
Abstract
Myocyte Enhancer Factor 2 C (MEF2C), one of the transcription factors of the MADS-BOX family, is involved in embryonic brain development, neuronal formation and differentiation, as well as in the growth and pruning of axons and dendrites. MEF2C is also involved in the development of various neuropsychiatric disorders, such as autism spectrum disorders (ASD), epilepsy, schizophrenia and Alzheimer’s disease (AD). Here, we review the relationship between MEF2C and neuropsychiatric disorders, and provide further insights into the mechanism of these diseases.
Similar content being viewed by others

Introduction
MEF2C is an important member of the myocyte enhancer factor 2 (MEF2). MEF2 is a subfamily of the MADS-BOX (MCM-1-agamous-deficiens-serum response factor) family of transcriptional regulatory factors, which play essential roles in embryogenesis and epigenetic modifications that control gene expressions during development and throughout adulthood [23]. The MEF2 family consist of four members MEF2A, MEF2B, MEF2C and MEF2D. MEF2C is the earliest expressed MEF2 isomer in the telencephalon of mouse embryos, and is the most expressed in the cerebral cortex of postnatal and adult mouse brains. Therefore, it is critical for proper nervous system development and functional maintenance [4].
The human MEF2C gene is located in the chromosome 5q14.3 region and its protein consists of five core domains; MADS domain, MEF2 domain, transcriptional activation domains 1 (TAD1) and 2 (TAD2), as well as the nuclear localization signal (NLS). The MADS and MEF2 domains mediate MEF2C dimerization, DNA binding as well as recruitment of cofactors. TAD is involved in the recruitment of cofactors, including co-activators such as histone acetyltransferase p300 and cAMP-response element-binding protein-binding protein (CBP), or co-repressors such as class II histone deacetylases (HDACs), that regulates transcription [4]. Besides, through alternative splicing at the mRNA level [85], and post-translational modifications such as phosphorylation or dephosphorylation [11], acetylation [3], sumoylation [63] and S-nitrosylation [52], MEF2C appears various expression pattern and transactivation functions. In addition, MEF2C mediates physiological processes such as cardiac morphogenesis, angiogenesis, muscle cell differentiation, bone development, and neural or lymphatic system development [44].
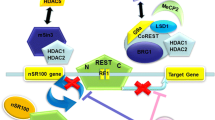
In the absence of external stimulation, the MEF2 protein in the central nervous system binds and inactivates target genes. Various stimuli, such as depolarization, neurotrophin or synaptic stimulation (e.g., glutamate synaptic Reelin) activates neurons, thereby triggering calcium signaling responses, including calmodulin-dependent protein kinase (CaMK), leading to phosphorylation of class IIa HDACs. These phosphorylation events cause to a nuclear output of HDACs, and their subsequent removal from the MEF2 target gene, allowing the recruitment of co-activators [46]. MiR-124 and miR-9 have been shown to co-inhibit HDACs 5 expressions, which activate the neuron membrane glycoprotein, GPM6A, a MEF2C target gene. These activities promote the development of axons in primary neurons (Fig. 1) [25].

The functional regulation model of MEF2C in neuronal cells, where MEF2C is particularly important for the regulation of dendritic spines. The blue italics are MEF2C-associated psychoneurotic disorders
In addition, protein phosphatase 2B or calcineurin can directly dephosphorylate MEF2, thereby affecting its affinity towards target DNA sequences, and promoting its transcription activities [43]. MEF2C is involved in regulation of neuronal migration, activity-dependent cell survival, neuronal differentiation, axon guidance and pruning, dendritic formation and remodeling, as well as in synaptic development and neuronal excitability [37, 39, 84]. In the last decade, human genome-wide association study (GWAS) and genome sequencing of large patient samples revealed that MEF2C is a candidate risk gene for various neuropsychiatric disorders, such as AD [67], ASD [83], schizophrenia [47], bipolar disorder [50], major depression [28], attention deficit and hyperactivity disorder [62], epilepsy [80] or Parkinson’s disease [59]. We review recent findings on MEF2C as well as its role in some neuropsychiatric diseases and provide theoretical and detailed insights that will inform future studies.
Alzheimer’s disease
AD is a neurodegenerative disease that is associated with progressive cognitive and memory deterioration. Inflammation is involved in AD pathogenesis [19]. As the first and main immune defense component of central nervous system (CNS), microglia act as macrophages, clearing metabolites and necrotic cells in the brain through phagocytosis. However, continuous activation of microglia leads to the secretion of various neurotoxic substances, which play an important role in AD development [54]. For instance, when interferon-β (IFN-β), a pro-inflammatory cytokine related to brain ageing, was injected into the cerebrospinal fluid of young wild type (WT) or MEF2C-knock out (MEF2C-KO) mice, suppressed social interaction outcomes were observed in the MEF2C-KO mice, compared to WT mice. Following immune activations, the expressions of pro-inflammatory chemokines (CCL2 and CCL5) and cytokines (IL-1b and TNF) in MEF2C-KO mice were significantly higher than in WT mice. Thus MEF2C enhances the resilience of microglia to immune challenges and promotes homeostasis in pre-inflammatory states. Type I interferon (IFN-I) accumulation down-regulates the MEF2C in microglia, leading to excess microglia responses under pro-inflammatory environments of AD or aging brains, and aggravates cognitive impairment and disease pathology [20]. As reported by Xue et al., the suppressed MEF2C nuclear translocation as an early and prominent microglial phenotype in a mouse model of brain amyloidosis (5 × FAD mice) implies an association between MEF2C deregulation and microglial phenotypes in AD-relative settings. Although they did not report the downregulation of MEF2C mRNA copies in the microglia of 5 × FAD mice, upregulated MEF2C in microglia after anti-IFNAR antibody administration implies a potential strategy for improving microglial elasticity by upregulation of MEF2C [77]. A significant decrease in MEF2C mRNA levels in leukocytes of AD patients has been positively correlated with total scores of Mini-Mental State Examination (MMSE). Therefore, expression levels of MEF2C mRNA in leukocytes may not only be a biomarker for AD diagnosis, but be a potential biomarker for early detection of cognitive decline, such as mild cognitive impairment (MCI) [60].
In addition, MEF2C plays an important role in amyloid-protein precursor protein (APP) -mediated anti-apoptotic neuroprotection [12]. MEF2C is a potential regulator of APP proteolysis, during which amyloid beta (Aβ), a central factor in AD initiation, is produced [13]. These findings elucidate on pathways involved in oligomeric Aβ-potentiated microglial activation.
Methyl CpG binding protein 2 (MeCP2; an important transcriptional inhibitor) levels have been shown to be increased in the cerebral cortex and hippocampus of AD patients. MeCP2-mediated MEF2C inhibition may be associated with cognitive decline in AD [33]. Interestingly, other studies have reported a significant correlation between cognitive functions and seasons among the elderly. There is a 4-year difference in cognitive functions between summer and autumn, compared to winter and spring, while the probability of meeting the criteria for MCI or dementia in winter and spring is nearly 30% higher, relative to that in the summer and autumn. MEF2C was found to be involved in this regulation of seasonal plasticity [40].
The high expressions of MEF2C in brain regions related to learning and memory, such as dentate gyrus, frontal cortex, entorhinal cortex or amygdala, strongly prompted the important role of MEF2C in cognition of AD [81]. Large meta-analysis studies identified that the variation of rs190982, a common single nucleotide polymorphism (SNP), in MEF2C is a protective factor against AD in a Caucasian population (OR = 0.93, 95% CI = 0.90 ~ 0.95) [66]. Besides, an association between rs190982 polymorphism and AD (OR = 0.885, 95% CI = 0.811 ~ 0.966) has been reported [10]. Based on analyses of pathological data on AD, among 22 genetic variations of AD, the rs190982 variation in MEF2C gene, was the only one that was found to exert significant effects on cognitive functions [42]. However, other studies have reported the negative association [8, 66]. A genome-wide association studies (GWAS) meta-analysis involving a Taiwanese population of China did not reveal positive association between MEF2Crs190982 polymorphism and AD, however, there were interactions between MEF2Crs9293506 and cognitive aging. The risk for cognitive aging in carriers with TC genotypes of MEF2C rs9293506 was 2.79 times higher than that of those with the CC genotype [41]. These variations may be attributed to genetic heterogeneities among different ethnic groups, such as differences in minor alleles, different frequencies of secondary alleles or heterogeneity of potential genetic structures.
In a 2013 meta-analysis, Lambert et al. found a genome-wide significant association between MEF2C and AD [35], however, the significant association was not replicated in a larger sample study performed in 2019 [34]. Moreover, Jansen et al. were unable to establish an association between MEF2C and AD in a large sample study from the UK Biobank (UKB) [29].
Although GWAS have been a valuable platform for identifying candidates for disease-related genetic variants, the confirmed risk loci for AD only explains a small portion of AD heritability (Table 1). To address some of the limitations encountered by univariate SNP-based analyses, advanced methods were developed to examine SNPs in aggregate. The aggregated SNP approach may reduce the total number of tests performed and increase power by exploiting linkage disequilibrium (LD) across multiple SNPS [76]. It hypothesized that variations in entire gene regions, rather than isolated single SNPS, play a role in cognitive decline [48]. Other strategies for improving detection effects should be evaluated in large sample sizes composed of different ethnic populations.
Epilepsy
Due to involvement of MEF2C in many processes during neural developmental stages, functional disruption of MEF2C results in various neurological symptoms, among which epilepsy is a common symptom. MEF2C conditional knockout mice revealed that MEF2C is involved in the migration of GABA and glutamate pyramidal neurons, as well as in the maintenance of synaptic stability and function [71]. Besides, MEF2C regulates inhibitory and excitatory states of neurons to maintain the balance in neural networks [26]. Disruption of this regulation may lead to abnormal synaptic activities, causing epileptic events.
MEF2C-related epilepsy has been reported [9, 53, 82] (Table 2). Its prevalence ranges between 54% and 82% [9, 53]. The associated genetic defects include MEF2C pathogenic variants, or microdeletions encompassing the MEF2C gene. MEF2C-related epilepsy usually occurs during infancy or early childhood. In a review involving 19 patients with MEF2C-related epilepsy, seizures were established to have occurred in the first 12 months of life in 12 patients (63%). The mean age of seizure onset was 13.5 months (median 12), ranging from 3 to 36 months [9]. Febrile epilepsy, tonic–clonic epilepsy and myoclonic epilepsy are the most common epilepsy types. Reported EEG abnormalities include epileptic activity, background activity disorder, and multifocal as well as generalized epileptiform discharge [9]. Although the frequency of early-onset epilepsy is high, some children with late onset, mild seizures or no epilepsy at all exhibit serious neurological defects, such as low muscle tone or hyperactivity. The severity of epilepsy is not always consistent with other neurological defects [49]. Certainly, patients with complete deletion of MEF2C have an increased risk for developing epilepsy, relative to those with partial deletion [57].
Etiopathogenic factors in other conditions are also associated with MEF2C in epilepsy. Among 73 patients with infantile spasm syndrome (ISS), an age-related epileptic syndrome, one patient with 3.24 Mb deletion in 5q14.3 located in 1 Mb upstream of the MEF2C gene was found [arr5q14.3q15 (890, 687, 77 ~ 923, 160, 85) x1, hg19] [10]. Bienvenu et al. identified a case of de novo MEF2C mutation in 50 patients with unexplained epileptic encephalopathy [7]. Moreover, Yu et al. and Zhou et al. reported cases of infantile spasm caused by a 5q14.3 microdeletion syndrome in China [79, 82]. Another study documented a 9.68% rare functional variation of MEF2C in ASD patients with epilepsy, but not in ASD patients without epilepsy, indicating that MEF2C functional variation significantly increased the risk of epilepsy in ASD [83].
In recent years, molecular studies have revealed further pathological mechanisms involved in epilepsy. For instance, truncated Salt-induced kinase 1 (SIK1) sequence variants p. (Glu347*) and p. (Gln633*) reduced the expressions of MEF2C. The SIK1 syndrome is a developmental epilepsy disorder that is caused by a heterozygous mutation in the SIK1 gene (OMIM no.616341). Decreased expressions of MEF2C proteins in neurons is correlated with abnormal expressions of target genes (ARC, NRG1 or NR4A1), which disrupts the balance in neuronal excitability, thereby reducing the epileptic threshold. This process is independent of HDAC5 phosphorylation, indicating that SIK1 may directly interact with MEF2C [55]. In addition, in rat models, the expressions of miR-203 in astrocytes were established to be up-regulated, which led to down-regulation of MEF2C, promotion of NF- κB, phosphorylation of I κ B/IKK and secretion of inflammatory effectors (IL-6 and TNF-α). Besides, LncRNAUCA1 (long-chain non-coding RNA urothelial carcinoma associated 1) inhibits inflammatory responses to epilepsy by modulating miR203-mediated regulation of the MEF2C/NF-κB signaling pathway, therefore, it may be a potential therapeutic target for epilepsy [80].
Nervous system tumor
The activities of MEF-2 are regulated by various factors, including alternative splicing, post-translational modification of C-terminal and dimerization of N-terminal with other transcription factors [44]. Not only are mutations in these regions associated with developmental abnormalities, they are also associated with tumors, leukemia and transcriptional abnormalities. MEF2C diversifies hematological tumors, pancreatic cancer or liver cancer, leading to exhibition of various tumor characteristics [17]. MEF2C plays an important role in tumor pathogenesis and development, however, a limited number of studies have evaluated its functions in nervous system tumors [21, 78].
A study on brain metastases of breast cancer (BCBM) demonstrated that MEF2C, as a target gene of miR-802-5p and miR-194-5p, is increased in metastatic tumor cells. Immunoreactivity analyses showed that MEF2C expression increased by 24% between the 3rd and 10th day of brain metastasis (p < 0.001), and by 20% between the 7th and 10th day (p < 0.001). Therefore, as a transcription factor, MEF2C promotes the development of metastatic tumors. It was also found that peritumoral astrocytes began to express MEF2C after exudation of tumor cells from tumor tissues, while non-peritumoral astrocytes did not show these expressions, suggesting that MEF2C is involved in the crosstalk between astrocytes and tumor cells during the development of BCBM [61].
In the cytoplasm, MEF2C was shown to impair β-catenin translocation into the nucleus, thereby inhibiting Wnt/β-catenin signaling during the early stages of metastases development. However, in advanced stages, MEF2C and Wnt/β-catenin translocated to the nucleus, which was accompanied by an increase in Ki-67 positive cells. Continuous expression of MEF2C and its translocation to the nucleus is associated with disease severity, and MEF2C may serve as a biomarker for BCBM development and prognosis as well as its potential therapeutic target [24].
The dual role of MEF2C in tumors has also been reported in liver cancer. In the nucleus of hepatocellular carcinoma, MEF2C promoted the invasion and angiogenesis of hepatocellular carcinoma cells, while cytoplasmic MEF2C isolated β-catenin in the cytoplasm and reduced the ability of β-catenin to promote cell proliferation. Subcellular distribution of MEF2C may determine the overall role of MEF2C [5]. On the other hand, after being assembled into multi-protein complexes as transcription factors, MEF2C can be transformed into transcription activators or inhibitors under the control of tumor microenvironments to produce the opposite effects [44]. Therefore, MEF2C may act as a “double-edged sword” (either as a proto-oncogene or a tumor suppressor) in tumor pathogenesis.
Autism spectrum disorders
Autism spectrum disorders (ASD) are a group of severe neurodevelopmental disorders that are characterized by impaired social interactions and communication skills, and/or narrow interests and repetitive stereotyped behaviors. Although there are many etiological models for ASD, such as gene mutations, abnormal synaptic development or signaling pathways, its pathogenesis has not been conclusively determined. One of the mechanisms involved in ASD pathogenesis is the imbalance in excitatory/inhibitory synaptic ratio [15]. Synaptic formation dominates the early stages of brain development, resulting in the generation of more synapses than are needed for brain functions. Consequently, the brain prunes the extra synapses [27]. MEF2C plays an important role in activity-dependent synaptic elimination. When depolarization and calcium influx occur in neurons after stimulation, MEF2C is activated, then it induces the transcription of protocadherin-10 (Pcdh10). Pcdh10 mediates the degradation of synaptic scaffold protein 95 (PSD-95) by binding ubiquitin PSD-95 to the proteasome, leading to elimination of excitatory synapses [64, 69].
Dysfunctions of the MEF2C gene may prevent the brain from eliminating unwanted excitatory synapses, leading to ASD-like syndromes (Table 3). A previous study reported a significant increase in the number of excitatory synapses and spinous processes in MEF2C-KO mice, as well as enhancement of basal and evoked synaptic transmission. This cascade of events led to development of hippocampal dependent learning and memory impairments, as well as ASD-like social behavior defects [6]. Conditional MEF2C KO in neural stem/progenitor cells expressing Nestin can affect neuronal differentiation, resulting in abnormal density and cell body sizes of cortical plate neurons, without affecting the proliferation as well as survival of neural stem cells. The conditioned MEF2C-KO mice that survived to adulthood showed more immature electrophysiological network characteristics and serious behavioral defects, indicating that MEF2C plays a key role in early programming of neuronal differentiation and distribution of neocortical layers in ASD [5]. Unlike the previous thought that MEF2C was only expressed in the cerebral cortex and hippocampus [2], recent studies have found that MEF2C was specifically expressed in the Purkinje cell layer of the cerebrum [32]. It selectively regulates the development of dendrites of Purkinje cells and prunes the synapses of climbing fibers. Similar with ASD findings, deletion or downregulation of MEF2C resulted in increased dendritic branches and spines in Purkinje cells, and changes in excitatory as well as inhibitory synaptic protein localization [37, 39]. Essentially, the increased spines are immature spines that are most likely to be pruned later, resulting in a decrease in the number of neurons and a decline in overall functions [45].
Studies on deletions of MEF2C in frontal brain regions of conditioned MEF2C-KO mice after birth, using the calcium/calmodulin-dependent protein kinase II (CaMKII)-Cre93 line, showed that although the number of spines in the hippocampus of mice increased significantly, these increases were not related with presentation of learning and memory, long-term potentiation (LTP) or social behaviors [16]. In spite of continuous roles of MEF2C in negative regulation of synaptogenesis, the functions of MEF2C, which involves the regulation of synaptic plasticity, learning and memory or behavioral expressions in ASD may depend on expressions of MEF2C during embryonic development, rather than just regulation of the number of synapses [1].
There is a significant overlap between the genes regulated by MEF2C and dozens of candidate ASD risk genes [26]. For instance, ASD-related symptoms have been reported in some patients with MEF2C haploid insufficiency syndrome (MCHS) [72]. MCHS, a neurodevelopmental disorder, is caused by microdeletion, missense or nonsense mutations in the MEF2C gene or copy number variations (CNVs). Chromosomal microarray data revealed that MEF2C mutations are scattered throughout the MEF2C protein, without thermal mutation regions. The patients showed varying degrees of intellectual disabilities, speech deficits, autism-like symptoms, seizures or motor abnormalities [36]. In general, phenotypic characteristics of MCHS may present dysplasia of multiple nerve cell populations at the transcriptional level, with ASD syndromes being the most pronounced [57].
Rett syndrome, one of the serious neurodevelopmental diseases, which occurs mostly in women. It is characterized by progressive decline in motor skills and intelligence. The MeCP2 mutation allele in region q28 on X chromosome has been shown to trigger the Rett syndrome [38]. Jiaping W et al. found 3 MEF2C heterozygous mutations in 44 patients with the Rett syndrome, without MeCP2 gene mutations, suggesting the MEF2C gene mutation is one of the risk factors for Rett syndrome [74]. Based on definitive pathogenesis of MeCP2 gene mutations, DSM-5 removed Rett syndrome from the diagnosis of ASD in 2013. However, this change has been questioned by many scholars. Since ASD is diagnosed via language as well as behavioral symptoms and is also caused by genetic variations, it may not remove a subtype of ASD when one pathogenic gene is found. According to current studies, some of the patients diagnosed with Rett syndromes do not have the MeCP2 gene mutation, but share common pathogenic genes with ASD. Therefore, more evidence is needed to distinguish Rett syndrome from ASD.
Schizophrenia
Schizophrenia is a severe, disabling mental disorder. Cognitive impairments in schizophrenia related to the prefrontal lobe are highly associated with disability. In a study involving 150,000 participants, the Psychiatric Genome Association identified 108 different genome loci associated with schizophrenia [51]. There are MEF2C target sites in the heritable risk factors for schizophrenia, which associate MEF2C with this disease.
An enrichment of MEF2C motifs in the SNP pool, with the top score being related to schizophrenia was reported [47]. Sequencing data of chromatin-associated histone methylation in the prefrontal neuronal chromatin of 17 schizophrenic patients showed that MEF2C-binding motifs were significantly overexpressed in about 1000 nucleosome sequences, affected by histone H3K4 hypermethylation. The hypermethylated sequence of trimethyl-histone H3-lysine 4 (H3K4me3) exhibited a strong neuronal footprint, with 6/12 of Gene Ontological (GO) categories being associated with synapses and neurons, and 8/18 of “drug” and “phenotypic” categories being matched with decreased cognition and abnormal behaviors. Down-regulation of MEF2C in cell culture models showed hypermethylation of H3K4 in affected nucleosomes, similar to the changes observed in prefrontal lobes of schizophrenia. Increasing expressions of MEF2C in the prefrontal lobe of cognitive impaired mice models with schizophrenia by using adeno-associated virus vectors significantly improved cognitive abilities. Following treatment with the NMDA receptor antagonist, MK-801, the cognitive performance of mice with up-regulated MEF2C levels were better compared to those of normal mice. Therefore, MEF2C transcription factors are promising targets for treatment of schizophrenia-associated cognitive impairment [30].
Summary
MEF2C plays a significant role in early brain development of humans or animals, and in normal development, distribution and electrical activity of neocortical neurons. Besides, it has a profound effect on neuropsychiatric phenotypes. Because of the important effects of MEF2C on synapses, MEF2C gene mutations or dysfunctions will lead to a series of syndromes, including intellectual deficiency, epilepsy and autism-like symptoms. The association between MEF2C and cognitive impairment coincides with the role of MEF2C in AD and ASD. In view of similar symptoms caused by MEF2C defects in various neuropsychiatric disorders, we recommend that it be described as MEF2C-related syndrome, which would be contribute to identify characteristic gene-related symptoms from these complex neuropsychiatric disorders. Further research on MEF2C will elucidate on the pathogenesis of neurological and mental disorders, as well as provide insights for improvement of symptoms associated with MEF2C gene deficiencies in neuropsychiatric disorders.
Tu et al. [70] reported that MEF2C heterozygous KO mice exhibited intellectual disabilities, autism-like symptoms, seizures, as well as motor abnormalities. After continuous injection of NitroSynapsin (a new antagonist of NMDA-type glutamate receptors) for 3 months, mice autism-like symptoms were significantly improved. These protective effects were associated with prevention of the loss of neurons and correcting the ratio of excitatory to inhibitory (E/I) neurotransmission imbalance [70]. Besides, in mice, neuronal MEF2C overexpression in adult prefrontal cortex improves working memory and object recognition memory, in conjunction with spinal remodeling in prefrontal projection neurons [47]. Therefore, these results have important implications in treatment of ASD, schizophrenia and other MEF2C-related neuropsychiatric disorders, at least improve the symptoms associated with MEF2C.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Adachi M, Lin PY, Pranav H, Monteggia LM. Postnatal loss of Mef2c results in dissociation of effects on synapse number and learning and memory. Biol Psychiatry. 2016;80(2):140–8. https://doi.org/10.1016/j.biopsych.2015.09.018.
Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, et al. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS ONE. 2012;7(4):e34863. https://doi.org/10.1371/journal.pone.0034863.
Angelelli C, Magli A, Ferrari D, Ganassi M, Matafora V, Parise F, et al. Differentiation-dependent lysine 4 acetylation enhances MEF2C binding to DNA in skeletal muscle cells. Nucleic Acids Res. 2008;36(3):915–28. https://doi.org/10.1093/nar/gkm1114.
Assali A, Harrington AJ, Cowan CW. Emerging roles for MEF2 in brain development and mental disorders. Curr Opin Neurobiol. 2019;59:49–58. https://doi.org/10.1016/j.conb.2019.04.008.
Bai XL, Zhang Q, Ye LY, Liang F, Sun X, Chen Y, et al. Myocyte enhancer factor 2C regulation of hepatocellular carcinoma via vascular endothelial growth factor and Wnt/beta-catenin signaling. Oncogene. 2015;34(31):4089–97. https://doi.org/10.1038/onc.2014.337.
Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, et al. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A. 2008;105(27):9391–6. https://doi.org/10.1073/pnas.0802679105.
Bienvenu T, Diebold B, Chelly J, Isidor B. Refining the phenotype associated with MEF2C point mutations. Neurogenetics. 2013;14(1):71–5. https://doi.org/10.1007/s10048-012-0344-7.
Boden KA, Barber IS, Clement N, Patel T, Guetta-Baranes T, Brookes KJ, et al. Methylation profiling RIN3 and MEF2C identifies epigenetic marks associated with sporadic early onset Alzheimer’s disease. J Alzheimers Dis Rep. 2017;1(1):97–108. https://doi.org/10.3233/ADR-170015.
Borlot F, Whitney R, Cohn RD, Weiss SK. MEF2C-related epilepsy: delineating the phenotypic spectrum from a novel mutation and literature review. Seizure. 2019;67:86–90. https://doi.org/10.1016/j.seizure.2019.03.015.
Boutry-Kryza N, Labalme A, Ville D, de Bellescize J, Touraine R, Prieur F, et al. Molecular characterization of a cohort of 73 patients with infantile spasms syndrome. Eur J Med Genet. 2015;58(2):51–8. https://doi.org/10.1016/j.ejmg.2014.11.007.
Brown FC, Still E, Koche RP, Yim CY, Takao S, Cifani P, et al. MEF2C phosphorylation is required for chemotherapy resistance in acute myeloid leukemia. Cancer Discov. 2018;8(4):478–97. https://doi.org/10.1158/2159-8290.CD-17-1271.
Burton TR, Dibrov A, Kashourd T, Amara FM. Anti-apoptotic wild-type Alzheimer amyloid precursor protein signaling involves the p38 mitogen-activated protein kinase/MEF2 pathway. Mol Brain Res. 2002;108(1–2):18. https://doi.org/10.1016/S0169-328X(02)00519-3.
Camargo LM, Zhang XD, Loerch P, Caceres RM, Marine SD, Uva P, et al. Pathway-based analysis of genome-wide siRNA screens reveals the regulatory landscape of APP processing. PLoS ONE. 2015;10(2):e0115369. https://doi.org/10.1371/journal.pone.0115369.
Cardoso C, Boys A, Parrini E, Mignon-Ravix C, McMahon JM, Khantane S, et al. Periventricular heterotopia, mental retardation, and epilepsy associated with 5q14.3-q15 deletion. Neurology. 2009;72(9):784–92. https://doi.org/10.1212/01.wnl.0000336339.08878.2d.
Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468(7321):263–9. https://doi.org/10.1038/nature09582.
Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27(3):327–31. https://doi.org/10.1038/85906.
Chen X, Gao B, Ponnusamy M, Lin Z, Liu J. MEF2 signaling and human diseases. Oncotarget. 2017;8(67):112152–65. https://doi.org/10.18632/oncotarget.22899.
Chen YC, Kuo HY, Bornschein U, Takahashi H, Chen SY, Lu KM, et al. Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci. 2016;19(11):1513–22. https://doi.org/10.1038/nn.4380.
Chidambaram H, Das R, Chinnathambi S. Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell Biosci. 2020;10:109. https://doi.org/10.1186/s13578-020-00474-4.
Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017;8(1):717. https://doi.org/10.1038/s41467-017-00769-0.
Di Giorgio E, Hancock WW, Brancolini C. MEF2 and the tumorigenic process, hic sunt leones. Biochim Biophys Acta Rev Cancer. 2018;1870(2):261–73. https://doi.org/10.1016/j.bbcan.2018.05.007.
Engels H, Wohlleber E, Zink A, Hoyer J, Ludwig KU, Brockschmidt FF, et al. A novel microdeletion syndrome involving 5q14.3-q15: clinical and molecular cytogenetic characterization of three patients. Eur J Hum Genet. 2009;17(12):1592–9. https://doi.org/10.1038/ejhg.2009.90.
Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60(6):1022–38. https://doi.org/10.1016/j.neuron.2008.11.029.
Galego S, Kauppila LA, Malho R, Pimentel J, Brito MA. Myocyte enhancer factor 2C as a new player in human breast cancer brain metastases. Cells. 2021. https://doi.org/10.3390/cells10020378.
Gu X, Fu C, Lin L, Liu S, Su X, Li A, et al. miR-124 and miR-9 mediated downregulation of HDAC5 promotes neurite development through activating MEF2C-GPM6A pathway. J Cell Physiol. 2018;233(1):673–87. https://doi.org/10.1002/jcp.25927.
Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, et al. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife. 2016. https://doi.org/10.7554/eLife.20059.
Hashimoto K, Kano M. Synapse elimination in the developing cerebellum. Cell Mol Life Sci. 2013;70(24):4667–80. https://doi.org/10.1007/s00018-013-1405-2.
Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48(9):1031–6. https://doi.org/10.1038/ng.3623.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–13. https://doi.org/10.1038/s41588-018-0311-9.
Javidfar B, Park R, Kassim BS, Bicks LK, Akbarian S. The epigenomics of schizophrenia, in the mouse. Am J Med Genet B Neuropsychiatr Genet. 2017;174(6):631–40. https://doi.org/10.1002/ajmg.b.32566.
Jung Y, Hsieh LS, Lee AM, Zhou Z, Coman D, Heath CJ, et al. An epigenetic mechanism mediates developmental nicotine effects on neuronal structure and behavior. Nat Neurosci. 2016;19(7):905–14. https://doi.org/10.1038/nn.4315.
Kamath S, Chen A. Myocyte enhancer factor 2c regulates dendritic complexity and connectivity of cerebellar Purkinje cells. Mol Neurobiol. 2019;56(6):4102–19. https://doi.org/10.1007/s12035-018-1363-7.
Kim B, Choi Y, Kim HS, Im HI. Methyl-CpG binding protein 2 in Alzheimer dementia. Int Neurourol J. 2019;23(Suppl 2):S72-81. https://doi.org/10.5213/inj.1938196.098.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Environmental Risk for Alzheimer’s Disease, C., et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30. https://doi.org/10.1038/s41588-019-0358-2.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8. https://doi.org/10.1038/ng.2802.
Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010;47(1):22–9. https://doi.org/10.1136/jmg.2009.069732.
Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD, et al. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc Natl Acad Sci U S A. 2008;105(27):9397–402. https://doi.org/10.1073/pnas.0802876105.
Li H, Zhong X, Chau KF, Williams EC, Chang Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat Neurosci. 2011;14(8):1001–8. https://doi.org/10.1038/nn.2866.
Li Z, McKercher SR, Cui J, Nie Z, Soussou W, Roberts AJ, et al. Myocyte enhancer factor 2C as a neurogenic and antiapoptotic transcription factor in murine embryonic stem cells. J Neurosci. 2008;28(26):6557–68. https://doi.org/10.1523/JNEUROSCI.0134-08.2008.
Lim ASP, Gaiteri C, Yu L, Sohail S, Swardfager W, Tasaki S, et al. Seasonal plasticity of cognition and related biological measures in adults with and without Alzheimer disease: analysis of multiple cohorts. PLoS Med. 2018;15(9):e1002647. https://doi.org/10.1371/journal.pmed.1002647.
Lin E, Tsai SJ, Kuo PH, Liu YL, Yang AC, Kao CF. Association and interaction effects of Alzheimer’s disease-associated genes and lifestyle on cognitive aging in older adults in a Taiwanese population. Oncotarget. 2017;8(15):24077–87. https://doi.org/10.18632/oncotarget.15269.
Liu G. Research on Alzheimer’s disease genetic variants using bioinformatics methods. (Doctor of Engineering), Harbin Institute of Technology, 2019.
Ma Q, Telese F. Genome-wide epigenetic analysis of MEF2A and MEF2C transcription factors in mouse cortical neurons. Commun Integr Biol. 2015;8(6):e1087624. https://doi.org/10.1080/19420889.2015.1087624.
Madugula K, Mulherkar R, Khan ZK, Chigbu DI, Patel D, Harhaj EW, Jain P. MEF-2 isoforms’ (A-D) roles in development and tumorigenesis. Oncotarget. 2019;10(28):2755–87. https://doi.org/10.18632/oncotarget.26763.
Martinez-Cerdeno V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev Neurobiol. 2017;77(4):393–404. https://doi.org/10.1002/dneu.22417.
McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408(6808):106–11. https://doi.org/10.1038/35040593.
Mitchell AC, Javidfar B, Pothula V, Ibi D, Shen EY, Peter CJ, et al. MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice. Mol Psychiatry. 2018;23(1):123–32. https://doi.org/10.1038/mp.2016.254.
Nettiksimmons J, Tranah G, Evans DS, Yokoyama JS, Yaffe K. Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age (Dordr). 2016;38(2):41. https://doi.org/10.1007/s11357-016-9885-2.
Novara F, Beri S, Giorda R, Ortibus E, Nageshappa S, Darra F, et al. Refining the phenotype associated with MEF2C haploinsufficiency. Clin Genet. 2010;78(5):471–7. https://doi.org/10.1111/j.1399-0004.2010.01413.x.
Nurnberger JI Jr, Koller DL, Jung J, Edenberg HJ, Foroud T, Guella I, Psychiatric Genomics Consortium Bipolar, G., et al. Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiat. 2014;71(6):657–64. https://doi.org/10.1001/jamapsychiatry.2014.176.
Ohi K, Shimada T, Nitta Y, Kihara H, Okubo H, Uehara T, Kawasaki Y. Specific gene expression patterns of 108 schizophrenia-associated loci in cortex. Schizophr Res. 2016;174(1–3):35–8. https://doi.org/10.1016/j.schres.2016.03.032.
Okamoto S, Nakamura T, Cieplak P, Chan SF, Kalashnikova E, Liao L, et al. S-nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep. 2014;8(1):217–28. https://doi.org/10.1016/j.celrep.2014.06.005.
Paciorkowski AR, Traylor RN, Rosenfeld JA, Hoover JM, Harris CJ, Winter S, et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics. 2013;14(2):99–111. https://doi.org/10.1007/s10048-013-0356-y.
Perea JR, Bolos M, Avila J. Microglia in Alzheimer’s disease in the context of tau pathology. Biomolecules. 2020. https://doi.org/10.3390/biom10101439.
Proschel C, Hansen JN, Ali A, Tuttle E, Lacagnina M, Buscaglia G, et al. Epilepsy-causing sequence variations in SIK1 disrupt synaptic activity response gene expression and affect neuronal morphology. Eur J Hum Genet. 2017;25(2):216–21. https://doi.org/10.1038/ejhg.2016.145.
Rajkovich KE, Loerwald KW, Hale CF, Hess CT, Gibson JR, Huber KM. Experience-dependent and differential regulation of local and long-range excitatory neocortical circuits by postsynaptic Mef2c. Neuron. 2017;93(1):48–56. https://doi.org/10.1016/j.neuron.2016.11.022.
Rocha H, Sampaio M, Rocha R, Fernandes S, Leao M. MEF2C haploinsufficiency syndrome: report of a new MEF2C mutation and review. Eur J Med Genet. 2016;59(9):478–82. https://doi.org/10.1016/j.ejmg.2016.05.017.
Ruiz A, Heilmann S, Becker T, Hernandez I, Wagner H, Thelen M, et al. Follow-up of loci from the International Genomics of Alzheimer’s disease project identifies TRIP4 as a novel susceptibility gene. Transl Psychiatry. 2014;4:e358. https://doi.org/10.1038/tp.2014.2.
Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155(6):1351–64. https://doi.org/10.1016/j.cell.2013.11.009.
Sao T, Yoshino Y, Yamazaki K, Ozaki Y, Mori Y, Ochi S, et al. MEF2C mRNA expression and cognitive function in Japanese patients with Alzheimer’s disease. Psychiatry Clin Neurosci. 2018;72(3):160–7. https://doi.org/10.1111/pcn.12618.
Sereno M, Hasko J, Molnar K, Medina SJ, Reisz Z, Malho R, et al. Downregulation of circulating miR 802–5p and miR 194–5p and upregulation of brain MEF2C along breast cancer brain metastasization. Mol Oncol. 2020;14(3):520–38. https://doi.org/10.1002/1878-0261.12632.
Shadrin AA, Smeland OB, Zayats T, Schork AJ, Frei O, Bettella F, et al. Novel loci associated with attention-deficit/hyperactivity disorder are revealed by leveraging polygenic overlap with educational attainment. J Am Acad Child Adolesc Psychiatry. 2018;57(2):86–95. https://doi.org/10.1016/j.jaac.2017.11.013.
Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311(5763):1012–7. https://doi.org/10.1126/science.1122513.
Shim JS, Min K, Lee SH, Park JE, Park SH, Kim M, Shim SH. MEF2C-related 5q14.3 microdeletion syndrome detected by array CGH: a case report. Ann Rehabil Med. 2015;39(3):482–7. https://doi.org/10.5535/arm.2015.39.3.482.
Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014;76(4):473–83. https://doi.org/10.1002/ana.24251.
Tang SS, Wang HF, Zhang W, Kong LL, Zheng ZJ, Tan MS, et al. MEF2C rs190982 polymorphism with late-onset Alzheimer’s disease in Han Chinese: a replication study and meta-analyses. Oncotarget. 2016;7(26):39136–42. https://doi.org/10.18632/oncotarget.9819.
Tansey KE, Cameron D, Hill MJ. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018;10(1):14. https://doi.org/10.1186/s13073-018-0523-8.
Tanteles GA, Alexandrou A, Evangelidou P, Gavatha M, Anastasiadou V, Sismani C. Partial MEF2C deletion in a Cypriot patient with severe intellectual disability and a jugular fossa malformation: review of the literature. Am J Med Genet A. 2015;167A(3):664–9. https://doi.org/10.1002/ajmg.a.36945.
Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, Huber KM. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151(7):1581–94. https://doi.org/10.1016/j.cell.2012.11.040.
Tu S, Akhtar MW, Escorihuela RM, Amador-Arjona A, Swarup V, Parker J, et al. NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nat Commun. 2017;8(1):1488. https://doi.org/10.1038/s41467-017-01563-8.
Vangoor VR, Reschke CR, Senthilkumar K, van de Haar LL, de Wit M, Giuliani G, et al. Antagonizing increased miR-135a levels at the chronic stage of experimental TLE reduces spontaneous recurrent seizures. J Neurosci. 2019;39(26):5064–79. https://doi.org/10.1523/JNEUROSCI.3014-18.2019.
Vidal S, Brandi N, Pacheco P, Maynou J, Fernandez G, Xiol C, et al. The most recurrent monogenic disorders that overlap with the phenotype of Rett syndrome. Eur J Paediatr Neurol. 2019;23(4):609–20. https://doi.org/10.1016/j.ejpn.2019.04.006.
Vrecar I, Innes J, Jones EA, Kingston H, Reardon W, Kerr B, et al. Further clinical delineation of the MEF2C haploinsufficiency syndrome: report on new cases and literature review of severe neurodevelopmental disorders presenting with seizures, absent speech, and involuntary movements. J Pediatr Genet. 2017;6(3):129–41. https://doi.org/10.1055/s-0037-1601335.
Wang J, Zhang Q, Chen Y, Yu S, Wu X, Bao X. Rett and Rett-like syndrome: expanding the genetic spectrum to KIF1A and GRIN1 gene. Mol Genet Genomic Med. 2019;7(11):e968. https://doi.org/10.1002/mgg3.968.
Wang J, Zhang Q, Chen Y, Yu S, Wu X, Bao X, Wen Y. Novel MEF2C point mutations in Chinese patients with Rett (-like) syndrome or non-syndromic intellectual disability: insights into genotype-phenotype correlation. BMC Med Genet. 2018;19(1):191. https://doi.org/10.1186/s12881-018-0699-1.
Wu MC, Kraft P, Epstein MP, Taylor DM, Chanock SJ, Hunter DJ, Lin X. Powerful SNP-set analysis for case-control genome-wide association studies. Am J Hum Genet. 2010;86(6):929–42. https://doi.org/10.1016/j.ajhg.2010.05.002.
Xue W, Chen J, Liu X, Gong W, Zheng J, Guo X, et al. PVT1 regulates the malignant behaviors of human glioma cells by targeting miR-190a-5p and miR-488–3p. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5 pt A):1783–94. https://doi.org/10.1016/j.bbadis.2018.02.022.
Yang Y, Ren M, Song C, Li D, Soomro SH, Xiong Y, et al. LINC00461, a long non-coding RNA, is important for the proliferation and migration of glioma cells. Oncotarget. 2017;8(48):84123–39. https://doi.org/10.18632/oncotarget.20340.
Yu D, Li S, Jiang N. Infantile spasm associated with 5q143 microdeletion syndrome: clinical and genetic characterization of a core family. Zhonghua Er Ke Za Zhi. 2015;53(2):140–2.
Yu Q, Zhao MW, Yang P. LncRNA UCA1 suppresses the inflammation via modulating miR-203-mediated regulation of MEF2C/NF-kappaB signaling pathway in epilepsy. Neurochem Res. 2020;45(4):783–95. https://doi.org/10.1007/s11064-019-02952-9.
Zhang ZG, Li Y, Ng CT, Song YQ. Inflammation in Alzheimer’s disease and molecular genetics: recent update. Arch Immunol Ther Exp (Warsz). 2015;63(5):333–44. https://doi.org/10.1007/s00005-015-0351-0.
Zhou T, Su W, Liang D, Xu Y, Luo Y, Tong G. Genetic analysis of a case with MEF2C deletion in association with 5q14.3 microdeletion syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38(8):779–82. https://doi.org/10.3760/cma.j.cn511374-20200608-00417.
Zhou WZ, Zhang J, Li Z, Lin X, Li J, Wang S, et al. Targeted resequencing of 358 candidate genes for autism spectrum disorder in a Chinese cohort reveals diagnostic potential and genotype-phenotype correlations. Hum Mutat. 2019;40(6):801–15. https://doi.org/10.1002/humu.23724.
Zhu B, Carmichael RE, Solabre Valois L, Wilkinson KA, Henley JM. The transcription factor MEF2A plays a key role in the differentiation/maturation of rat neural stem cells into neurons. Biochem Biophys Res Commun. 2018;500(3):645–9. https://doi.org/10.1016/j.bbrc.2018.04.125.
Zhu B, Gulick T. Phosphorylation and alternative pre-mRNA splicing converge to regulate myocyte enhancer factor 2C activity. Mol Cell Biol. 2004;24(18):8264–75. https://doi.org/10.1128/MCB.24.18.8264-8275.2004.
Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E, et al. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat. 2010;31(6):722–33. https://doi.org/10.1002/humu.21253.
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
YX-Z provided ideas and supervision for the review. ZK-Z wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Both authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Z., Zhao, Y. Progress on the roles of MEF2C in neuropsychiatric diseases. Mol Brain 15, 8 (2022). https://doi.org/10.1186/s13041-021-00892-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-021-00892-6