
Abstract
Alzheimer’s disease (AD) is defined by the excessive accumulation of toxic peptides, such as beta amyloid (Aβ) plaques and intracellular neurofibrillary tangles (NFT). The risk factors associated with AD include genetic mutations, aging, insulin resistance, and oxidative stress. To date, several studies that have demonstrated an association between AD and diabetes have revealed that the common risk factors include insulin resistance, sleep disturbances, blood brain barrier (BBB) disruption, and altered glucose homeostasis. Many researchers have discovered that there are mechanisms common to both diabetes and AD. AD that results from insulin resistance in the brain is termed “type 3 diabetes”. Melatonin synthesized by the pineal gland is known to contribute to circadian rhythms, insulin resistance, protection of the BBB, and cell survival mechanisms. Here, we review the relationship between melatonin and type 3 diabetes, and suggest that melatonin might regulate the risk factors for type 3 diabetes. We suggest that melatonin is crucial for attenuating the onset of type 3 diabetes by intervening in Aβ accumulation, insulin resistance, glucose metabolism, and BBB permeability.
Similar content being viewed by others

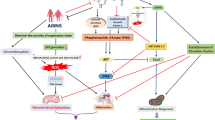

Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder that is characterized by the abnormal aggregation and accumulation of toxic peptides resulting in beta amyloid (Aβ) plaques and intracellular neurofibrillary tangles (NFT) [1]. According to recent reports, the number of patients with AD will be over 13.8 million by 2050, which will place a tremendous burden on society globally [2,3,4]. The onset of AD is linked to various causes, such as genetic mutations [5, 6], sex [7], lipid metabolism [8,9,10,11], aging [12,13,14], and diet [9, 15]. AD pathology results from excessive oxidative stress, synaptic loss, neuronal cell death, impaired insulin signaling, and abnormal glucose metabolism [16,17,18]. Cohort studies have demonstrated that type 2 diabetes (T2DM) increases the risk of dementia and results from common risk factors associated with dementia, including insulin resistance and hyperglycemia [19]. Many patients with metabolic diseases, such as cardiovascular disease, diabetes, and obesity, are reported to have a progressive decline in cognitive function, leading to the development of AD [20, 21]. One meta-analysis showed that diabetes significantly increases the risk for AD in elderly people [22]. Owing to the common risk factors between diabetes and AD, recent studies have proposed that AD is a brain-specific type of diabetes, which they termed “type 3 diabetes” [17, 23,24,25].
Melatonin (N-acetyl-5-methoxytryptamine) is mainly secreted as a neurohormone by the pineal gland [26]. It plays a role in various physiological functions, including circadian rhythm regulation, antioxidant activities, and the regulation of mitochondrial function [27,28,29,30]. Given that sleep disorders frequently occur in up to 45% of patients with AD [31,32,33], melatonin is an important hormone for the treatment of AD since it corrects abnormal sleep patterns [34, 35]. In AD, melatonin levels are decreased in the cerebrospinal fluid (CSF) compared to those in the normal population [36, 37]. Several studies have demonstrated that melatonin reduces Aβ accumulation [38], tau hyperphosphorylation [39], synaptic dysfunction [40], and blood brain barrier (BBB) permeability [41]. Moreover, melatonin attenuates insulin resistance [42], and regulates glucose homeostasis [43, 44]. In this review, we summarize the therapeutic functions of melatonin in type 3 diabetes from various perspectives.
The risk factors for diabetes contribute to the onset and progression of Alzheimer’s disease
Insulin resistance leads to cognitive decline
Diabetes is characterized by insulin resistance, diminished pancreatic beta-cell function, and abnormally high glucose levels [45]. Diabetes is commonly classified into two types, namely, type 1 (T1DM) and T2DM [45]. T2DM occurs more frequently in the global population than T1DM and is accompanied by insulin resistance, hyperglycemia, cognitive decline, and impaired circadian rhythms [46, 47]. T2DM is known to be associated with cognitive impairments [48], and is commonly used as an index for the development of vascular dementia [49], and AD [50, 51]. The high prevalence of central nervous system (CNS) diseases in patients with diabetes has already been revealed by global reports [52,53,54,55]. The onset and progression of AD is associated with the capacity of the brain to utilize glucose for energy production [56, 57]. In the CNS, insulin signaling plays central roles in the cognitive dysfunction found in AD [58]. Insulin is known to be neuroprotective and has powerful effects on memory [59]. Previous studies have shown that deficiencies in insulin receptors (IRs) in the brain, a factor implicated in insulin resistance, leads to memory dysfunction [18, 60]. IRs are localized in cerebral regions, such as the hippocampus, amygdala, and septum [61, 62]. AD patients show an 80% reduction in IRs in their brains compared to normal subjects [17]. Consequently, insulin signaling is abnormal [63]. Some studies have demonstrated that the hippocampus regulates the consolidation of memory via insulin signaling [64, 65]. Based on this evidence, decreased insulin levels were subsequently found in the CSF of patients with AD and mild cognitive impairment (MCI) [60, 66, 67]. Aβ accumulation, abnormalities in the cholinergic system, tau hyperphosphorylation, and damage to neuronal cells contributes to impaired insulin signaling [68, 69]. Insulin receptors deficiency in the AD brain results in insulin resistance in AD neuropathology [18, 70]. For these reasons, reduced levels of insulin receptor genes may contribute to the progression of AD [23, 71]. Moreover, tau pathology in AD is mediated by impaired tau gene expression owing to the attenuation in insulin signaling [72, 73]. Insulin resistance in the AD brain reduces the phosphorylation of phosphoinositol-3-kinase (PI3K), and Akt [72, 73], which ordinarily function to promote neuronal growth and survival, and promotes GABAergic transmission involved in learning and memory [74], and blocks the accumulation of Aβ [75]. Additionally, insulin resistance increases the activation of glycogen synthase kinase (GSK-3) [76, 77], which is related to the hyperphosphorylation of tau and the acceleration of tau misfolding [78]. Indeed, owing to deficiencies in insulin, the change of GSK-3 activity leads to the hyperphosphorylation of tau [79], perhaps unsurprising given what we know about the aberrant activation of GSK-3β and the resultant Aβ accumulation and tau protein phosphorylation [80, 81]. Moreover, several clinical studies have demonstrated a positive correlation between diabetes and AD [57, 82], and suggested that the central reasons for this include aberrant insulin signaling and dementia [58, 83,84,85]. In in vivo studies, an AD mouse model showed insulin resistance [24], reduced glucose metabolism, oxidative stress, and cognitive impairments [86] following injections of streptozotocin (STZ). In addition, insulin resistance leads to hippocampal neuronal loss owing to amyloid neurotoxicity [68], reduced glucose uptake by inhibiting the expression of glucose transporters in cell membrane [87], and accelerated amyloid aggregation during early AD [88]. Consequently, insulin resistance and impaired insulin signaling are significantly related to tau hyperphosphorylation and Aβ deposition in AD, and ultimately contribute to cognitive decline [69] (Fig. 1).

Insulin resistance triggers cognitive decline. Insulin resistance increases p-GSK3β phosphorylation, tau hyperphosphorylation, Aβ aggregation and reduces p-AKT phosphorylation, leading to cognitive deficits
Hyperglycemia triggers BBB disruption leading to cognitive dysfunction
According to previous studies, hyperglycemia in T2DM leads to cognitive dysfunction [89,90,91]. An abnormal glycemic condition is one of the main causes of BBB breakdown in patients with diabetes [92, 93]. Several studies demonstrated that the loss of tight junction proteins which make up the BBB and the activation of matrix metalloproteinases (MMPs) was shown in hyperglycemia in vivo model [94] and in patients [95, 96]. The BBB is comprised of brain endothelial cells lining the cerebral microvessels with astrocytic end-feet processes. The BBB endothelium is characterized by specific transmembrane transport systems that control the trafficking of small molecules in and out of the brain parenchyma [97]. Glucose, the primary energetic source in the brain, can cross the BBB through transporter proteins, such as facilitative sodium independent transporters (e.g., the glucose transporter [GLUT]) [98, 99]. One animal study has shown downregulation of BBB glucose transporters in hyperglycemic mice compared to wild-type mice [100]. In chronic hyperglycemia conditions, GLUT1 and GLUT3 expression was attenuated in diabetic animal brain and subsequently aberrant GLUT’s expression triggers neuronal cell damage [100]. In addition, many studies have reported that the BBB in the diabetic brain has increased permeability owing to the activation of hypoxia-inducible factor-1α (HIF-1α) and increased levels of vascular endothelial growth factor (VEGF) [101, 102]. Hyperglycemia promotes the production of reactive oxygen species (ROS) [103, 104] and downregulates glucose transporters in brain endothelial cells [105]. Moreover, hyperglycemia aggravates amyloid toxicity, independent of insulin resistance [106]. Numerous studies have demonstrated that diet-induced hyperglycemia triggers an increase in BBB permeability and BBB damage [107]. The expression of IgG as the marker of BBB permeability was increased and tight junction proteins were attenuated in a hyperglycemia model [107]. In AD, BBB disruption promotes tau hyperphosphorylation [108, 109]. BBB disruption decreases the expression of glucose transporters [110], promotes ROS production [111] and increases infiltration of inflammatory mediators [112]. Tau aggregation is associated with increase of inflammation [112] and reduction of glucose transporters [110]. In addition, BBB dysfunction in AD contributes to Aβ clearance, activates glial cells, and aggravates inflammation by recruiting leukocytes to the brain [113]. Given this evidence, hyperglycemia-induced BBB disruption might play an important role in the onset and progression of AD (Fig. 2).

Hyperglycemia leads to cognitive decline. Hyperglycemia aggravate BBB breakdown, and increase the generation of ROS, inflammatory response, and Aβ aggregation, ultimately leading to cognitive decline
Melatonin in AD
Melatonin has been shown to have neuroprotective effects in a mouse model of AD [114, 115], since it attenuates Aβ accumulation and synaptic dysfunction by stabilizing the mitochondria function and inhibiting DNA damage [38, 40]. Melatonin controls several molecular signaling pathways, such as PI3/Akt/GSk3β and hemooxygenase-1 [39, 116, 117], and free radical scavenging mechanisms [118, 119] in the AD brain. A recent study demonstrated that melatonin improves synapse dysfunction via the Notch1/Hes1 signaling pathway in the hippocampus [120]. Another study suggested that melatonin inhibits apoptotic mediators and promotes pro-survival signaling in a model of AD [121]. An animal study demonstrated that chronic melatonin treatment for 30 days improves memory impairments in the AD mouse model [117]. Moreover, in patients with AD, melatonin levels were significantly decreased in the serum and CSF, and levels of melatonin were considered as a candidate risk factor for diagnosis of AD [37, 122]. Clinically, melatonin and its agonist have been regarded as treatments for AD [123, 124]. As mentioned above, melatonin has the potential to attenuate AD pathology via numerous mechanisms including PI3K/Akt/GSK3β [37] and Notch1 signaling [120], and RAGE/NF-κB/JNK signaling pathway [117]. Future study of the specific mechanisms of melatonin in the CNS is necessary to identify potential therapeutic solutions for AD.
The relationship between melatonin and type 3 diabetes
Melatonin protects cells against Aβ toxicity and inhibits tau hyperphosphorylation
Aβ, the main component of amyloid plaques, is believed to cause memory dysfunction [125]. Melatonin improves soluble Aβ-induced memory dysfunction and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway [120], suggesting that the modulation of Notch1 could restore neurogenesis and cognitive function in AD models [126]. According to the results of an in vivo study, melatonin administration inhibits the expression of amyloid precursor protein-cleaving secretases in the hippocampus [127]. In addition, melatonin attenuates the memory impairments induced by Aβ accumulation in a sporadic AD model [38, 128, 129]. Melatonin inhibits the transcription of β-secretases via the melatonin receptors in SH-SY5Y neuronal cells [130]. Melatonin attenuates Aβ-induced memory dysfunction and tau hyperphosphorylation via the PI3/Akt/GSK3β pathway in the mouse brain [39]. Melatonin suppresses the activity of GSK3β through activation of p-GSK3β (Ser9) in Aβ in vitro model [131]. Moreover, it improves Aβ-induced impairments in hippocampal long-term potentiation (LTP) in rats [132]. Melatonin inhibits superoxide anion production in microglia under conditions of Aβ toxicity [115]. In addition, it inhibits memory dysfunction and tau phosphorylation in rats [133]. Considering the effect of melatonin on Aβ toxicity and tau hyperphosphorylation in AD, melatonin may be a key to improving memory function by suppressing the cell damage induced by Aβ toxicity and tau hyperphosphorylation.
Melatonin protects cells against insulin resistance and hyperglycemia
Diabetes is accompanied by dysregulation of the circadian system [134]. This is interesting given that glucose metabolism is regulated by the circadian system [135, 136]. In animals and humans with diabetes, increased insulin levels and abnormal glucose metabolism triggers aberrant circadian rhythms [42, 137]. One study demonstrated that a reduction of melatonin levels in serum is linked with high insulin levels in T2DM rats [42]. Moreover, Sakotnik et al. suggested that polymorphisms in the melatonin receptor gene are related to fasting blood glucose levels and increases in the prevalence of T2DM [138]. Several genome wide studies have shown that specific single nucleotide polymorphisms of the melatonin receptor 2 (MTNR1B) locus are related to the high glucose levels found in T2DM [139,140,141]. Genome-wide studies have shown that allelic variations in the melatonin receptor 2 (MT2) contribute to the elevations in fasting glucose levels in plasma, insulin resistance, and ultimately the risk for type 2 diabetes [142, 143]. Type 3 diabetes is related to the prevalence of T2DM and results from insulin resistance and hyperglycemia [144,145,146]. Therefore, a method of reducing the cell damage induced by insulin resistance and hyperglycemia is crucial in both diabetes and AD. Melatonin activates the expression of the MT2 receptor, which can inhibit the secretion of insulin from pancreatic β-cells [147, 148]. Numerous studies have shown that melatonin contributes to glucose homeostasis and that low glucose levels are present in patients with T2DM [137, 149]. A recent study has shown that loss of the melatonin receptor contributes to the activation of pancreatic islet hormones, and hepatic glucose transporters (Glut1 and 2) [150]. Melatonin attenuates the glucose-mediated release of insulin from pancreatic cells [151]. The reduction in melatonin secretion induced by nocturnal light exposure is a crucial factor for T2DM development [136, 152, 153]. Furthermore, the melatonin receptor 1 (MT1) is involved in the regulation of glucose homeostasis and stimulates the secretion of insulin to induce glucose uptake [43]. In humans, melatonin administration attenuates glucose tolerance and insulin resistance [44]. Melatonin could suppress mitochondrial dysfunction against insulin resistance in Male Zucker diabetic fatty rats [154]. Furthermore, melatonin attenuates the secretion of pro inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor (TNF)-α, interferon (IFN)-gamma under insulin resistance condition in high fat diet mouse [155]. Melatonin is associated with metabolic pathways involved with the insulin pathway [156,157,158]. The phosphorylation of IRS-1, leading to the activation of phosphoinositide 3-kinase (PI-3 K), and SHP-2 protein was increased by melatonin [159, 160]. In the AD brain, the disturbance of insulin signaling is linked to the senile plaques formation [80, 161]. An impaired insulin receptor signaling triggers the decrease of insulin-mediated activation of PI-3 K/Akt signaling, resulting in hyperactivity of GSK-3 that induces tau hyperphosphorylation and Aβ accumulation [162]. The administration of melatonin rescues insulin receptor mechanisms and increases the activity of PI-3 K/Akt signaling and less Aβ accumulation and less tau hyperphosphorylation [163] (Fig. 3). One study suggested that the lack of melatonin by pinealectomy reduced insulin sensitivity [164]. The reduction of insulin levels in T1DM are linked to high melatonin levels in plasma [165]. Taking these results together, melatonin appears to be involved in the genesis of diabetes [42] accompanied by insulin resistance and high glucose, and may influence the cognitive dysfunction in diabetes-induced AD (Fig. 4). In this sense, melatonin may be a key molecule in the pathogenesis of Type 3 diabetes.

Melatonin restores the disruption of insulin signaling in AD. In insulin resistance condition, melatonin activates PI3K/Akt signaling, leading to the decrease of tau hyperphosphorylation and Aβ accumulation

Melatonin improve cognitive decline by regulating insulin resistance and hyperglycemia. Melatonin controls insulin resistance and glucose metabolism. Melatonin increases the expression of glucose transporters such as Glut1 and Glut2. Also, melatonin increase the secretion of insulin and protects cell damage, and reduces glial reactivity. Finally, melatonin improves cognitive decline in AD brain
Melatonin protects the BBB against hyperglycemia
Several studies have shown that disruption of the BBB is strongly associated with cognitive dysfunction in AD [166, 167]. The BBB is a heterogeneous structure that consists of various cells important for transferring nutrients and oxygen into brain, and disruption of the BBB has been observed in patients with T2DM [168, 169]. Increases in glucose levels in the blood leads to impaired neurovascular coupling [170, 171], and increased vascular permeability [172]. Hyperglycemia-induced increases in BBB permeability lead to cognitive decline and the development of AD [101]. Hyperglycemia-induced ROS results in BBB disruption and triggers cognitive decline [101]. Dysfunction of metabolic pathways, owing to BBB disruption in diabetes, leads to cognitive deficits [173, 174]. In an in vivo study, STZ-induced diabetes results in increased BBB permeability [101]. Impaired BBB function in diabetes may be a strong risk factor for the development of AD [175, 176]. The excessive generation of ROS in T2DM has been shown to increase BBB permeability by changing tight junction protein expression [177, 178]. According to recent studies, melatonin protects BBB integrity in brain microvascular endothelial cells against inflammation [179], and protects against cerebral endothelial cell dysfunction via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 [180]. Moreover, melatonin prevents the increase in BBB permeability by inhibiting matrix metalloproteinase-9 expression [41]. In addition, melatonin protects against the loss of tight junction proteins and BBB disruption by promoting anti-inflammatory and antioxidant mediators, and axonal regrowth [29]. Melatonin reduces the oxidative stress-induced generation of ROS in brain endothelial cells [181], and ameliorates BBB permeability and nitric oxide levels caused by oxidative stress [182, 183]. In addition, melatonin protects against the degradation of tight junction proteins, BBB disruption, serves as an anti-inflammatory and angiogenesis regulator, and promotes axonal regrowth under high glucose conditions [29, 184]. Based on previous reports, melatonin might alleviate BBB breakdown in diabetes-induced AD by inhibiting the loss of tight junction and the increase of BBB permeability (Fig. 5).

Melatonin improves cognitive decline by inhibiting BBB breakdown. Melatonin inhibits the production of ROS and the vascular permeability and increases axonal regrowth. Melatonin suppresses BBB breakdown and enhances cognitive decline
Conclusions and prospects
Diabetes-induced AD has been called “type 3 diabetes” owing to the common risk factors, which include insulin resistance and hyperglycemia. Here, we reviewed the effect of melatonin in type 3 diabetes from various angles. Melatonin influences type 3 diabetes by 1) suppressing Aβ toxicity and tau hyperphosphorylation, 2) controlling insulin resistance and hyperglycemia, and 3) preventing hyperglycemia-induced BBB disruption. Hence, we suggest that melatonin would be a key in attenuating the pathogenesis of type 3 diabetes.
Change history
08 December 2017
In this article, published on 1 August 2017, Fig. 3 contains a typo. In the Correction the incorrect and correct version of Fig. 3 are shown.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Beta amyloid
- BBB:
-
Blood brain barrier
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- GLUT:
-
Glucose transporter
- Glut1:
-
Glucose transporter 1
- GSK-3:
-
Glycogen synthase kinase
- HIF-1α:
-
Hypoxia-inducible factor-1α
- IFN-γ:
-
interferon –gamma
- IL-6:
-
interleukin-6
- IRs:
-
Insulin receptors
- MCI:
-
Mild cognitive impairment
- MT1:
-
Melatonin receptor 1
- NFT:
-
Intracellular neurofibrillary tangles
- PI3K:
-
Phosphoinositol-3-kinase
- ROS:
-
Reactive oxygen species
- STZ:
-
Streptozotocin
- T1DM:
-
Type 1 diabetes
- T2DM:
-
Type 2 diabetes
- TNF-α:
-
tumor necrosis factor-α
- VEGF:
-
Vascular endothelial growth factor
References
Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66.
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet. 2011;377(9770):1019–31.
Alzheimer's Association National Plan Milestone W, Fargo KN, Aisen P, Albert M, Au R, Corrada MM, et al. 2014 report on the milestones for the US National Plan to address Alzheimer's disease. Alzheimers Dement. 2014;10(5 Suppl):S430–52.
Gulland A. Number of people with dementia will reach 65.7 million by 2030, says report. BMJ. 2012;344:e2604.
Zhang G, Xie Y, Wang W, Feng X, Jia J. Clinical characterization of an APP mutation (V717I) in five Han Chinese families with early-onset Alzheimer's disease. J Neurol Sci. 2016; doi:10.1016/j.jns.2016.10.039.
Cuccaro ML, Carney RM, Zhang Y, Bohm C, Kunkle BW, Vardarajan BN, et al. SORL1 mutations in early- and late-onset Alzheimer disease. Neurol Genet. 2016;2(6):e116.
Canerina-Amaro A, Hernandez-Abad LG, Ferrer I, Quinto-Alemany D, Mesa-Herrera F, Ferri C, et al. Lipid raft ER signalosome malfunctions in menopause and Alzheimer's disease. Front Biosci (Schol Ed). 2017;9:111–26.
Dong HK, Gim JA, Yeo SH, Kim HS. Integrated late onset Alzheimer's disease (LOAD) susceptibility genes: cholesterol metabolism and trafficking perspectives. Gene. 2016; doi:10.1016/j.gene.2016.10.022.
Loffler T, Flunkert S, Temmel M, Hutter-Paier B. Decreased plasma Abeta in Hyperlipidemic APPSL transgenic mice is associated with BBB dysfunction. Front Neurosci. 2016;10:232.
Zhou X, Li Y, Shi X, Ma C. An overview on therapeutics attenuating amyloid beta level in Alzheimer's disease: targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am J Transl Res. 2016;8(2):246–69.
Thomas J, Thomas CJ, Radcliffe J, Itsiopoulos C. Omega-3 fatty acids in early prevention of inflammatory neurodegenerative disease: a focus on Alzheimer's disease. Biomed Res Int. 2015;2015:172801.
Domenico FD, Tramutola A, Butterfield DA. Role of 4-Hydroxy-2-Nonenal (Hne) in the pathogenesis of Alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2016; doi:10.1016/j.freeradbiomed.2016.10.490.
Li H, Lv CL, Yang CS, Wei DF, Chen KW, Li SW, et al. SORL1 rs1699102 polymorphism modulates age-related cognitive decline and gray matter volume reduction in non-demented individuals. Eur J Neurol. 2016; doi:10.1111/ene.13182.
Na HK, Kang DR, Kim S, Seo SW, Heilman KM, Noh Y, et al. Malignant progression in parietal-dominant atrophy subtype of Alzheimer's disease occurs independent of onset age. Neurobiol Aging. 2016;47:149–56.
Dominguez LJ, Barbagallo M. Dietary approaches and supplements in the prevention of cognitive decline and Alzheimer's disease. Curr Pharm Des. 2016;22(6):688–700.
Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66(5):631–45.
de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis. 2005;7(1):45–61.
Craft S. Alzheimer disease: insulin resistance and AD--extending the translational path. Nat Rev Neurol. 2012;8(7):360–2.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5(1):64–74.
Huang CC, Chung CM, Leu HB, Lin LY, Chiu CC, Hsu CY, et al. Diabetes mellitus and the risk of Alzheimer's disease: a nationwide population-based study. PLoS One. 2014;9(1):e87095.
Ding J, Strachan MW, Reynolds RM, Frier BM, Deary IJ, Fowkes FG, et al. Diabetic retinopathy and cognitive decline in older people with type 2 diabetes: the Edinburgh type 2 diabetes study. Diabetes. 2010;59(11):2883–9.
Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010;67(6):505–12.
de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2(6):1101–13.
Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J Alzheimers Dis. 2006;9(1):13–33.
Lin B, Hasegawa Y, Takane K, Koibuchi N, Cao C, Kim-Mitsuyama S. High-Fat-Diet Intake Enhances Cerebral Amyloid Angiopathy and Cognitive Impairment in a Mouse Model of Alzheimer's Disease, Independently of Metabolic Disorders. J Am Heart Assoc. 2016;5(6):e003154. doi:10.1161/JAHA.115.003154.
Wiechmann AF, Sherry DM. Role of melatonin and its receptors in the vertebrate retina. Int Rev Cell Mol Biol. 2013;300:211–42.
Chen CY, Logan RW, Ma T, Lewis DA, Tseng GC, Sibille E, et al. Effects of aging on circadian patterns of gene expression in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2016;113(1):206–11.
Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, et al. Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res. 2015;59(4):403–19.
Moretti R, Zanin A, Pansiot J, Spiri D, Manganozzi L, Kratzer I, et al. Melatonin reduces excitotoxic blood-brain barrier breakdown in neonatal rats. Neuroscience. 2015;311:382–97.
Hardeland R, Cardinali DP, Brown GM, Pandi-Perumal SR. Melatonin and brain inflammaging. Prog Neurobiol. 2015;127–128:46–63.
Pistacchi M, Gioulis M, Contin F, Sanson F, Marsala SZ. Sleep disturbance and cognitive disorder: epidemiological analysis in a cohort of 263 patients. Neurol Sci. 2014;35(12):1955–62.
Cipriani G, Lucetti C, Danti S, Nuti A. Sleep disturbances and dementia. Psychogeriatrics. 2015;15(1):65–74.
Song Q, Feng G, Huang Z, Chen X, Chen Z, Ping Y. Aberrant axonal Arborization of PDF neurons induced by Abeta42-mediated JNK activation underlies sleep disturbance in an Alzheimer's model. Mol Neurobiol. 2016; doi:10.1007/s12035-016-0165-z.
Ooms S, Ju YE. Treatment of sleep disorders in dementia. Curr Treat Options Neurol. 2016;18(9):40.
Zhang W, Chen XY, Su SW, Jia QZ, Ding T, Zhu ZN, et al. Exogenous melatonin for sleep disorders in neurodegenerative diseases: a meta-analysis of randomized clinical trials. Neurol Sci. 2016;37(1):57–65.
Wu YH, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer's disease. J Pineal Res. 2005;38(3):145–52.
Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer's changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35(2):125–30.
Rudnitskaya EA, Muraleva NA, Maksimova KY, Kiseleva E, Kolosova NG, Stefanova NA. Melatonin attenuates memory impairment, amyloid-beta accumulation, and neurodegeneration in a rat model of sporadic Alzheimer's disease. J Alzheimers Dis. 2015;47(1):103–16.
Ali T, Kim MO. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3beta pathway in the mouse hippocampus. J Pineal Res. 2015;59(1):47–59.
Stefanova NA, Maksimova KY, Kiseleva E, Rudnitskaya EA, Muraleva NA, Kolosova NG. Melatonin attenuates impairments of structural hippocampal neuroplasticity in OXYS rats during active progression of Alzheimer's disease-like pathology. J Pineal Res. 2015;59(2):163–77.
Alluri H, Wilson RL, Anasooya Shaji C, Wiggins-Dohlvik K, Patel S, Liu Y, et al. Melatonin preserves blood-brain barrier integrity and permeability via matrix metalloproteinase-9 inhibition. PLoS One. 2016;11(5):e0154427.
Peschke E, Frese T, Chankiewitz E, Peschke D, Preiss U, Schneyer U, et al. Diabetic Goto Kakizaki rats as well as type 2 diabetic patients show a decreased diurnal serum melatonin level and an increased pancreatic melatonin-receptor status. J Pineal Res. 2006;40(2):135–43.
Contreras-Alcantara S, Baba K, Tosini G. Removal of melatonin receptor type 1 induces insulin resistance in the mouse. Obesity (Silver Spring). 2010;18(9):1861–3.
Rubio-Sastre P, Scheer FA, Gomez-Abellan P, Madrid JA, Garaulet M. Acute melatonin administration in humans impairs glucose tolerance in both the morning and evening. Sleep. 2014;37(10):1715–9.
Kendall DM, Bergenstal RM. Comprehensive management of patients with type 2 diabetes: establishing priorities of care. Am J Manag Care. 2001;7(10 Suppl):S327–43–quiz S44–8.
Bixler E. Sleep and society: an epidemiological perspective. Sleep Med. 2009;10(Suppl 1):S3–6.
Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 2009;106(11):4453–8.
Mogi M, Horiuchi M. Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circ J. 2011;75(5):1042–8.
Cheng G, Huang C, Deng H, Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies. Intern Med J. 2012;42(5):484–91.
Umegaki H. Neurodegeneration in diabetes mellitus. Adv Exp Med Biol. 2012;724:258–65.
Holscher C. Diabetes as a risk factor for Alzheimer's disease: insulin signalling impairment in the brain as an alternative model of Alzheimer's disease. Biochem Soc Trans. 2011;39(4):891–7.
Tsuruta R, Fujita M, Ono T, Koda Y, Koga Y, Yamamoto T, et al. Hyperglycemia enhances excessive superoxide anion radical generation, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Brain Res. 2010;1309:155–63.
Fukasawa R, Hanyu H, Namioka N, Hatanaka H, Sato T, Sakurai H. Elevated inflammatory markers in diabetes-related dementia. Geriatr Gerontol Int. 2014;14(1):229–31.
Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther. 2012;136(1):82–93.
Shao B, Bayraktutan U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-beta. Diabetes Obes Metab. 2013;15(11):993–9.
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol. 2011;68(1):51–7.
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–38.
de la Monte SM, Longato L, Tong M, Wands JR. Insulin resistance and neurodegeneration: roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr Opin Investig Drugs. 2009;10(10):1049–60.
Nelson TJ, Sun MK, Hongpaisan J, Alkon DL. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur J Pharmacol. 2008;585(1):76–87.
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80.
Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature. 1978;272(5656):827–9.
Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296(4):E581–91.
Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm (Vienna). 1998;105(4–5):423–38.
Zhao WQ, Alkon DL. Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol. 2001;177(1–2):125–34.
Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology. 2001;74(4):270–80.
Gil-Bea FJ, Solas M, Solomon A, Mugueta C, Winblad B, Kivipelto M, et al. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer's disease. J Alzheimers Dis. 2010;22(2):405–13.
Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, et al. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003;278(29):26929–37.
Schioth HB, Craft S, Brooks SJ, Frey WH 2nd, Benedict C. Brain insulin signaling and Alzheimer's disease: current evidence and future directions. Mol Neurobiol. 2012;46(1):4–10.
Ahmed S, Mahmood Z, Zahid S. Linking insulin with Alzheimer's disease: emergence as type III diabetes. Neurol Sci. 2015;36(10):1763–9.
Yin F, Jiang T, Cadenas E. Metabolic triad in brain aging: mitochondria, insulin/IGF-1 signalling and JNK signalling. Biochem Soc Trans. 2013;41(1):101–5.
Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004;490(1–3):115–25.
Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23(18):7084–92.
Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. 2004;101(9):3100–5.
Balasubramanian AS. Role of phosphorylation and dephosphorylation in the functions of mannose-6-phosphate/insulin like growth factor II receptor. Indian J Biochem Biophys. 1997;34(1–2):178–80.
Dricu A, Carlberg M, Wang M, Larsson O. Inhibition of N-linked glycosylation using tunicamycin causes cell death in malignant cells: role of down-regulation of the insulin-like growth factor 1 receptor in induction of apoptosis. Cancer Res. 1997;57(3):543–8.
Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116(Pt 7):1175–86.
Grilli M, Ferrari Toninelli G, Uberti D, Spano P, Memo M. Alzheimer's disease linking neurodegeneration with neurodevelopment. Funct Neurol. 2003;18(3):145–8.
Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278(46):45937–45.
Watson GS, Craft S. The role of insulin resistance in the pathogenesis of Alzheimer's disease: implications for treatment. CNS Drugs. 2003;17(1):27–45.
de la Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res. 2012;9(1):35–66.
de la Monte S, Derdak Z, Wands JR. Alcohol, insulin resistance and the liver-brain axis. J Gastroenterol Hepatol. 2012;27(Suppl 2):33–41.
Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer's disease. Discov Med. 2013;16(90):277–86.
Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer's disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71(3):365–76.
Tong M, de la Monte SM. Mechanisms of ceramide-mediated neurodegeneration. J Alzheimers Dis. 2009;16(4):705–14.
de la Monte SM, Tong M. Mechanisms of nitrosamine-mediated neurodegeneration: potential relevance to sporadic Alzheimer's disease. J Alzheimers Dis. 2009;17(4):817–25.
de la Monte SM, Tong M, Lester-Coll N, Plater M Jr, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer's disease. J Alzheimers Dis. 2006;10(1):89–109.
Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169–78.
Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015;11(5):504–10. e1
Ravona-Springer R, Moshier E, Schmeidler J, Godbold J, Akrivos J, Rapp M, et al. Changes in glycemic control are associated with changes in cognition in non-diabetic elderly. J Alzheimers Dis. 2012;30(2):299–309.
Ravona-Springer R, Heymann A, Schmeidler J, Moshier E, Godbold J, Sano M, et al. Trajectories in glycemic control over time are associated with cognitive performance in elderly subjects with type 2 diabetes. PLoS One. 2014;9(6):e97384.
Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7(2):108–14.
Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2003;74(1):70–6.
Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50(1):202–11.
Chehade JM, Haas MJ, Mooradian AD. Diabetes-related changes in rat cerebral occludin and zonula occludens-1 (ZO-1) expression. Neurochem Res. 2002;27(3):249–52.
de la Monte SM. Therapeutic targets of brain insulin resistance in sporadic Alzheimer's disease. Front Biosci (Elite Ed). 2012;4:1582–605.
de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer's disease. Drugs. 2012;72(1):49–66.
Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–85.
Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298(2):E141–5.
Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain and Development. 2009;31(7):545–52.
Hou WK, Xian YX, Zhang L, Lai H, Hou XG, Xu YX, et al. Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin Med J. 2007;120(19):1704–9.
Huber JD, VanGilder RL, Houser KA. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am J Physiol Heart Circ Physiol. 2006;291(6):H2660–8.
Acharya NK, Levin EC, Clifford PM, Han M, Tourtellotte R, Chamberlain D, et al. Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis. 2013;35(1):179–98.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–70.
Okouchi M, Okayama N, Alexander JS, Aw TY. NRF2-dependent glutamate-L-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia- induced brain endothelial cell apoptosis. Curr Neurovasc Res. 2006;3(4):249–61.
Duelli R, Maurer MH, Staudt R, Heiland S, Duembgen L, Kuschinsky W. Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res. 2000;858(2):338–47.
Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam study. Neurology. 2010;75(22):1982–7.
Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, et al. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer's disease. J Gerontol A Biol Sci Med Sci. 2014;69(10):1212–26.
Kovac A, Zilkova M, Deli MA, Zilka N, Novak M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis. 2009;18(4):897–906.
Blair LJ, Frauen HD, Zhang B, Nordhues BA, Bijan S, Lin YC, et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun. 2015;3:8.
Sabzichi M, Samadi N, Mohammadian J, Hamishehkar H, Akbarzadeh M, Molavi O. Sustained release of melatonin: a novel approach in elevating efficacy of tamoxifen in breast cancer treatment. Colloids Surf B Biointerfaces. 2016;145:64–71.
Zetner D, Andersen LP, Rosenberg J. Pharmacokinetics of alternative administration routes of melatonin: a systematic review. Drug Res (Stuttg). 2016;66(4):169–73.
El-Gammal MY, Salem AS, Anees MM, Tawfik MA. Clinical and radiographic evaluation of immediate loaded dental implants with local application of melatonin: a preliminary randomized controlled clinical trial. J Oral Implantol. 2016;42(2):119–25.
Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer's disease. Neurobiol Dis. 2016; doi:10.1016/j.nbd.2016.07.007.
Olcese JM, Cao C, Mori T, Mamcarz MB, Maxwell A, Runfeldt MJ, et al. Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J Pineal Res. 2009;47(1):82–96.
Zhou J, Zhang S, Zhao X, Wei T. Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J Pineal Res. 2008;45(2):157–65.
Buendia I, Egea J, Parada E, Navarro E, Leon R, Rodriguez-Franco MI, et al. The melatonin-N,N-dibenzyl(N-methyl)amine hybrid ITH91/IQM157 affords neuroprotection in an in vitro Alzheimer's model via hemo-oxygenase-1 induction. ACS Chem Neurosci. 2015;6(2):288–96.
Ali T, Badshah H, Kim TH, Kim MO. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J Pineal Res. 2015;58(1):71–85.
Galano A, Tan DX, Reiter RJ. On the free radical scavenging activities of melatonin's metabolites. AFMK and AMK J Pineal Res. 2013;54(3):245–57.
Hardeland R, Tan DX, Reiter RJ. Kynuramines, metabolites of melatonin and other indoles: the resurrection of an almost forgotten class of biogenic amines. J Pineal Res. 2009;47(2):109–26.
Zhang S, Wang P, Ren L, Hu C, Bi J. Protective effect of melatonin on soluble Abeta1-42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimers Res Ther. 2016;8(1):40.
Gutierrez-Cuesta J, Tajes M, Jimenez A, Coto-Montes A, Camins A, Pallas M. Evaluation of potential pro-survival pathways regulated by melatonin in a murine senescence model. J Pineal Res. 2008;45(4):497–505.
Wu YH, Feenstra MG, Zhou JN, Liu RY, Torano JS, Van Kan HJ, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88(12):5898–906.
Cardinali DP, Vigo DE, Olivar N, Vidal MF, Furio AM, Brusco LI. Therapeutic application of melatonin in mild cognitive impairment. Am J Neurodegener Dis. 2012;1(3):280–91.
Wilhelmsen M, Amirian I, Reiter RJ, Rosenberg J, Gogenur I. Analgesic effects of melatonin: a review of current evidence from experimental and clinical studies. J Pineal Res. 2011;51(3):270–7.
Thal DR, Walter J, Saido TC, Fandrich M. Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer's disease. Acta Neuropathol. 2015;129(2):167–82.
Mozaffari S, Abdollahi M. Melatonin, a promising supplement in inflammatory bowel disease: a comprehensive review of evidences. Curr Pharm Des. 2011;17(38):4372–8.
Mukda S, Panmanee J, Boontem P, Govitrapong P. Melatonin administration reverses the alteration of amyloid precursor protein-cleaving secretases expression in aged mouse hippocampus. Neurosci Lett. 2016;621:39–46.
Polimeni G, Esposito E, Bevelacqua V, Guarneri C, Cuzzocrea S. Role of melatonin supplementation in neurodegenerative disorders. Front Biosci (Landmark Ed). 2014;19:429–46.
Di Domenico F, Barone E, Perluigi M, Butterfield DA. Strategy to reduce free radical species in Alzheimer's disease: an update of selected antioxidants. Expert Rev Neurother. 2015;15(1):19–40.
Panmanee J, Nopparat C, Chavanich N, Shukla M, Mukda S, Song W, et al. Melatonin regulates the transcription of betaAPP-cleaving secretases mediated through melatonin receptors in human neuroblastoma SH-SY5Y cells. J Pineal Res. 2015;59(3):308–20.
Devine C. Melatonin: a non-benzodiazepine hypnotic in an elderly blind female with dementia. Ther Adv Psychopharmacol. 2011;1(3):89–90.
Liu XJ, Yuan L, Yang D, Han WN, Li QS, Yang W, et al. Melatonin protects against amyloid-beta-induced impairments of hippocampal LTP and spatial learning in rats. Synapse. 2013;67(9):626–36.
Zhu LQ, Wang SH, Ling ZQ, Wang DL, Wang JZ. Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J Pineal Res. 2004;37(2):71–7.
Pulimeno P, Mannic T, Sage D, Giovannoni L, Salmon P, Lemeille S, et al. Autonomous and self-sustained circadian oscillators displayed in human islet cells. Diabetologia. 2013;56(3):497–507.
Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Curr Biol. 2013;23(5):372–81.
Fonken LK, Nelson RJ. The effects of light at night on circadian clocks and metabolism. Endocr Rev. 2014;35(4):648–70.
Peschke E, Stumpf I, Bazwinsky I, Litvak L, Dralle H, Muhlbauer E. Melatonin and type 2 diabetes - a possible link? J Pineal Res. 2007;42(4):350–8.
Sakotnik A, Liebmann PM, Stoschitzky K, Lercher P, Schauenstein K, Klein W, et al. Decreased melatonin synthesis in patients with coronary artery disease. Eur Heart J. 1999;20(18):1314–7.
Staiger H, Machicao F, Schafer SA, Kirchhoff K, Kantartzis K, Guthoff M, et al. Polymorphisms within the novel type 2 diabetes risk locus MTNR1B determine beta-cell function. PLoS One. 2008;3(12):e3962.
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. 2009;41(1):89–94.
Lyssenko V, Nagorny CL, Erdos MR, Wierup N, Jonsson A, Spegel P, et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet. 2009;41(1):82–8.
Andersson EA, Holst B, Sparso T, Grarup N, Banasik K, Holmkvist J, et al. MTNR1B G24E variant associates with BMI and fasting plasma glucose in the general population in studies of 22,142 Europeans. Diabetes. 2010;59(6):1539–48.
Sparso T, Bonnefond A, Andersson E, Bouatia-Naji N, Holmkvist J, Wegner L, et al. G-allele of intronic rs10830963 in MTNR1B confers increased risk of impaired fasting glycemia and type 2 diabetes through an impaired glucose-stimulated insulin release: studies involving 19,605 Europeans. Diabetes. 2009;58(6):1450–6.
Kothari V, Luo Y, Tornabene T, O'Neill AM, Greene MW, Thangiah G, et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta. 2016; doi:10.1016/j.bbadis.2016.10.006.
FangFang LH, Qin T, Li M, Ma S. Thymol improves high-fat diet-induced cognitive deficits in mice via ameliorating brain insulin resistance and upregulating NRF2/HO-1 pathway. Metab Brain Dis. 2016; doi:10.1007/s11011-016-9921-z.
Guo C, Zhang S, Li JY, Ding C, Yang ZH, Chai R, et al. Chronic hyperglycemia induced via the heterozygous knockout of Pdx1 worsens neuropathological lesion in an Alzheimer mouse model. Sci Rep. 2016;6:29396.
Stumpf I, Muhlbauer E, Peschke E. Involvement of the cGMP pathway in mediating the insulin-inhibitory effect of melatonin in pancreatic beta-cells. J Pineal Res. 2008;45(3):318–27.
Stumpf I, Bazwinsky I, Peschke E. Modulation of the cGMP signaling pathway by melatonin in pancreatic beta-cells. J Pineal Res. 2009;46(2):140–7.
Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41(1):77–81.
Bazwinsky-Wutschke I, Bieseke L, Muhlbauer E, Peschke E. Influence of melatonin receptor signalling on parameters involved in blood glucose regulation. J Pineal Res. 2014;56(1):82–96.
Peschke E, Bahr I, Muhlbauer E. Melatonin and pancreatic islets: interrelationships between melatonin, insulin and glucagon. Int J Mol Sci. 2013;14(4):6981–7015.
Gottlieb DJ, Punjabi NM, Newman AB, Resnick HE, Redline S, Baldwin CM, et al. Association of sleep time with diabetes mellitus and impaired glucose tolerance. Arch Intern Med. 2005;165(8):863–7.
Yaggi HK, Araujo AB, McKinlay JB. Sleep duration as a risk factor for the development of type 2 diabetes. Diabetes Care. 2006;29(3):657–61.
Agil A, El-Hammadi M, Jimenez-Aranda A, Tassi M, Abdo W, Fernandez-Vazquez G, et al. Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J Pineal Res. 2015;59(1):70–9.
Cano Barquilla P, Pagano ES, Jimenez-Ortega V, Fernandez-Mateos P, Esquifino AI, Cardinali DP. Melatonin normalizes clinical and biochemical parameters of mild inflammation in diet-induced metabolic syndrome in rats. J Pineal Res. 2014;57(3):280–90.
Piccinetti CC, Migliarini B, Olivotto I, Simoniello MP, Giorgini E, Carnevali O. Melatonin and peripheral circuitries: insights on appetite and metabolism in Danio Rerio. Zebrafish. 2013;10(3):275–82.
Ostrowska Z, Kos-Kudla B, Swietochowska E, Marek B, Kajdaniuk D, Ciesielska-Kopacz N. Influence of pinealectomy and long-term melatonin administration on GH-IGF-I axis function in male rats. Neuro Endocrinol Lett. 2001;22(4):255–62.
Vriend J, Sheppard MS, Borer KT. Melatonin increases serum growth hormone and insulin-like growth factor I (IGF-I) levels in male Syrian hamsters via hypothalamic neurotransmitters. Growth Dev Aging. 1990;54(4):165–71.
Fasshauer M, Klein J, Ueki K, Kriauciunas KM, Benito M, White MF, et al. Essential role of insulin receptor substrate-2 in insulin stimulation of Glut4 translocation and glucose uptake in brown adipocytes. J Biol Chem. 2000;275(33):25494–501.
Kaburagi Y, Satoh S, Tamemoto H, Yamamoto-Honda R, Tobe K, Veki K, et al. Role of insulin receptor substrate-1 and pp60 in the regulation of insulin-induced glucose transport and GLUT4 translocation in primary adipocytes. J Biol Chem. 1997;272(41):25839–44.
O'Neill C, Kiely AP, Coakley MF, Manning S, Long-Smith CM. Insulin and IGF-1 signalling: longevity, protein homoeostasis and Alzheimer's disease. Biochem Soc Trans. 2012;40(4):721–7.
Hildreth KL, Van Pelt RE, Schwartz RS. Obesity, insulin resistance, and Alzheimer's disease. Obesity (Silver Spring). 2012;20(8):1549–57.
Hardeland R, Cardinali DP, Srinivasan V, Spence DW, Brown GM, Pandi-Perumal SR. Melatonin--a pleiotropic, orchestrating regulator molecule. Prog Neurobiol. 2011;93(3):350–84.
Zanquetta MM, Seraphim PM, Sumida DH, Cipolla-Neto J, Machado UF. Calorie restriction reduces pinealectomy-induced insulin resistance by improving GLUT4 gene expression and its translocation to the plasma membrane. J Pineal Res. 2003;35(3):141–8.
Peschke E, Wolgast S, Bazwinsky I, Ponicke K, Muhlbauer E. Increased melatonin synthesis in pineal glands of rats in streptozotocin induced type 1 diabetes. J Pineal Res. 2008;45(4):439–48.
Zeevi N, Pachter J, McCullough LD, Wolfson L, Kuchel GA. The blood-brain barrier: geriatric relevance of a critical brain-body interface. J Am Geriatr Soc. 2010;58(9):1749–57.
Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J NeuroImmune Pharmacol. 2006;1(3):223–36.
Lovell MA, Markesbery WR. Oxidative damage in mild cognitive impairment and early Alzheimer's disease. J Neurosci Res. 2007;85(14):3036–40.
Osawa A, Maeshima S, Shimamoto Y, Maeshima E, Sekiguchi E, Kakishita K, et al. Relationship between cognitive function and regional cerebral blood flow in different types of dementia. Disabil Rehabil. 2004;26(12):739–45.
Fouillioux C, Contreras F, Lares M, Cano R, Leal E, Arraiz N, et al. Metabolic and hemodynamic markers of endothelial dysfunction in patients with hypertension and patients with type 2 diabetes during the cold pressor test. Am J Ther. 2008;15(4):389–96.
Riva CE, Logean E, Falsini B. Visually evoked hemodynamical response and assessment of neurovascular coupling in the optic nerve and retina. Prog Retin Eye Res. 2005;24(2):183–215.
Oltman CL, Kleinschmidt TL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Treatment of cardiovascular dysfunction associated with the metabolic syndrome and type 2 diabetes. Vasc Pharmacol. 2008;48(1):47–53.
Ryan CM, Geckle MO, Orchard TJ. Cognitive efficiency declines over time in adults with type 1 diabetes: effects of micro- and macrovascular complications. Diabetologia. 2003;46(7):940–8.
Ryan CM. Diabetes, aging, and cognitive decline. Neurobiol Aging. 2005;26(Suppl 1):21–5.
Nelson PT, Smith CD, Abner EA, Schmitt FA, Scheff SW, Davis GJ, et al. Human cerebral neuropathology of type 2 diabetes mellitus. Biochim Biophys Acta. 2009;1792(5):454–69.
Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med (Berl). 2004;82(8):510–29.
Haorah J, Schall K, Ramirez SH, Persidsky Y. Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: novel mechanism for neurodegeneration associated with alcohol abuse. Glia. 2008;56(1):78–88.
Balakumar P, Chakkarwar VA, Krishan P, Singh M. Vascular endothelial dysfunction: a tug of war in diabetic nephropathy? Biomed Pharmacother. 2009;63(3):171–9.
Jumnongprakhon P, Govitrapong P, Tocharus C, Tocharus J. Melatonin promotes blood-brain barrier integrity in methamphetamine-induced inflammation in primary rat brain microvascular endothelial cells. Brain Res. 1646;2016:182–92.
Jumnongprakhon P, Govitrapong P, Tocharus C, Tocharus J. Inhibitory effect of melatonin on cerebral endothelial cells dysfunction induced by methamphetamine via NADPH oxidase-2. Brain Res. 1650;2016:84–92.
Song J, Kang SM, Lee WT, Park KA, Lee KM, Lee JE. The beneficial effect of melatonin in brain endothelial cells against oxygen-glucose deprivation followed by reperfusion-induced injury. Oxidative Med Cell Longev. 2014;2014:639531.
Turgut M, Erdogan S, Ergin K, Serter M. Melatonin ameliorates blood-brain barrier permeability, glutathione, and nitric oxide levels in the choroid plexus of the infantile rats with kaolin-induced hydrocephalus. Brain Res. 2007;1175:117–25.
Chen TY, Lee MY, Chen HY, Kuo YL, Lin SC, Wu TS, et al. Melatonin attenuates the postischemic increase in blood-brain barrier permeability and decreases hemorrhagic transformation of tissue-plasminogen activator therapy following ischemic stroke in mice. J Pineal Res. 2006;40(3):242–50.
Xie M, Hu A, Luo Y, Sun W, Hu X, Tang S. Interleukin-4 and melatonin ameliorate high glucose and interleukin-1beta stimulated inflammatory reaction in human retinal endothelial cells and retinal pigment epithelial cells. Mol Vis. 2014;20:921–8.
Acknowledgements
This study was supported by the Brain Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning NRF-2016M3C7A1905469 (B.K.) and a grant from 2016R1D1A1B03930394 (J.S.).
Funding
All sources of funding (the Ministry of Science, ICT & Future Planning NRF-2016M3C7A1905469 & - 2016R1D1A1B03930394) for the research declare the support of the funding in collecting references and in writing the manuscript.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
JS contributed to writing the preliminary draft of this manuscript. DJW revised the overall manuscript with a logical argument. BCK revised the manuscript as a whole. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Song, J., Whitcomb, D.J. & Kim, B.C. The role of melatonin in the onset and progression of type 3 diabetes. Mol Brain 10, 35 (2017). https://doi.org/10.1186/s13041-017-0315-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-017-0315-x