
Abstract
Background
De novo acute myeloid leukemia (AML) with concurrent DNMT3A, FLT3 and NPM1 mutations (AMLDNMT3A/FLT3/NPM1) has been suggested to represent a unique AML subset on the basis of integrative genomic analysis, but the clinical features of such patients have not been characterized systematically.
Methods
We assessed the features of patients (n = 178) harboring mutations in DNMT3A, FLT3 and/or NPM1, including an index group of AMLDNMT3A/FLT3/NPM1 patients.
Results
Patients with AMLDNMT3A/FLT3/NPM1 (n = 35) were significantly younger (median, 56.0 vs. 62.0 years; p = 0.025), mostly women (65.7% vs. 46.9%; p = 0.045), and presented with a higher percentage of bone marrow blasts (p < 0.001) and normal cytogenetics (p = 0.024) in comparison to patients within other mutation groups in this study. Among patients <60 years old, those with AMLDNMT3A/FLT3/NPM1 had a shorter event-free survival (EFS) (p = 0.047). DNMT3A mutations and not FLT3 or NPM1 mutations were independently associated with overall survival (OS) (p = 0.026). Within mutation subgroups, patients with AMLDNMT3A/NPM1 had a significantly shorter OS compared to those with AMLFLT3-ITD/NPM1 (p = 0.047) suggesting that the adverse impact of DNMT3A mutations is more pronounced than that of FLT3- ITD among patients with NPM1 mutation.
Conclusions
DNMT3A has a significant dominant effect on the clinical features and outcomes of de novo AML patients with concurrent DNMT3A, FLT3 and NPM1 mutations.
Similar content being viewed by others

Background
DNMT3A, FLT3, and NPM1 mutations are among the most common genomic alterations in de novo acute myeloid leukemia (AML) and play a key role in the pathogenesis and evolution of the disease, particularly in the absence of AML-associated recurrent cytogenetic abnormalities [1],[2]. DNMT3A encodes a DNA methyltransferase that catalyzes the addition of a methyl group to cytosine residues in CpG islands resulting in reduced expression of downstream genes. DNMT3A mutations, most commonly DNMT3AR882, are significantly enriched in patients with intermediate risk cytogenetics [1]-[4]. Mutations resulting in constitutive FLT3 activation include internal tandem duplication (FLT3-ITD) mutations and tyrosine kinase domain (FLT3-TKD) point mutations; FLT3-ITD in particular is a known adverse prognostic indicator in AML patients [2],[5]-[13]. NPM1 mutations are generally associated with more favorable outcomes in cytogenetically normal (CN)-AML in the absence of FLT3-ITD [6],[14].
Integrative genomic analysis of de novo AML by The Cancer Genome Atlas (TCGA) consortium recently identified a subset of AML patients in which DNMT3A, FLT3, and NPM1 mutations coexist at a higher frequency than would be expected for a chance occurrence [1]. Importantly, while this AML subset (herein referred to as AMLDNMT3A/FLT3/NPM1) seems to have unique characteristics at the mRNA, miRNA, and epigenetic levels [1], its clinical features are not known. However, whereas the clinical implications of FLT3 and NPM1 mutations in de novo AML are largely well established, the impact of DNMT3A mutations on NPM1 and/or FLT3 mutations remains poorly understood especially in patients with AMLDNMT3A/FLT3/NPM1[15],[16]. Several studies have suggested that the presence of DNMT3A mutations in AML is associated with poor clinical outcomes [3],[4],[17],[18]. In one study, patients with AML harboring both DNMT3A and FLT3-ITD mutations relapsed more often than patients with either DNMT3A or FLT3-ITD alone [19]. Yet, in other studies DNMT3A mutations were not independently predictive of clinical outcomes in the overall AML population or specific subgroups [2],[15],[19]-[21].
In this study, we assess the clinical and outcome characteristics of patients with AMLDNMT3A/FLT3/NPM1 as an index group and compare them to those with de novo AML harboring other mutation combinations involving DNMT3A, FLT3 and/or NPM1 to further elucidate the clinical effects of these mutations when they occur concurrently.
Results
Study group characteristics
The study group consisted of 178 de novo AML patients with one, two, or three mutations involving the DNMT3A, FLT3 and/or NPM1 genes. The composition of the mutation subgroups is illustrated in Figure 1. The MD Anderson Cancer Center (MDACC) group (n = 85) consisted of 43 women and 42 men with a median age of 64 years (range, 23 to 91 years). They included 18 patients with AMLDNMT3A/FLT3/NPM1 and 67 patients with the following mutations: FLT3 and NPM1 (AMLFLT3/NPM1, n = 22); NPM1 only (AMLNPM1, n = 15); DNMT3A only (AMLDNMT3A, n = 13); DNMT3A and FLT3 (AMLDNMT3A/FLT3, n = 8); DNMT3A and NPM1 (AMLDNMT3A/NPM1, n = 7); and, FLT3 only (AMLFLT3, n = 2). The TCGA group (n = 93) consisted of 47 women and 46 men with a median age of 58 years (range, 21 to 83 years). They included 17 patients with AMLDNMT3A/FLT3/NPM1 and 76 patients with the following mutations: FLT3 and NPM1 (AMLFLT3/NPM1, n = 12); NPM1 only (AMLNPM1, n = 14); DNMT3A only (AMLDNMT3A, n = 19); DNMT3A and FLT3 (AMLDNMT3A/FLT3, n = 4); DNMT3A and NPM1 (AMLDNMT3A/NPM1, n = 10); and, FLT3 only (AMLFLT3, n = 17).

Mutation subgroups of de novo acute myeloid leukemia included in the study.
Characteristics of AMLDNMT3A/FLT3/NPM1
The characteristics of patients within mutation subgroups are summarized in Table 1. Since patients with concomitant mutations in DNMT3A, FLT3 and NPM1 (index group) had been reported to possibly represent a distinctive genomic subset of de novo AML with unique characteristics at the mRNA, miRNA, and epigenetic levels [1], their features were first compared to those of patients in other mutation groups. Patients with AMLDNMT3A/FLT3/NPM1 were significantly younger (median 56.0 vs. 62.0 years; p = 0.025). Most were women (65.7% vs. 46.9%; p = 0.045) who presented with a heavy disease burden, namely an elevated WBC (50.9 vs. 19.0 × 103/μL; p = 0.006) and BM blast counts (83.5% vs. 71.0%, p < 0.001).
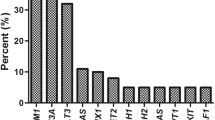
Morphologically, AMLDNMT3A/FLT3/NPM1 tended to be associated with myelomonocytic blast morphology, which corresponds to the M4 and M5 categories in the French American British (FAB) classification [22]. Among 32 AMLDNMT3A/FLT3/NPM1 cases with FAB classification data, 10 (31%) were M4; 8 (25%) were M5; 6 (19%) were M1; 6 (19%) were M2; 1 (3%) was M0; and, 1 (3%) was refractory anemia with excess blasts in transformation. None of these cases displayed sufficient dysplasia in background hematopoietic elements to warrant classification as AML with myelodysplasia-related changes. In most cases blasts showed "cup-like" nuclear morphology, which had been described previously in association with FLT3-ITD and NPM1 mutations [23],[24]. Namely, blasts with cup-like nuclear morphology were identified in 17/18 (94.4%) AMLDNMT3A/FLT3/NPM1 cases and comprised a variable subset of total blasts, ranging from 3-53% (median 9%; mean 16.1%). In 10/18 (55.5%) cases, such blasts comprised >10% of total blasts.
By flow cytometry immunophenotyping (MDACC group), all AMLDNMT3A/FLT3/NPM1 had a definite myeloid immunophenotype. In all cases, blasts were positive for CD13, CD33, and CD123 and negative for surface and cytoplasmic CD3 expression. Myeloperoxidase expression was detected in 11/14 (78.6%) cases. Blasts were positive for CD34 in 13/18 (72.2%) cases. In accordance with myelomonocytic morphology, blasts expressed one or more monocyte-associated antigens (CD4, CD14, CD64) in 11/18 (61.1%) cases. In one case, myeloid blasts co-expressed the B-cell marker CD19. (Additional file 1: Figure S1).
A significant association was observed between AMLDNMT3A/FLT3/NPM1 and diploid karyotype (CN-AML) (p = 0.024). Among 33 AMLDNMT3A/FLT3/NPM1 patients with available cytogenetics data, 32 (97%) had intermediate-risk cytogenetics and included 28 (85%) CN-AML. Only 1 (3%) patient had poor-risk cytogenetics. Notably, none of the patients had recurrent AML-associated cytogenetic abnormalities.
A summary of mutations in genes assessed in both the MDACC and TCGA groups is provided in Figure 2a. All DNMT3A variants detected in patients with AMLDNMT3A/FLT3/NPM1 were missense substitutions in exon 23, and most involved codon 882 resulting in the substitution of arginine by histidine (14/35; 40%) or cysteine (14/35; 40%). Mutations in other codons - 882 (arginine > serine), 901 (leucine > proline), 874 (tyrosine-stop), 803 (arginine to serine), 855 (lysine to threonine) and 723 (frameshift) - were detected with less frequency. (Figure 2b) (Additional file 2: Table S1).

Schematic representation of mutations. (a) Mutation status of genes assessed in all patients in the study group. Each column represents an individual case. The bottom rows represent, respectively, the cytogenetic risk group (CG) and mutation group (MG) per corresponding color designation. Assessed genes in which mutations were not detected are not included in this diagram. Mutation detection techniques in the MDACC and TCGA groups were different (see Methods section). (b) Distribution of genomic variants within DNMT3A detected in the study group. PWWP represents a region encoding for a highly conserved proline-tryptophan-tryptophan-proline motif. ZNF represents the ADD (ATRX, DNMT3, and DNMT3L)-type zinc finger domain.
FLT3 mutations in the AMLDNMT3A/FLT3/NPM1 group included FLT3-ITD in 24/35 (69%), FLT3-TKDD835 in 9/35 (26%), and simultaneous FLT3-ITD/TKDD835 mutations in 2/35 (6%). There was no significant difference in FLT3 mutation types between the AMLDNMT3A/FLT3/NPM1 and the other mutations groups (p = 0.108) (Additional file 2: Table S2). NPM1 mutations uniformly involved exon 12 in all MDACC cases. Other mutations observed in AMLDNMT3A/FLT3/NPM1 included IDH1 (17.1%), NRAS (8.5%), KIT (2.8%), IDH2 (2.8%), CEBPA (2.8%) and NOTCH1 (2.8%). None of the cases had mutations in TP53.
To assess the relative dominance of mutations in individual cases within the AMLDNMT3A/FLT3/NPM1 group, we estimated the variant frequency of each of the mutant DNMT3A, FLT3, and NPM1 alleles in a subset MDACC samples and found them to be generally commensurate, suggesting that they commonly co-occur within a dominant clone (Figure 3). In addition, the estimated variant frequency suggested that these mutations are heterozygous.

Estimated DNMT3A, FLT3 , and NPM1 variant (allelic) frequencies in a subset of de novo acute myeloid leukemia samples harboring all three mutations.
Prognostic impact of concurrent DNMT3A, FLT3, and NPM1 mutations
Follow-up data was available for 84/85 patients in the MDACC group with a median follow up of 11.8 months (range, 0.6-69.0 months). Of these patients, 50/84 presented to our institution at diagnosis prior to induction chemotherapy (4/50 with AMLDNMT3A/FLT3/NPM1), 9 presented with primary refractory disease (2/9 with AMLDNMT3A/FLT3/NPM1), and 20 presented in relapse (10/20 with AMLDNMT3A/FLT3/NPM1). An additional 5 patients (2/5 with AMLDNMT3A/FLT3/NPM1) were in remission at the time of referral to our institution. Allogeneic SCT therapy was administered to 25 patients (7/25 with AMLDNMT3A/FLT3/NPM1) in the MDACC group. When the actual last follow up date was considered, no difference in OS was observed between those who received allo-SCT and those who did not (p = 0.203); similarly, no difference in OS was observed between AMLDNMT3A/FLT3/NPM1 patients who received allo-SCT and those who did not (p = 0.186). At last follow up, 32 MDACC patients (11 AMLDNMT3A/FLT3/NPM1) were dead of disease. Importantly, there was no identifiable bias in survival outcomes on the basis of therapy selection (high-intensity vs. low-intensity, and "3 + 7" vs. "non-3 + 7") for any of the prognostic parameters assessed by univariate or multivariate modeling in the MDACC group. Survival data was available for 87/93 patients in the TCGA group with a median follow up of 9.2 months (range, 0.9-62.8 months). At last follow up, 60 patients (13 AMLDNMT3A/FLT3/NPM1) in the TCGA group were dead of disease, the majority during first relapse.
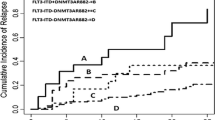
Younger (<60 years) patients with AMLDNMT3A/FLT3/NPM1 had a significantly shorter EFS (p = 0.047) and a tendency towards shorter OS (p = 0.095) compared to those in the other mutation groups (Figure 4a-b). Although patients with AMLDNMT3A/FLT3/NPM1 tended to have shorter OS compared to those in the other mutation groups, the difference was not significant in the entire group as well as within the CN-AML group (p = 0.211 and p = 0.209, respectively). Importantly, there was no significant difference in OS among AMLDNMT3A/FLT3/NPM1 patients with FLT3-ITD and FLT3-TKD mutations types (p = 0.822).

Survival analysis by mutation status. Overall survival (a) and event-free survival (b) of de novo acute myeloid leukemia patients younger than 60 years of age with concomitant mutations in DNMT3A, FLT3, and NPM1 (AMLDNMT3A/FLT3/NPM1) in comparison to those within other mutation subgroups in this study. Overall survival (c) and event-free survival (d) of patients with AML harboring NPM1 mutation in conjunction with wild-type DNMT3A (NPM1 group) or mutant DNMT3A (NPM1/DNMT3A group). Overall survival (e) of patients with CN-AML harboring NPM1 mutations with concomitant FLT3 (NPM1/FLT3 group) or DNMT3A (NPM1/DNMT3A group).
To begin understanding the differential clinical impact of DNMT3A mutations relative to FLT3 and NPM1 on outcomes in de novo AML, including AMLDNMT3A/FLT3/NPM1, we then turned our attention to multivariate analysis. The latter demonstrated that age (p = 0.019), DNMT3A mutations (p = 0.026), IDH1 mutation (p = 0.002), and cytogenetics (poor-risk vs. intermediate/better-risk) (p = 0.018) were independently associated with OS. Poor-risk cytogenetics followed by DNMT3A mutations were the strongest unfavorable factors, while IDH1 mutation was favorable (Table 2). The adverse effect of DNMT3A mutations (p = 0.039) persisted when only younger (<60years) patients were considered in multivariate analysis. To control for the impact of cytogenetics, we proceeded to assess OS and EFS as a function of DNMT3A mutation status among patients with CN-AML. Indeed, DNMT3A mutations within the CN-AML subset were associated with significantly shorter OS and EFS (p = 0.044 and p = 0.051, respectively). (Additional file 3: Figure S2A-B) The adverse impact of DNMT3A mutations on OS was particularly notable among CN-AML patients <60 years of age (p = 0.014). These data suggest that in the absence of high-risk cytogenetics, DNMT3A mutations exert an independent impact on de novo AML outcomes including those of the AMLDNMT3A/FLT3/NPM1 group.
We then asked whether the dominant effect of DNMT3A mutations seen in AMLDNMT3A/FLT3/NPM1 is observed in AML with either NPM1 or FLT3 mutations, which are important factors in risk stratification particularly for younger patients with CN-AML [26],[27]. Notably, among patients with NPM1 mutation, those with CN-AMLNPM1/DNMT3A had significantly shorter OS and EFS compared to those with CN-AMLNPM1 and wild-type DNMT3A (p = 0.022 and p = 0.040, respectively). These differences in OS and EFS were also observed within the entire AML group (p = 0.017 and p = 0.025, respectively) (Figure 4c-d). Importantly, the adverse impact of DNMT3A mutations on OS among AMLNPM1 patients was particularly pronounced in those <60 years of age, within both the entire group and the CN-AML group (p = 0.008 and p = 0.014, respectively). Furthermore, patients with CN-AMLDNMT3A/NPM1 had shorter OS and EFS compared to those with CN-AMLFLT3-ITD/NPM1 (p = 0.047 and p = 0.072, respectively). This effect on OS and EFS persisted when all FLT3- mutated (ITD and TKD) cases were included (p = 0.054 and p = 0.051, respectively) (Figure 4e) and suggests that the impact of DNMT3A mutations is more pronounced than that of FLT3 (ITD and TKD) in CN-AMLNPM1. Conversely, among all patients with DNMT3A, there was no significant difference in OS or EFS between AMLDNMT3A/NPM1 and AMLDNMT3A/FLT3 (p = 0.301 and p = 0.989, respectively), suggesting that the dominant effect of DNMT3A mutations is not modulated by the presence of NPM1 or FLT3 mutations. Finally, when we compared OS and EFS for patients with AMLDNMT3A/FLT3/NPM1 (n = 35) to that of patients with AMLFLT3/NPM1 (n = 34) we noticed that the former group tended to have worse OS and EFS, although the difference was not statistically significant (Additional file 4: Figure S3).
Discussion
Integrative genomic analysis data from TCGA suggest that AMLDNMT3A/FLT3/NPM1 is a unique disease subset with distinctive features at the mRNA, miRNA, and epigenetic levels, but the clinical features and outcome characteristics of this AML subset remain unknown. By simultaneously harboring DNMT3A, FLT3 and NPM1 mutations, AMLDNMT3A/FLT3/NPM1 offers a unique advantage to study the differential impact of these genes in AML. This is particularly important because although the clinical implications of FLT3 and NPM1 mutations in de novo AML have been well characterized, their prognostic impact in the presence of concurrent DNMT3A mutations has not been assessed systematically. Accordingly, we designed this study to investigate the clinical and outcome characteristics of patients with AMLDNMT3A/FLT3/NPM1 as an index group and compare them to those with de novo AML harboring other mutation combinations involving DNMT3A, FLT3 and/or NPM1. Since the incidence of AMLDNMT3A/FLT3/NPM1 is low, cases seen at our institution were combined with those in the TCGA data set to increase the statistical power of our analysis. The inclusion of patients from two different cohorts and the combination of MDACC patients with untreated disease along with those who presented in relapse or with persistent leukemia for statistical purposes are acknowledged limitations of this retrospective study. Notwithstanding, our data demonstrate a significant association between AMLDNMT3A/FLT3/NPM1 and the following clinical features: young age, female gender, diploid cytogenetics, poor EFS and OS particularly in patients <60 years of age, and a heavy disease burden manifested by elevated peripheral blood and bone marrow blast counts. These features of AMLDNMT3A/FLT3/NPM1 are reminiscent of the salient clinical attributes described broadly for AML patients with DNMT3A mutations [1]-[4].
In the NCCN [26] and ELN [27] risk classification systems for AML, due importance is placed on NPM1 and FLT3 but not DNMT3A mutations in CN-AML. In our study group, patients with DNMT3A mutations had worse outcomes than those with wild-type DNMT3A. Hou et al.[28] and Ribeiro et al.[29] have similarly demonstrated that DNMT3A mutations are independent predictors of shorter OS in patients with de novo AML. Shirarov et al.[30] also demonstrated in their study that DNMT3A mutations were associated with worse outcome including significantly shorter OS and EFS and are an independent determinate of worse outcome in younger patients with CN-AML. In addition, recent data demonstrate that DNMT3A mutation status has an impact on therapy selection; in one study, patients with DNMT3A mutations who were treated with high-dose daunorubicin compared to standard-dose therapy were shown to have longer OS [2].
Our findings suggest that in the absence of high-risk cytogenetics, DNMT3A mutation status has an impact on outcome in the presence of FLT3 and/or NPM1. In this study group, mutant DNMT3A is associated with adverse outcomes among CN-AML patients that have NPM1 mutations. This effect appears more pronounced than that of either FLT3-ITD or FLT3-TKD mutations and is not mitigated by NPM1 or dependent on the presence of cytogenetic abnormalities. However, by current risk stratification schemes CN-AMLDNMT3A/NPM1 patients would be regarded as a better/favorable risk group whereas they likely should be reclassified in higher risk categories. Along similar lines, Kihara et al suggested recently that ELN risk stratification can be enhanced by reassigning patients in the Favorable risk category to the Intermediate-I group if they have DNMT3A mutations [31]. Data from this study suggests that among patients with de novo CN-AML, particularly those <60 years, AMLDNMT3A/FLT3/NPM1 patients seem to have the worst clinical outcomes, followed by those with AMLFLT3/DNMT3A and then those with AMLNPM1/DNMT3A.
Methods
Patient study group
This study was approved by the MDACC Institutional Review Board. We searched our records for patients with de novo AML and known mutation status for each of DNMT3A, FLT3 and NPM1 who presented to our institution between 3/2009 and 12/2013. All patients met the following study inclusion requirements: 1) positive for mutations in DNMT3A, FLT3 and/or NPM1; 2) 18 years of age or older at diagnosis; 3) acute myeloid leukemia diagnosis as defined in the World Health Organization classification [32]. Patients with therapy-related AML, history of myelodysplastic syndrome or myeloproliferative neoplasms, and AML with t(15;17) were excluded.
The demographic, laboratory, and clinical data on patients in the study group were collected by chart review with emphasis on variables of demonstrated prognostic utility in AML [33]. Flow cytometry immunophenotyping and cytogenetic analyses were performed as part of routine clinical workup using previously described techniques [34],[35]. Patients were allocated to intermediate- and high-risk cytogenetic risk groups per NCCN guidelines (version 2.2013) [26]. A detailed review of treatment history for all patients in the MDACC study group was undertaken, and patients were divided into high-intensity (cytarabine and anthracycline based induction regimens, further subdivided into "3 + 7" and "non-3 + 7") and low-intensity therapy groups.
As reported previously [1], the TCGA cohort consisted of 200 adult patients with various subtypes of de novo AML. To increase the statistical power of our analysis and encompass a broader cohort of de novo AML patients, TCGA patients who had mutations in DNMT3A, FLT3 and/or NPM1 were included in our study; those with t(15;17) were excluded. Publicly available annotated exome and whole-genome sequencing data as well as associated patient clinical data files were downloaded from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/) at various time points between September 2013 and January 2014. Patients were allocated to cytogenetic risk groups as described above. Patients in the TCGA group were treated per NCCN guidelines [1]. Patient enrollment and utilization of data were conducted in accordance with TCGA Human Subjects Protection and Data Access Policies [36].
Mutation analysis
For NGS-based mutation screening of MDACC cases, analysis of hotspot genomic areas was performed by paired-end sequencing using the MiSeq (Illumina, San Diego, CA) sequencer and the TruSeq Amplicon Cancer Panel kit (Illumina) with 5 additional customized probe pairs targeting DNMT3A (exon 23, codons 866-913), XPO1, KLHL6, MYD88 and EZH2 as described previously [37]. Variant (allelic) frequency was estimated as the percentage of variant reads over total reads at a specific allelic location. Matched germline DNA was not available for comparison. FLT3 (ITD and TKD) and NPM1 mutations were also screened using polymerase chain reaction (PCR) followed by capillary electrophoresis on Genetic Analyzer (Applied Biosystems, Foster City, CA), as described previously [38]. NGS-based mutation analysis and testing for FLT3 and NPM1 status are routinely performed on all patients with new onset or relapsed acute leukemia at our institution irrespective of their cytogenetic risk groups. The TCGA mutation analysis data was derived from whole genome and exome NGS and analyzed alongside matched germline DNA [1].
Statistical analysis
Categorical and continuous variables were analyzed using Pearson Chi-square and Kruskal-Wallis rank sum tests, respectively. Overall survival (OS) was estimated by the Kaplan-Meier method using as a reference time point the date of initial AML diagnosis until death from any cause or date of last follow-up. Event-free survival (EFS) represented the duration of disease between the initial diagnosis and first relapse. No EFS data was available for TCGA patients. For MDACC patients who received allogeneic hematopoietic stem cell transplant (allo-SCT), the date of transplant was assigned as the last follow-up date. Survival curves were compared by the log rank test. Differences between two groups were considered significant if p-values were <0.05 in a two-tailed test. Multivariate analysis was performed by Cox proportional regression model. Significant factors were identified by Wald backward stepwise elimination.
References
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ, Laird PW, Baty JD, Fulton LL, Fulton R, Heath SE, Kalicki-Veizer J, Kandoth C, Klco JM, Koboldt DC, Kanchi KL, Kulkarni S, Lamprecht TL, Larson DE, Lin L, Lu C, McLellan MD, McMichael JF, Payton J, Schmidt H, Spencer DH, Tomasson MH, Wallis JW: Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013, 368: 2059-2074. 10.1056/NEJMoa1301689.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O: Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012, 366: 1079-1089. 10.1056/NEJMoa1112304.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O'Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ: DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010, 363: 2424-2433. 10.1056/NEJMoa1005143.
Thol F, Damm F, Ludeking A, Winschel C, Wagner K, Morgan M, Yun H, Gohring G, Schlegelberger B, Hoelzer D, Lubbert M, Kanz L, Fiedler W, Kirchner H, Heil G, Krauter J, Ganser A, Heuser M: Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011, 29: 2889-2896. 10.1200/JCO.2011.35.4894.
Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, Dohner H, Dohner K: Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002, 100: 4372-4380. 10.1182/blood-2002-05-1440.
Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Dohner H: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008, 358: 1909-1918. 10.1056/NEJMoa074306.
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Saito H, Ueda R, Ohno R, Naoe T: Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001, 97: 2434-2439. 10.1182/blood.V97.8.2434.
Whitman SP, Maharry K, Radmacher MD, Becker H, Mrozek K, Margeson D, Holland KB, Wu YZ, Schwind S, Metzeler KH, Wen J, Baer MR, Powell BL, Carter TH, Kolitz JE, Wetzler M, Moore JO, Stone RM, Carroll AJ, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD: FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010, 116: 3622-3626. 10.1182/blood-2010-05-283648.
Whitman SP, Ruppert AS, Radmacher MD, Mrozek K, Paschka P, Langer C, Baldus CD, Wen J, Racke F, Powell BL, Kolitz JE, Larson RA, Caligiuri MA, Marcucci G, Bloomfield CD: FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008, 111: 1552-1559. 10.1182/blood-2007-08-107946.
Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S: Prognostic relevance of FLT3-TKD mutations in AML: the combination matters–an analysis of 3082 patients. Blood. 2008, 111: 2527-2537. 10.1182/blood-2007-05-091215.
Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE: FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007, 110: 1262-1270. 10.1182/blood-2006-04-015826.
Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Loffler H, Sauerland CM, Serve H, Buchner T, Haferlach T, Hiddemann W: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002, 100: 59-66. 10.1182/blood.V100.1.59.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, Wermke M, Bornhauser M, Ritter M, Neubauer A, Ehninger G, Illmer T: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002, 99: 4326-4335. 10.1182/blood.V99.12.4326.
Haferlach C, Mecucci C, Schnittger S, Kohlmann A, Mancini M, Cuneo A, Testoni N, Rege-Cambrin G, Santucci A, Vignetti M, Fazi P, Martelli MP, Haferlach T, Falini B: AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood. 2009, 114: 3024-3032. 10.1182/blood-2009-01-197871.
Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE, Boyiadzis M: DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies.Leukemia 2014.,
Cagnetta A, Adamia S, Acharya C, Patrone F, Miglino M, Nencioni A, Gobbi M, Cea M: Role of genotype-based approach in the clinical management of adult acute myeloid leukemia with normal cytogenetics. Leuk Res. 2014, 38: 649-659. 10.1016/j.leukres.2014.03.006.
Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, Radmacher MD, Kohlschmidt J, Nicolet D, Whitman SP, Wu YZ, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Moore JO, Caligiuri MA, Larson RA, Bloomfield CD: Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol. 2012, 30: 742-750. 10.1200/JCO.2011.39.2092.
Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, Cayuela JM, Hayette S, Reman O, Contentin N, Bordessoule D, Pautas C, Botton S, Revel T, Terre C, Fenaux P, Thomas X, Castaigne S, Dombret H, Preudhomme C: Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia. 2012, 26: 1247-1254. 10.1038/leu.2011.382.
Markova J, Michkova P, Burckova K, Brezinova J, Michalova K, Dohnalova A, Maaloufova JS, Soukup P, Vitek A, Cetkovsky P, Schwarz J: Prognostic impact of DNMT3A mutations in patients with intermediate cytogenetic risk profile acute myeloid leukemia. Eur J Haematol. 2012, 88: 128-135. 10.1111/j.1600-0609.2011.01716.x.
Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, Schmid C, Wendtner CM, Staib P, Serve H, Kreuzer KA, Kern W, Haferlach T, Haferlach C: A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012, 120: 2963-2972. 10.1182/blood-2012-03-419622.
Gaidzik VI, Schlenk RF, Paschka P, Stolzle A, Spath D, Kuendgen A, von Lilienfeld-Toal M, Brugger W, Derigs HG, Kremers S, Greil R, Raghavachar A, Ringhoffer M, Salih HR, Wattad M, Kirchen HG, Runde V, Heil G, Petzer AL, Girschikofsky M, Heuser M, Kayser S, Goehring G, Teleanu MV, Schlegelberger B, Ganser A, Krauter J, Bullinger L, Dohner H, Dohner K: Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG). Blood. 2013, 121: 4769-4777. 10.1182/blood-2012-10-461624.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C: Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982, 51: 189-199. 10.1111/j.1365-2141.1982.tb08475.x.
Kussick SJ, Stirewalt DL, Yi HS, Sheets KM, Pogosova-Agadjanyan E, Braswell S, Norwood TH, Radich JP, Wood BL: A distinctive nuclear morphology in acute myeloid leukemia is strongly associated with loss of HLA-DR expression and FLT3 internal tandem duplication. Leukemia. 2004, 18: 1591-1598. 10.1038/sj.leu.2403458.
Chen W, Konoplev S, Medeiros LJ, Koeppen H, Leventaki V, Vadhan-Raj S, Jones D, Kantarjian HM, Falini B, Bueso-Ramos CE: Cuplike nuclei (prominent nuclear invaginations) in acute myeloid leukemia are highly associated with FLT3 internal tandem duplication and NPM1 mutation. Cancer. 2009, 115: 5481-5489. 10.1002/cncr.24610.
Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, Gale RE: The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011, 118: 409-412. 10.1182/blood-2010-12-322479.
Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Lowenberg B, Bloomfield CD, European L: Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010, 115: 453-474. 10.1182/blood-2009-07-235358.
O'Donnell MR, Abboud CN, Altman J, Appelbaum FR, Arber DA, Attar E, Borate U, Coutre SE, Damon LE, Goorha S, Lancet J, Maness LJ, Marcucci G, Millenson MM, Moore JO, Ravandi F, Shami PJ, Smith BD, Stone RM, Strickland SA, Tallman MS, Wang ES, Naganuma M, Gregory KM: Acute myeloid leukemia. J Natl Compr Canc Netw. 2012, 10: 984-1021.
Hou HA, Lin CC, Chou WC, Liu CY, Chen CY, Tang JL, Lai YJ, Tseng MH, Huang CF, Chiang YC, Lee FY, Kuo YY, Lee MC, Liu MC, Liu CW, Lin LI, Yao M, Huang SY, Ko BS, Hsu SC, Wu SJ, Tsay W, Chen YC, Tien HF: Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014, 28: 50-58. 10.1038/leu.2013.236.
Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, Figueroa ME, Zeilemaker A, Melnick A, Lowenberg B, Valk PJ, Delwel R: Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012, 119: 5824-5831. 10.1182/blood-2011-07-367961.
Shivarov V, Gueorguieva R, Stoimenov A, Tiu R: DNMT3A mutation is a poor prognosis biomarker in AML: results of a meta-analysis of 4500 AML patients. Leukemia research. 2013, 37: 1445-1450. 10.1016/j.leukres.2013.07.032.
Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, Imai K, Uike N, Kimura F, Kitamura K, Nakaseko C, Onizuka M, Takeshita A, Ishida F, Suzushima H, Kato Y, Miwa H, Shiraishi Y, Chiba K, Tanaka H: Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014, 28: 1586-1595. 10.1038/leu.2014.55.
Arber DA, Brunning RD, Orazi A, Porwit A, Peterson L, Thiele J, Le Beau MM: Acute Myeloid Leukemia, Not Otherwise Specified. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Edited by: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. 2008, IARC, Lyon, 130-139.
Estey EH: Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013, 88: 318-327. 10.1002/ajh.23404.
Khoury JD, Sen F, Abruzzo LV, Hayes K, Glassman A, Medeiros LJ: Cytogenetic findings in blastoid mantle cell lymphoma. Hum Pathol. 2003, 34: 1022-1029. 10.1053/S0046-8177(03)00412-X.
Giordano A, Gao H, Cohen EN, Anfossi S, Khoury J, Hess K, Krishnamurthy S, Tin S, Cristofanilli M, Hortobagyi GN, Woodward WA, Lucci A, Reuben JM: Clinical relevance of cancer stem cells in bone marrow of early breast cancer patients. Ann Oncol. 2013, 24: 2515-2521. 10.1093/annonc/mdt223.
TCGA Human Subjects Protection and Data Access Policies. URL: http://cancergenome.nih.gov/PublishedContent/Files/pdfs/6.3.1_TCGA_Human_Subjects_and_Data_Access_policies_FINAL_011211.pdf In Book TCGA Human Subjects Protection and Data Access Policies. The Cancer Genome Atlas Project; URL: ., [http://cancergenome.nih.gov/PublishedContent/Files/pdfs/6.3.1_TCGA_Human_Subjects_and_Data_Access_policies_FINAL_011211.pdf]
Zhang L, Singh RR, Patel KP, Stingo F, Routbort M, You MJ, Miranda RN, Garcia-Manero G, Kantarjian HM, Medeiros LJ, Luthra R, Khoury JD: BRAF kinase domain mutations are present in a subset of chronic myelomonocytic leukemia with wild-type RAS. Am J Hematol. 2014, 89: 499-504. 10.1002/ajh.23652.
Warren M, Luthra R, Yin CC, Ravandi F, Cortes JE, Kantarjian HM, Medeiros LJ, Zuo Z: Clinical impact of change of FLT3 mutation status in acute myeloid leukemia patients. Mod Pathol. 2012, 25: 1405-1412. 10.1038/modpathol.2012.88.
Acknowledgements
The authors thank Sherry Pierce for assistance with data acquisition. This work was supported in part by the MD Anderson Cancer Center Support Grant (CA016672) from the National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JDK: Conception and design of study, data analysis, and manuscript preparation. SL and ZZ: Data collection, data analysis, and manuscript preparation. ZZ: Statistical analysis. KPP, RRS, LZ, LJM, JC, MR, FRK, HMK, CBR, and RL: Data analysis and manuscript preparation. All authors read and approved the final manuscript.
Electronic supplementary material
13045_2014_74_MOESM1_ESM.jpeg
Additional file 1: Figure S1.: Immunophenotypic features of AML with DNMT3A, FLT3, and NPM1 mutations as assessed by flow cytometry (n = 18). Color legend: red = positive; nude = partial positive, blue = negative; blank = not assessed. (JPEG 271 KB)
13045_2014_74_MOESM2_ESM.docx
Additional file 2: Table S1.: DNMT3A mutation types in the study group. Table S2. FLT3 mutation types in the study group. (DOCX 18 KB)
13045_2014_74_MOESM3_ESM.jpeg
Additional file 3: Figure S2.: Overall survival and event-free survival of de novo acute myeloid leukemia patients with mutant DNMT3A compared to those with wild-type DNMT3A.(JPEG 80 KB)
13045_2014_74_MOESM4_ESM.jpeg
Additional file 4: Figure S3.: Overall survival of de novo acute myeloid leukemia patients with DNMT3A, FLT3, and NPM1 mutations compared to those with FLT3 and NPM1 mutations and wild-type DNMT3A.(JPEG 53 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Loghavi, S., Zuo, Z., Ravandi, F. et al. Clinical features of De Novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J Hematol Oncol 7, 74 (2014). https://doi.org/10.1186/s13045-014-0074-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-014-0074-4