. Systematic analysis of AU-rich element expression in cancer reveals common functional clusters regulated by key RNA-binding proteins. Cancer Res. 2016;76:4068–80.
Khabar KS. Hallmarks of cancer and AU-rich elements. Wiley Interdiscip Rev RNA. 2017;8.
Ge Z, Quek BL, Beemon KL, Hogg JR. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. Elife. 2016;5.
Filippova N, Yang X, Wang Y, Gillespie GY, Langford C, King PH, Wheeler C, Nabors LB. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol Cancer Res. 2011;9:648–59.
Guo X, Connick MC, Vanderhoof J, Ishak MA, Hartley RS. MicroRNA-16 modulates HuR regulation of cyclin E1 in breast cancer cells. Int J Mol Sci. 2015;16:7112–32.
Stöhr N, Köhn M, Lederer M, Glass M, Reinke C, Singer RH, Hüttelmaier S. IGF2BP1 promotes cell migration by regulating MK5 and PTEN signaling. Genes Dev. 2012;26:176–89.
Gutschner T, Hämmerle M, Pazaitis N, Bley N, Fiskin E, Uckelmann H, Heim A, Groβ M, Hofmann N, Geffers R, et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology. 2014;59:1900–11.
Dai N, Ji F, Wright J, Minichiello L, Sadreyev R, Avruch J. IGF2 mRNA binding protein-2 is a tumor promoter that drives cancer proliferation through its client mRNAs IGF2 and HMGA1. Elife. 2017;6.
Ye S, Song W, Xu X, Zhao X, Yang L. IGF2BP2 promotes colorectal cancer cell proliferation and survival through interfering with RAF-1 degradation by miR-195. FEBS Lett. 2016;590:1641–50.
Lu W, Ning H, Gu L, Peng H, Wang Q, Hou R, Fu M, Hoft DF, Liu J. MCPIP1 selectively destabilizes transcripts associated with an antiapoptotic gene expression program in breast cancer cells that can elicit complete tumor regression. Cancer Res. 2016;76:1429–40.
Hämmerle M, Gutschner T, Uckelmann H, Ozgur S, Fiskin E, Gross M, Skawran B, Geffers R, Longerich T, Breuhahn K, et al. Posttranscriptional destabilization of the liver-specific long noncoding RNA HULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology. 2013;58:1703–12.
Mizutani R, Imamachi N, Suzuki Y, Yoshida H, Tochigi N, Oonishi T, Suzuki Y, Akimitsu N. Oncofetal protein IGF2BP3 facilitates the activity of proto-oncogene protein eIF4E through the destabilization of EIF4E-BP2 mRNA. Oncogene. 2016;35:3495–502.
Mofatteh M, Bullock SL. SnapShot: subcellular mRNA Localization. Cell. 2017;169:178–178.e171.
Shav-Tal Y, Singer RH. RNA localization. J Cell Sci. 2005;118:4077–81.
Medioni C, Mowry K, Besse F. Principles and roles of mRNA localization in animal development. Development. 2012;139:3263–76.
Díaz-Muñoz MD, Kiselev VY, Le Novère N, Curk T, Ule J, Turner M. Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells. Nat Commun. 2017;8:530.
Hamdollah Zadeh MA, Amin EM, Hoareau-Aveilla C, Domingo E, Symonds KE, Ye X, Heesom KJ, Salmon A, D'Silva O, Betteridge KB, et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol Oncol. 2015;9:167–78.
Liu Y, Liu R, Yang F, Cheng R, Chen X, Cui S, Gu Y, Sun W, You C, Liu Z, et al: miR-19a promotes colorectal cancer proliferation and migration by targeting TIA1. Mol Cancer 2017, 16:53.
Wächter K, Köhn M, Stöhr N, Hüttelmaier S. Subcellular localization and RNP formation of IGF2BPs (IGF2 mRNA-binding proteins) is modulated by distinct RNA-binding domains. Biol Chem. 2013;394:1077–90.
Huang X, Zhang H, Guo X, Zhu Z, Cai H, Kong X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J Hematol Oncol. 2018;11:88.
Hüttelmaier S, Zenklusen D, Lederer M, Dictenberg J, Lorenz M, Meng X, Bassell GJ, Condeelis J, Singer RH. Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature. 2005;438:512–5.
Bell JL, Wächter K, Mühleck B, Pazaitis N, Köhn M, Lederer M, Hüttelmaier S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. 2013;70:2657–75.
Gu W, Pan F, Singer RH. Blocking beta-catenin binding to the ZBP1 promoter represses ZBP1 expression, leading to increased proliferation and migration of metastatic breast-cancer cells. J Cell Sci. 2009;122:1895–905.
Hamilton KE, Noubissi FK, Katti PS, Hahn CM, Davey SR, Lundsmith ET, Klein-Szanto AJ, Rhim AD, Spiegelman VS, Rustgi AK. IMP1 promotes tumor growth, dissemination and a tumor-initiating cell phenotype in colorectal cancer cell xenografts. Carcinogenesis. 2013;34:2647–54.
Hamilton KE, Chatterji P, Lundsmith ET, Andres SF, Giroux V, Hicks PD, Noubissi FK, Spiegelman VS, Rustgi AK. Loss of stromal IMP1 promotes a tumorigenic microenvironment in the colon. Mol Cancer Res. 2015;13:1478–86.
Gu W, Katz Z, Wu B, Park HY, Li D, Lin S, Wells AL, Singer RH. Regulation of local expression of cell adhesion and motility-related mRNAs in breast cancer cells by IMP1/ZBP1. J Cell Sci. 2012;125:81–91.
Rebucci M, Sermeus A, Leonard E, Delaive E, Dieu M, Fransolet M, Arnould T, Michiels C. miRNA-196b inhibits cell proliferation and induces apoptosis in HepG2 cells by targeting IGF2BP1. Mol Cancer. 2015;14:79.
Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young-Pearse TL, Flanagan JG. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell. 2014;158:368–82.
Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14:261–78.
Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016;352:1413–6.
Truitt ML, Ruggero D. New frontiers in translational control of the cancer genome. Nat Rev Cancer. 2017;17:332.
Piserà A, Campo A, Campo S. Structure and functions of the translation initiation factor eIF4E and its role in cancer development and treatment. J Genet Genomics. 2018;45:13–24.
Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D. Differential requirements for eIF4E dose in normal development and cancer. Cell. 2015;162:59–71.
Holcik M, Korneluk RG. Functional characterization of the X-linked inhibitor of apoptosis (XIAP) internal ribosome entry site element: role of La autoantigen in XIAP translation. Mol Cell Biol. 2000;20:4648–57.
Petz M, Them N, Huber H, Beug H, Mikulits W. La enhances IRES-mediated translation of laminin B1 during malignant epithelial to mesenchymal transition. Nucleic Acids Res. 2012;40:290–302.
Petz M, Them NC, Huber H, Mikulits W. PDGF enhances IRES-mediated translation of Laminin B1 by cytoplasmic accumulation of La during epithelial to mesenchymal transition. Nucleic Acids Res. 2012;40:9738–49.
Miles WO, Tschöp K, Herr A, Ji JY, Dyson NJ. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012;26:356–68.
Miles WO, Lembo A, Volorio A, Brachtel E, Tian B, Sgroi D, Provero P, Dyson N. Alternative polyadenylation in triple-negative breast tumors allows NRAS and c-JUN to bypass PUMILIO posttranscriptional regulation. Cancer Res. 2016;76:7231–41.
Bao X, Guo X, Yin M, Tariq M, Lai Y, Kanwal S, Zhou J, Li N, Lv Y, Pulido-Quetglas C, et al. Capturing the interactome of newly transcribed RNA. Nat Methods. 2018;15:213–20.
Kung JT, Kesner B, An JY, Ahn JY, Cifuentes-Rojas C, Colognori D, Jeon Y, Szanto A, del Rosario BC, Pinter SF, et al. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol Cell. 2015;57:361–75.
Kai M. Roles of RNA-binding proteins in DNA damage response. Int J Mol Sci. 2016;17:310.
Wei C, Xiao R, Chen L, Cui H, Zhou Y, Xue Y, Hu J, Zhou B, Tsutsui T, Qiu J, et al. RBFox2 binds nascent RNA to globally regulate polycomb complex 2 targeting in mammalian genomes. Mol Cell. 2016;62:875–89.
Xiao R, Chen JY, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X, et al. Pervasive chromatin-RNA binding protein interactions enable RNA-based regulation of transcription. Cell. 2019;178:107–21 e118.
Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell. 2009;35:228–39.
Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14:318–28.
Chand SN, Zarei M, Schiewer MJ, Kamath AR, Romeo C, Lal S, Cozzitorto JA, Nevler A, Scolaro L, Londin E, et al. Posttranscriptional regulation of PARG mRNA by HuR facilitates DNA repair and resistance to PARP inhibitors. Cancer Res. 2017;77:5011–25.
Lin MZ, Marzec KA, Martin JL, Baxter RC. The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents. Oncogene. 2014;33:85–96.
Abbotts R, Wilson DM 3rd. Coordination of DNA single strand break repair. Free Radic Biol Med. 2017;107:228–44.
Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389:2430–42.
Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329:18–25.
Nowsheen S, Bonner JA, Yang ES. The poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99:331–8.
Lee M, Kim B, Kim VN. Emerging roles of RNA modification: m(6)A and U-tail. Cell. 2014;158:980–7.
Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–24.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69:1028–1038.e1026.
Jaffrey SR, Kharas MG. Emerging links between m(6)A and misregulated mRNA methylation in cancer. Genome Med. 2017;9:2.
Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, Zhao JC. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci. 2018;21:195–206.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Roos M, Pradère U, Ngondo RP, Behera A, Allegrini S, Civenni G, Zagalak JA, Marchand JR, Menzi M, Towbin H, et al. A small-molecule inhibitor of Lin28. ACS Chem Biol. 2016;11:2773–81.
Carabet LA, Leblanc E, Lallous N, Morin H, Ghaidi F, Lee J, Rennie PS, Cherkasov A. Computer-aided discovery of small molecules targeting the RNA splicing activity of hnRNP A1 in castration-resistant prostate cancer. Molecules. 2019;24.
He T, Jin M, Xu C, Ma Z, Wu F, Zhang X. The homeostasis-maintaining metabolites from bacterial stress response to bacteriophage infection suppress tumor metastasis. Oncogene. 2018;37:5766–79.
Wu X, Lan L, Wilson DM, Marquez RT, Tsao WC, Gao P, Roy A, Turner BA, McDonald P, Tunge JA, et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem Biol. 2015;10:1476–84.
Lang M, Berry D, Passecker K, Mesteri I, Bhuju S, Ebner F, Sedlyarov V, Evstatiev R, Dammann K, Loy A, et al. HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis. Cancer Res. 2017;77:2424–38.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10:818–27.
Kudinov AE, Karanicolas J, Golemis EA, Boumber Y. Musashi RNA-binding proteins as cancer drivers and novel therapeutic targets. Clin Cancer Res. 2017;23:2143–53.
Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–406.
Wang T, Xiao G, Chu Y, Zhang MQ, Corey DR, Xie Y. Design and bioinformatics analysis of genome-wide CLIP experiments. Nucleic Acids Res. 2015;43:5263–74.
Sundararaman B, Zhan L, Blue SM, Stanton R, Elkins K, Olson S, Wei X, Van Nostrand EL, Pratt GA, Huelga SC, et al. Resources for the comprehensive discovery of functional RNA elements. Mol Cell. 2016;61:903–13.
Blackinton JG, Keene JD. Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin Cell Dev Biol. 2014;34:44–54.
Acknowledgements
Not applicable
Funding
This work was supported by the National Natural Science Foundation of China, No. 81903857, 81702957; the Special Postdoctoral Funding Scheme of China, No. 2019 T120485; the Postdoctoral Science Foundation of China, No. 2017 M620230; the Basic Scientific Research Business Expense Project of China Pharmaceutical University, No. 2632020ZD10; and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
Hai Qin, Xiaoman Li, Yaqin Yuan, Haiwei Ni, and Yichen Liu wrote this paper. Tao Xi and Lufeng Zheng reviewed this paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qin, H., Ni, H., Liu, Y. et al. RNA-binding proteins in tumor progression. J Hematol Oncol 13, 90 (2020). https://doi.org/10.1186/s13045-020-00927-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00927-w